

Abstract
Myosin VI has challenged the lever arm hypothesis of myosin movement because of its ability to take ~36-nm steps along actin with a canonical lever arm that seems to be too short to allow such large steps. Here we demonstrate that the large step of dimeric myosin VI is primarily made possible by a medial tail in each monomer that forms a rare single α-helix of ~10 nm, which is anchored to the calmodulin-bound IQ domain by a globular proximal tail. With the medial tail contributing to the ~36-nm step, rather than dimerizing as previously proposed, we show that the cargo binding domain is the dimerization interface. Furthermore, the cargo binding domain seems to be folded back in the presence of the catalytic head, constituting a potential regulatory mechanism that inhibits dimerization.
Myosin VI (M6) is a class of unconventional myosins that translocates along actin filaments to move and localize components within eukaryotic cells. Dimeric M6 undergoes processive motion, with a single molecule taking large, ~36-nm steps along an actin filament and traveling hundreds of nanometers without dissociation1-3. M6 is unique in the myosin superfamily as it moves to the (–) end of actin filaments4, as a result of the addition of a unique insert of ~40 residues that forms a hairpin turn inserted in its lever arm just as it exits the catalytic head at the converter domain5-7.
The processive step of M6 (refs. 1-3,8-11) is similar in size to that of myosin V12-14, even though its lever arm is composed of only two calmodulin light chains15, three times fewer than the six light chains of myosin V. It was therefore unclear whether M6 could work by the lever arm hypothesis16 given the apparent lack of reach. This dilemma was partially resolved when it was shown that the M6 lever arm swings through ~180°6, compared to only ~70° for myosins II and V17,18, thus allowing for a large stroke size despite its short lever arm. This ~180° swing requires an unexpected change in the conformation of the converter, and the movement of the end of the IQ domain should be ~12 nm19. How the M6 reaches the additional ~24 nm to achieve a stride of ~36 nm remains to be understood. One possibility is the presence of a flexible element located C-terminal to the lever arm, presumably just proximal to it, which allows the free head to diffuse forward the additional ~24 nm. This flexibility was suggested to be in the ~70-residue region just proximal to the lever arm, possibly configured as a random coil, with the next ~70 residues or medial tail region forming a coiled coil for dimerization8,20. Here we show that the proximal tail is actually structured, and the medial tail does not dimerize but provides the additional needed reach as a single α-helix, as was suggested by previous work that revealed an analogous structure in the tail of myosin X21.
Whereas full-length M6 is monomeric22, recent studies indicate that M6 loaded onto vesicles is dimeric23,24 and that increasing the effective concentration of M6 leads to dimerization25, showing that dimerization is possible. With the determination that the medial tail does not act as a dimerization domain, the assembly of M6 dimers became perplexing. Here we demonstrate that the cargo binding domain dimerizes the tail, but only in the absence of the M6 head. Additionally, the monomeric full-length protein has the cargo binding domain folded back onto the head, potentially in a regulated state that inhibits dimerization. This is consistent with previous results showing that full-length chicken M6 is monomeric and compact22.
RESULTS
The M6 tail is composed of four distinct domains
The sequence of the ~450 residues immediately following the catalytic head and the known lever arm of M6 can be divided into four regions (Fig. 1 and Supplementary Fig. 1 online). The characteristic heptad repeat pattern of a coiled coil with hydrophobic residues dominating the a and d positions is identifiable only in the first ~70 residues, which we call the proximal tail (PT, Pro835–Glu907; numbering is based on the human sequence). A transition to the second domain, also ~70 residues, occurs somewhere between Glu907 and Gln915. After Gln915, an extended run of alternating charge appears with only a few hydrophobic residues, the medial tail (MT, Glu908–Arg980) (Fig. 1). This alternating-charge pattern abruptly ends at residue Arg980, and the C-terminal tail sequence of ~50 residues just before the cargo binding domain has no obvious primary-sequence motifs; we call this region the distal tail (DT, Ile981–Arg1035). We refer to the fourth and final region of ~250 residues as the cargo binding domain (CBD, Arg1036–Lys1285).
Figure 1.
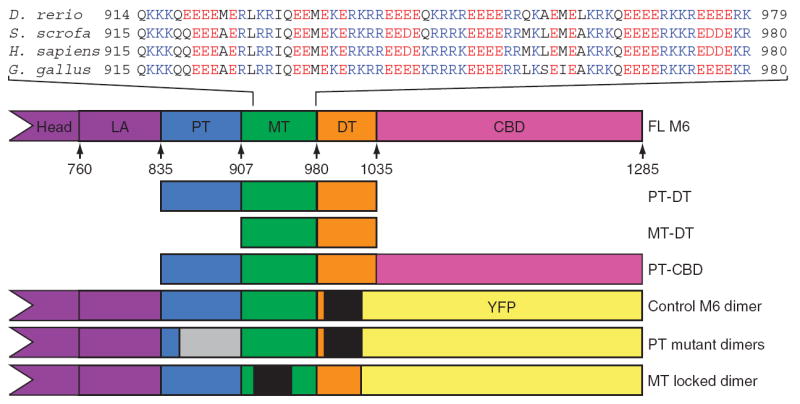
M6 tail domains and experimental constructs. The tail domains of M6 are indicated in the context of the full-length protein, with the position of the first residue of each domain in the human sequence annotated. The calmodulin binding domains are the heretofore known elements of the lever arm (LA); the end of the IQ helix is residue 835. Sequences from the MT from four species are presented to show the repeating-charge pattern, which switches approximately every four residues. The E. coli – expressed tail fragments are shown along with the construct name. The control M6 dimer, the MT locked mutant and the PT mutant constructs were modified by insertion of a GCN4 segment (black regions) to ensure dimerization at the low concentrations used for single-molecule analyses and by replacing the cargo binding domain (CBD) with YFP to provide a specific surface-attachment point via a YFP monoclonal antibody. The location of the randomized PT is gray.
The PT is a folded domain required for long steps
The PT has been proposed to be a random coil acting as the flexible element in the M6 tail that spans the remaining ~24-nm gap20. To explore this model, we created three M6 mutants with altered PT domains (Supplementary Fig. 2a online). The three constructs changed the sequence to varying degrees ranging from point mutations that removed nine core hydrophobic residues, proposed to be a and d positions on the basis of coiled-coil prediction algorithms26 (altered A&D), to randomization of up to 58 residues (850–907 random and 857–907 random). These changes were not expected to impair function if the proximal tail is a random coil, and hence these mutants would be expected to show the same stepping behavior of the control M6 dimer1; however, they would be expected to disrupt any organized tertiary structure. By labeling the motors with fluorophores, the motion of single molecules along actin filaments on the surface can be observed in a total internal reflection fluorescence (TIRF) microscope, allowing the rate of motion to be determined. Each of the three mutant constructs showed velocities in TIRF motility assays at 80 μM ATP that were only ~50% of that of the control M6 dimer (Supplementary Fig. 2b,c). Because velocities can be altered by changing either step size or the rate of stepping, detailed optical trap assays were undertaken for one of the mutants. In this case, two beads are attached to an actin filament, one at each end, and held in two separate optical traps. The filament is then positioned above a single motor on the surface, and individual interactions between the motor and the filament are observed. Processive motors produce a characteristic staircase pattern where the displacement between plateaus measures the step size and the duration of the dwell measures the rate of stepping. These assays showed that the altered velocities result from a shortened step size and not altered stepping rates (Supplementary Fig. 2d,e). Therefore, the model of the PT as a random coil is unlikely.
CD spectroscopy was used to determine what secondary-structure elements were present in the PT. CD revealed that the PT has a classic α-helical spectrum with minima at 222 nm and 208 nm and a maximum at 194 nm (Fig. 2a). We estimated that ~80% of the residues in the PT are in an α-helical conformation. The strong minimum at 222 nm was measured as a function of temperature to track the stability of the PT. It unfolds in a cooperative manner characteristic of compact proteins, where tertiary contacts stabilize the secondary structure (Fig. 2a). These results indicate that the PT is a compact domain with a defined structure. This conclusion agrees with the hydrodynamic radius determination from gel filtration, which is that predicted for a spherical compact protein27 (Table 1). An estimate of the PT structure (residues 835–915, in the human sequence) from the Rosetta 2.2.0 structure prediction algorithm28 consistently yielded (7 out of 10 runs) a three-helix bundle with an end-to-end distance of ~3 nm, which is in good agreement with all experimental data and is considered to be a good estimate of the PT structure (Supplementary Fig. 3a,b online).
Figure 2.
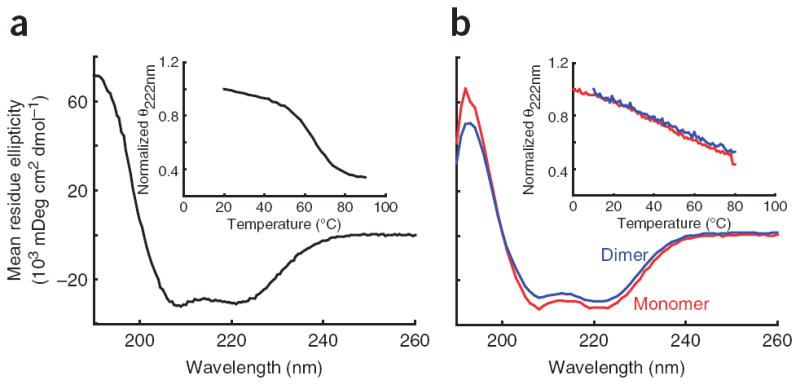
CD spectra for the PT and MT-DT domains. (a) A typical CD spectrum of the PT showing the characteristic double minima of an α-helical protein. Inset, the thermal melt of the PT showing the cooperative melt typical of a folded protein. (b) CD spectra of the MT-DT as a monomer (red) or as an artificial dimer held together by a C-terminal disulfide bridge (blue). Inset, thermal melt curves showing that both constructs have a broad thermal unfolding transition as expected for a single α-helix. The similarity of the spectra and melt curves of the monomer and dimer indicates that no structural changes occur when the MT-DT is placed in conditions mimicking high concentrations. This indicates an inability of the MT-DT to dimerize.
Table 1.
Molecular weights and dimensions of various myosin VI constructs
| Construct | Predicted MW (kDa) | MALDIa (kDa) | MALS (kDa) | Maximum concentration in MALS (μM) | Oligomerization state | Rh (nm) | Rgb (nm) |
|---|---|---|---|---|---|---|---|
| PTc | 9.0 | 9.0 | 16.3 ± 0.1 | 167 | Dimer | 1.6 ± 0.1d, 2.2 ± 0.1d | 2.2 ± 0.2 |
| PT to DT | 22.9 | 23.2 | 23.3 ± 0.1 | 45.6 | Monomer | 4.1 (11.6%)e | 4.4 ± 0.2 |
| MT to DT | 15.0 | 15.2 | 17.1 ± 0.3 | 18.8 | Monomer | 3.6 (12%)e | 4.3 ± 0.2 |
| PT to CBD | 53.5 | 52.9 | 110.8 ± 4.4 | 0.4 | Dimer | 5.7 (13.7%)e | ND |
| Full length | 181.7f | ND | 202.6 ± 4.0 | 1.0 | Monomer | 5.3 ± 0.3d | 4.6 ± 0.2 |
Standard errors are shown, unless otherwise indicated. ND, not determined.
MALDI MS polypeptide chain molecular weight.
Radii of gyration from Guinier analysis from SAXS data.
It should be noted that, at the high concentration used in these studies, PT alone dimerizes, presumably artificially, with a weak Kd of ~150 μM (Supplementary Fig. 3c); when purified as PT-DT, PT remains monomeric even at these high concentrations. The Rh for both monomer and dimer can be measured by gel filtration and are presented, but only the dimer form is seen in MALS and SAXS analyses due to the high concentration of protein used in those measurements (Supplementary Fig. 3a,b).
By gel filtration ± s.d.
By dynamic light scattering with polydispersity in parentheses.
Combined molecular weight of one heavy chain and two calmodulin light chains.
The MT is a single α-helix providing reach for 36-nm steps
Two previous models have been put forward for the MTof M6. First, it has been suggested that at high concentrations M6 dimerizes via coiled-coil formation25. Second, M6 contains a charge-repeat pattern homologous to a domain in myosin X that has been shown to be a single α-helix in solution21, suggesting that M6 may take the same conformation. To test these models, the MT and DT domains were expressed together in Escherichia coli and purified to homogeneity. A clear difference between the two models is that the coiled coil would be dimeric and the single α-helix would be monomeric. Multiple-angle light scattering (MALS) is capable of determining molecular weights in solution by measuring scattering as a function of angle while simultaneously measuring the protein concentration. Using MALS, we found that the MT-DT is monomeric, consistent with the single α-helix hypothesis. This was corroborated by dynamic light scattering, which measures light scattering as a function of time to determine the hydrodynamic radii (Rh). The measured Rh of 3.6 nm is consistent with an extended structure rather than a globular one (1.8 nm predicted27; Table 1). Furthermore, the CD spectrum and thermal melt curves of the MT-DT are those expected of a single α-helix, with a strongly α-helical spectrum and a broad thermal unfolding transition spanning ~70 °C, as shown for charged synthetic peptides29 (Fig. 2b). To directly assess the global structure of the domains, we recorded small-angle X-ray scattering (SAXS) profiles and used ab initio reconstructions to develop low-resolution structural models of the MT-DT construct. The reconstruction for MT-DT (Fig. 3a), as well as that for the PT-DT construct (Fig. 3b), shows a long, bent, slender cylinder, similar in length to that expected with the 70-residue MT configured as a single α-helix.
Figure 3.
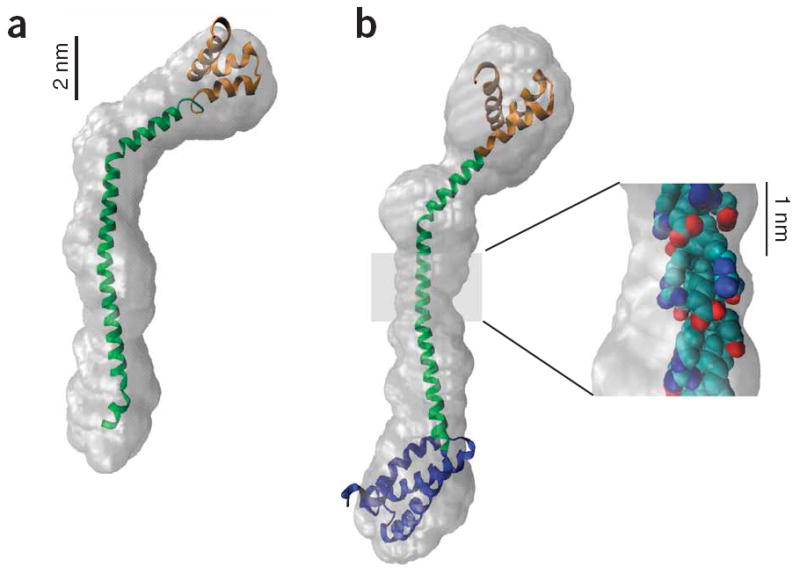
SAXS envelope reconstructions of tail domains. (a) A model of the MT-DT structure with the MT structure (green) derived from the single α-helix prediction and the DT structure (orange) from a Rosetta prediction28. The model was constructed by aligning the peptide backbone manually to the consensus best GASBOR reconstruction and then docking the model into the filtered GASBOR reconstruction envelope using the Situs software package51. (b) A model of the PT-DT structure was constructed by adding a Rosetta prediction for the PT structure (blue) to the N terminus of the MT-DT model in a. This model was docked as above into the filtered GASBOR reconstruction. Note that even though the number of residues has increased from 129 to 201 (56%), the envelope is only ~3 nm longer, indicating a compact PT. Inset, the structure of a segment of the highly charged MT (residues 935–955), with the side chain atoms color coded by charge, revealing bands of charge circling the helix and providing stabilizing i to i+4 charge-charge interactions.
Although the MT-DT is monomeric under these conditions, it is possible that it was not assayed at a high enough concentration to dimerize. To increase the effective concentration, a Gly-Gly-Cys sequence was placed at the C terminus of the natively cysteine-free MT-DT construct, so that two copies could be held in close proximity by a specific disulfide bridge. This mechanism, which has been used in the study of the GCN4 coiled coil30, does not inhibit coiled-coil formation and results in an apparent concentration of at least 4 mM31. The CD spectrum and melt profile of this oxidized dimer were essentially identical to that of the MT-DT monomer (Fig. 2b), showing that even under these conditions the MT-DT is unable to form a dimeric coiled coil.
Constraining the MT eliminates processive stepping
The above results are consistent with the MT providing the reach needed for the 36-nm step of M6. If the MT-DT domain is the long element that allows the two heads of M6 to separate during processive stepping, removing that reach will alter that processive stepping. A GCN4 sequence was placed just beyond the PT-MT boundary at residue 920 (Fig. 1, MT locked). This locks the two copies of M6 together in a coiled coil at this position and eliminates any contribution of the MT to processive stepping. The impact on motion was tested in gliding filament assays, where fluorescently labeled actin filaments glide across a motor-covered surface. This geometry is useful as it can assay both processive and nonprocessive motors, whereas the TIRF motility assay can assay only processive motors. Control M6 dimer at 2 mM ATP moved at 110 ± 21 nm s−1, and MT locked moved at 75 ± 26 nm s−1, significantly different results as assessed by a t-test (n = 20 per group, P-value > 0.001). The 75 nm s−1 velocity is that expected for a stroke size of ~20 nm, which derives from the ~180° swing of the lever arm extended by the globular PT.
Gliding filament assays are capable of testing more than just the velocity of a motor. Other parameters, such as the rate at which filaments land on the surface and the fraction of filaments that move greater than their length, can also be counted. The theoretical framework of the landing and continuous movement assays allows for quantitative predictions of the landing rate and the fraction of filaments that will move greater than their length as a function of motor density and the number of motors it takes to attach a filament to the surface32. We created models in which either one motor (processive movement) or two motors (nonprocessive movement) were needed for filament attachment. Data from the control M6 dimer were more consistent with the processive model, whereas the MT locked construct was more consistent with the nonprocessive model in both assays (Fig. 4a,b). Optical trap assays also indicate that a single motor of MT locked is incapable of producing the characteristic staircases of a processive motor. Instead, single bind-and-release events, characteristic of nonprocessive motors, were seen with a mean displacement of 23 ± 26 nm (Fig. 4c) and kinetics similar to the control M6 dimer with a mean dwell of 0.9 ± 0.02 s. From these data, we conclude that the MT is the primary element in M6 that allows the heads to sample a large space and processively step along the 36-nm pseudorepeat of the actin filament.
Figure 4.
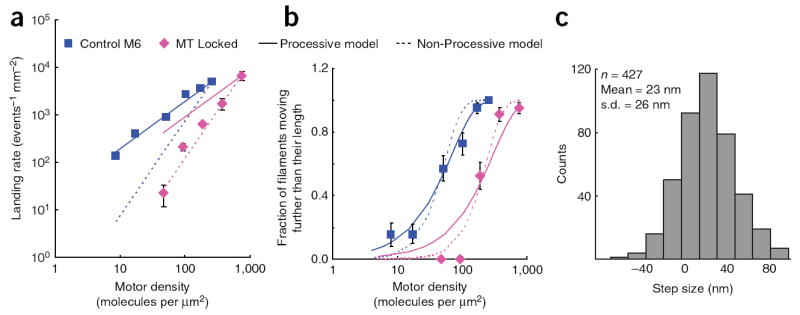
Motility assays for the MT locked mutant compared to control M6 dimer. (a) Plots of log landing rate versus log motor density with lines depicting the processive model and the nonprocessive model superimposed on the data. The control M6 dimer data match the processive model (number of motors for attachment = 1.0), and the MT locked data more closely match the nonprocessive model (number of motors for attachment = 2.0). (b) Plots of the probability of a filament moving further than its length versus log motor density. As described for a, models for processive and nonprocessive movement were superimposed over the data, with the control M6 dimer being in better agreement with the one-motor model (r2 processive model = 0.98, r2 nonprocessive model = 0.95) and the MT locked mutant being in better agreement with the two-motor model (r2 processive model = 0.93, r2 nonprocessive model = 0.97). (c) Histogram of measured nonprocessive displacements for the MT locked mutant in a dual-beam optical trap assay. The mean displacement is indicated and represents the length of the power stroke of the motor. All error bars are standard errors.
The CBD regulates dimerization of M6
Neither the PT nor the MT dimerize at ~1 μM (Table 1). To further explore M6 dimerization, we examined the role of the CBD. Indeed, MALS analysis showed that a construct extending from the beginning of the PT to the end of the CBD is dimeric at concentrations in the micromolar range, which indicates reasonable affinity (Table 1). Because the PT and the MT-DT do not self associate at these concentrations, we conclude that the CBD is needed for this dimerization. However, under the same conditions, the full-length M6 is monomeric (Table 1), in agreement with previous work22. From these results, we conclude that the CBD dimerization must be inhibited by the M6 head. This is corroborated by the compactness of the SAXS envelope of full-length M6 (Fig. 5). If the model for the PT-MT (Fig. 3b) is fused to the structure of the M6 head and a globular CBD, the resultant full-length M6 monomer cannot fit into the experimental SAXS envelope without a high degree of bending (Fig. 5). This implies that the full-length M6 monomer is in a folded-back state, with an interaction occurring between the CBD and the M6 head, and that this interaction inhibits dimerization. It is likely that the binding of cargo to the CBD helps regulate the equilibrium between this folded-back monomeric state and the dimeric form of M6, especially given the orientation of the CBD when bound to the membrane23 and the observation that M6 dimerizes when binding to membrane24.
Figure 5.
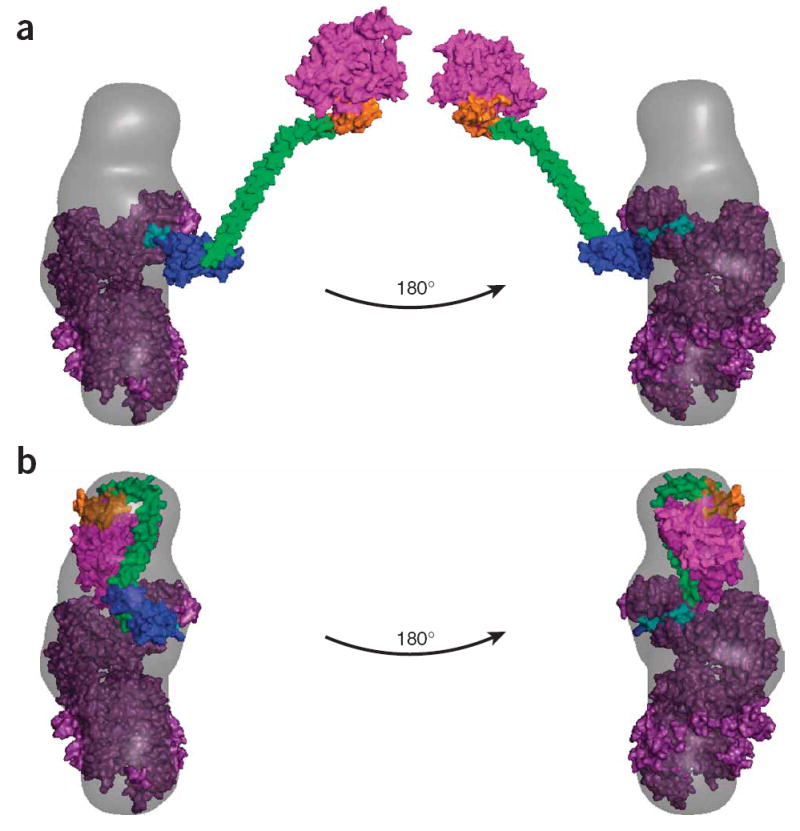
SAXS envelope and models for full-length myosin VI. (a) A model of an extended full-length M6 containing the published post-stroke crystal structure of the catalytic domain5 with the calmodulin-bound unique insert and IQ regions (purple), the PT-DT model from Figure 3b and the Rosetta prediction of the CBD (magenta) docked into the corresponding SAXS envelope. With the head aligned to one end of the reconstruction and the PT fused to the IQ such that it extends the lever arm in the same conformation as in Figure 6, the rest of the tail lies well outside the calculated scattering envelope. (b) A model of an alternate compact state for monomeric M6, with the CBD folded back onto the lever arm calmodulins, docked into the same SAXS envelope as in a.
DISCUSSION
The average spacing between the two heads of the processively stepping M6 dimer is 36 nm, yet the lever arm structure that encompasses the two calmodulin binding domains swinging through an angle of ~180° explains the protein structure for only ~12 nm of this distance19. This leaves a large 24-nm gap that needs to be spanned by relatively few residues. One possibility is that this gap is filled by both the PT and the MT, and the CBD self-associates to hold the two copies of the full-length M6 together. Because PT is a globular folded domain that immediately follows the calmodulin binding domains, the PT could extend the lever arm by as much as ~3 nm. Owing to the ~180° lever arm rotation, this ~3-nm extension would increase the stroke size of the motor by ~6 nm to a total of 18 nm6. This would leave an ~18-nm gap in the stride distance of the two heads of the stepping dimer that must be filled by the MT-DT. The MT, owing to its stable single α-helix, is ~10 nm long, with the DT adding an additional ~3 nm, assuming a somewhat folded DT structure (Fig. 3a). Thus, two copies of MT-DT are more than sufficient to bridge the gap between the two heads and would also allow step sizes larger than 36 nm, as has been reported1-3 (Fig. 6). This model is supported by the linear arrangement of the PT-MT-DT domains suggested by the SAXS envelope (Fig. 3b), where the IQ-based lever arm is simply being extended by an additional rod-like segment.
Figure 6.
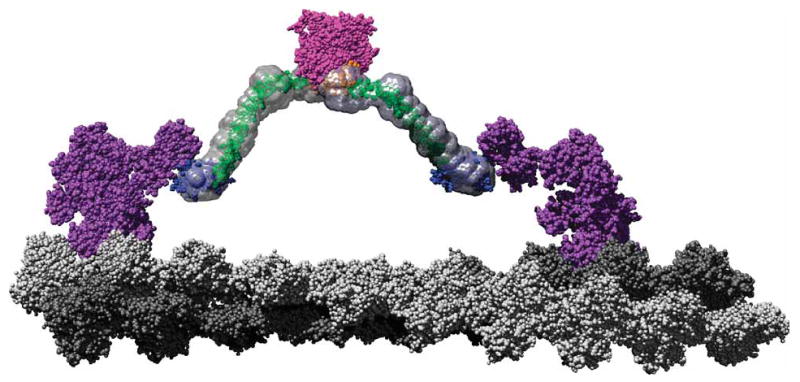
A scale model of a M6 dimer moving along an actin filament. The F-actin–docked myosin model is based on that proposed by Holmes et al.54 Monomers corresponding to the two protofilaments are colored in light and dark gray, respectively, to emphasize the pseudorepeat at 36 nm. Using a structural alignment in PyMOL (http://pymol.sourceforge.net), the post-stroke structure of the M6 head (PDB 2BKI5) was docked onto the filament. The prestroke structure (PDB 2V2619) was also docked onto the actin filament 13 monomers removed from the post-stroke head. These structures along with the associated light chains are shown in purple. The tail model presented in Figure 3b, with the same color scheme, has been fused to the end of the IQ domains such that the PT projects along the same vector as the IQ helix (cyan) and then rotates around the Gly839 to remove steric clashes. This represents an orientation where the PT maximally contributes to the M6 stroke, which is one of many potential angles at which it meets the IQ domain. The cargo binding domains are shown in close association, as their dimerization suggests. The SAXS envelopes were then superimposed on the model to place the data in context of a working motor using Chimera55. This model shows that the proposed roles for the tail domains are clearly compatible with a 36-nm processive step for a M6 dimer.
It should be emphasized that the angle between the folded PT and the canonical calmodulin-IQ lever arm is unknown, and the PT may be orientated such that it does not add its full-length to the working stroke of the motor. In fact, it is even possible that the PT may actually shorten the stroke if it is able to fold back onto the IQ-bound calmodulin, in an arrangement made possible because the MT is long enough to bridge the gap between the heads without help from the PT. This model of the PT folding back onto the IQ-calmodulin lever arm actually leads to a better fit into the monomeric full-length SAXS envelope (Fig. 5). This raises the possibility that the purpose of the PT may not be to extend the lever arm, but rather to mediate interactions with other proteins. This fits with the observation that the Drosophila melanogaster protein Echinoid was identified as binding M6 at the PT33. It is also possible that the PT is part of the binding site for the M6 CBD in the folded-back structure in Figure 5. The role of the PT is most likely to be some hybrid of the two orientations described, emphasizing the need for additional structural studies to completely characterize the IQ and PT junction and the role of the PT.
The ~10-nm single α-helical structure of the MT is remarkable, with few other natural examples21,34,35 in known protein structures. The characteristic i to i+4 alternating-charge pattern (Fig. 3b, inset) has been shown previously to stabilize α-helical peptides in solution34; it has also been noted that the MT is similar to the single stable α-helix that has been described for myosin X and is proposed to extend the myosin X lever arm21. Studies on the material properties of other single α-helices indicate that the Young’s modulus for lateral bending is ~2.5 GPa36,37, which would lead to a persistence length of ~12 nm. This suggests that the MT would not be a rigid rod at the scale of these lengths, which is supported by the observation that synthetic α-helical peptides have radii of gyration shorter than expected for rigid rods38. This seems contradictory with the MT adding to the stroke of M6, but the previously studied peptides have only one stabilizing i to i+4 interaction per helical turn, whereas the MT has as many as four of these interactions, potentially enhancing its rigidity and enabling it to extend the lever arm. A rigid MT is supported by the Rh measurement of 4.3 nm, which is in agreement with the predicted 3.7-nm radius of gyration from the suggested structure, indicating that the static model has similar properties to the dynamic object in solution. The hypothesis of the rigid tail domain is made more plausible with the recent observations that the M6 head can adopt many lever angles that could explain the highly variable step size of M6 (refs. 19,39), without requiring a flexible tail domain. Further experimentation is needed to clarify the material properties of the MT and its exact contribution to the M6 stroke.
With the PT and MT spanning the distance between the two heads, the CBD does not need to add to the reach of the M6 dimer. Here we show that at ~1 μM the PT-DT is monomeric, whereas the CBD can initiate dimerization of the tail. Notably, it was previously reported that, when the effective M6 concentration is increased using monoclonal antibody binding to the C terminus or rigor binding of M6 monomers to F-actin, a construct ending at residue 992 in the DT seems to dimerize, as judged by rotary-shadowing EM and single-molecule processive movement25. Remarkably, this dimerization persists under dilute conditions (less than nanomolar concentrations) used in single-molecule assays. Further experiments are necessary to explain the persistent dimerization observed in their construct, given that our studies show that PT-DT does not dimerize at 1 μM. However, our experiments suggest that the CBD is the preferred dimerization interface for the M6 helical tail, as depicted in our model (Fig. 6).
We find that the full-length M6 structure is compact, resulting from a folding back of the CBD onto the M6 head. It has recently been observed that a high degree of flexibility exists in the M6 motor domain7,19,39. This flexibility exists before the beginning of the lever arm and presumably relates to the ability of the lever arm to adopt multiple angles relative to the head. This variability in the lever arm angle could influence the folded-back state that we observe, but it is clear that a separate new flexibility is needed to create the folded-back state proposed here. It is unclear from where this new flexibility originates, given that all of the tail fragments seem rigid. One possibility is that the joints between the domains are flexible, enabling the domains to bend with respect to each other. Another possibility is that the energy released upon CBD docking to another part of the structure may be enough to induce a change in one of the tail domains, creating a bend. Thus, there could be two possible IQ-PT connection angles: one in the monomeric folded state (Fig. 5) and another in the extended dimeric state (Fig. 6). Alternatively, the MT might rearrange as there are several places where the repeating-charge pattern is less robust and the MT is presumably more likely to bend.
We propose a model for M6 regulation where the cytoplasmic CBD dimerization is inhibited by binding of the CBD to the M6 head, and upon binding to cargo the CBD releases the head and initiates dimerization. This allows us to draw a scale model of stepping M6 (Fig. 6) that successfully accounts for the behavior of the motor and assigns the necessary functions to each domain.
METHODS
Expression of tail domain constructs
Further details for all of the methods are included in the Supplementary Methods online. The temperature was 22 °C, unless otherwise noted. The appropriate sequences from the human M6 cDNA were cloned into a modified pET28a vector (EMD Chemicals). The modified vector contained a hexahistidine (His6) tag, maltose binding protein (MBP) and a tobacco etch virus (TEV) protease cleavage site 5′ to the cloning site. Proteins were expressed in E. coli Rosetta (DE3) cells (EMD Chemicals) at room temperature, with all subsequent purification steps being conducted at 4 °C. Cells were lysed by sonication. Lysates were clarified by centrifugation at 100,000g for 30 min and bound to nickel–nitrilotriacetic acid (Ni-NTA) resin (Qiagen). The eluted proteins were dialyzed in the presence of TEV protease (1:100 by weight) overnight to cleave the His6 and MBP portions from the M6 domains. M6 domains were further purified by running the cleavage reaction over a Ni-NTA column to remove His6-containing fragments, running a MonoQ column (GE Healthcare) and running a Superdex 200 column (GE Healthcare) in either CD buffer (10 mM phosphate, pH 7.4, 25 mM NaCl) or scattering buffer (10 mM phosphate, pH 7.4, 150 mM NaCl).
Expression of motor domain–containing constructs
The porcine control M6 dimer construct described previously40 was modified to create the mutants, and full-length M6 was made using the human M6 cDNA. M6 altered A&D, 850–907 random, and 857–907 random sequences were synthesized by DNA 2.0 (Menlo Park). The altered protein sequences are described in Supplementary Figure 2. M6 MT locked was made with a GCN4 sequence inserted in place of residues 919–950 of the control dimer. All sequences were placed under the control of the polyhedron promoter of the pFastBac Dual vector (Invitrogen), with sea urchin calmodulin (P05934) under the control of the p10 promoter. Recombinant baculoviruses were created as per the Invitrogen protocol. Proteins were purified as described previously40. Briefly, Sf-9 cells were infected with recombinant virus at 28 °C, with all subsequent purification steps being conducted at 4 °C. Cell lysates were clarified by centrifugation for 1 h at 200,000g, and proteins were bound to Flag M2–affinity gel (Sigma) and eluted with Flag peptide. For scattering analysis, full-length M6 was gel filtered using a Superdex 200 column in scattering buffer. For all constructs, absorbance at 280 nm in 6 M guanidinium chloride was used to determine the protein concentration.
Circular dichroism
CD spectra were acquired using an Aviv 62DS instrument (Aviv Biomedical) with a 1-mm path-length cell in CD buffer. Spectra were taken at 10 °C, with data collected every 1 nm with a 20-s averaging time; results are the average of 3 repeat scans. Concentrations ranged from 1–17 μM. Melt data was collected every 1 °C with a 30-s averaging time and a 2-min equilibration. In all cases, the reverse melt showed at least 90% reversibility. The percent helical content was determined as described previously41.
Multiple-angle light scattering
In solution, molecular weights were determined using a size-exclusion chromatography system coupled to a MALS detector. Protein concentrations were determined with an Optilab rEX refractive index detector, and scattering was detected with a Dawn 18 angle MALS light-scattering instrument (Wyatt Technology Corporation).
MALDI-TOF mass spectrometry
Protein samples at ~10 μM in CD buffer were mixed in a 2:1 ratio with sinapinic acid and spotted onto a stainless steel MALDI plate. Analysis was conducted on a Voyager-DE RP (Applied Biosystems) instrument in positive linear mode.
Dynamic light scattering
Dynamic light scattering measurements were made using a DynaPro instrument (Protein Solutions) running Dynamics version 6 software. Samples at 1–10 mg ml−1 in scattering buffer were assayed at 25 °C with an acquisition interval of 10 s. Samples were spun at ~15,000g for 10 min immediately before analysis. Results were derived using a regularization fit and with PBS buffer settings.
Analytical gel filtration
Proteins were concentrated to 10–300 μM and loaded onto a 25-ml Superdex 200 equilibrated in scattering buffer at 4 °C. The elution volume was determined from the average of at least three runs and converted to Rh.
SAXS measurements
SAXS measurements were carried out at the XOR/BESSRC undulator beamline 12-ID of the Advanced Photon Source, using a sample-detector distance of 2 m and CCD detector readout (MAR USA). The data were collected using a custom-made sample cell42 at an X-ray energy of 12 keV. Details of the beamline are as described previously42-44.
Samples were centrifuged at 10,000g for 10 min before measurement, and three 0.5-s exposures were obtained. Data were image corrected, normalized by incident flux and circularly averaged. The three profiles for each condition were averaged to improve signal quality. Buffer profiles were collected using identical procedures and subtracted for background correction, and the data showed no signs of radiation damage, based on comparison of consecutive scattering profiles from the same sample (data not shown).
Small-angle X-ray scattering data analysis
Scattering intensities as a function of the momentum transfer were obtained at different protein concentrations. The SAXS profiles for the MT-DT, PT-DT and full-length M6 constructs are superimposable after scaling by forward-scattering intensity, suggesting that there are no detectable aggregation or interparticle interference effects (Supplementary Fig. 4 online). Radii of gyration were determined from Guinier analysis of the small momentum transfer scattering data45 (Supplementary Fig. 5 online). The radii of gyration obtained from Guinier analysis agree within experimental errors with the values from the real-space distribution function P(r) computed using the regularized transform method implemented in the program GNOM46.
Small-angle X-ray scattering structure reconstructions
The programs DAM-MIN47 and GASBOR48 were used to construct three-dimensional bead models that fit the scattering data (Supplementary Fig. 6 online). Both programs use a simulated annealing procedure and a compactness criterion. We performed ten independent DAMMIN and GASBOR runs for each scattering profile, using default parameters, the ‘slow’ mode for DAMMIN, no symmetry assumptions (P1 symmetry) and the full recorded scattering profiles. The models resulting from independent runs were superimposed and compared using the program SUPCOMB49 based on the normalized spatial discrepancy (NSD) criterion. Models with NSD values <1 are considered similar. For all data presented in the main text, the ten independent repeat runs yielded models with pairwise NSD values <1, indicating that the algorithms converged reproducibly to similar structures. The ten independent structures for each scattering profile were subsequently averaged, and ‘filtered’ consensus models were computed using the program DAMAVER with default settings50. Consensus models constructed with DAMMIN and GASBOR gave similar results. For visualization, the reconstructed bead models were converted to electron-density maps with the program Situs51.
In vitro motility, landing and continuous movement assays
Assays were conducted as described previously32. Briefly, motors were attached with monoclonal anti-GFP antibody (Chemicon MAB3580) to nitrocellulose-coated cover slips. Tetramethyl rhodamine iso-thiocyanate (TRITC) phalloidin-labeled actin was observed using a total internal reflection microscope52. Movies were scored for the number of filaments landing and moving more than 0.5 μm, the fraction of filaments running greater than their length and the velocity with which filaments moved.
Total internal reflection fluorescence motility assays
Assays were conducted as described previously52. Briefly, actin biotinylated at 1 in approximately every 25 monomers was attached to a coverslip using steptavidin and nonspecifically absorbed biotinylated BSA. Motors were labeled at a 1:1 ratio with anti-GFP antibody conjugated with multiple Cy3 fluorophores to enhance signal were assayed at 80 μM ATP. Movies were collected on the microscope described in ref. 52 and analyzed using imageJ (National Institutes of Health).
Optical trap assays
Assays were conducted as described previously3,53 with a few exceptions, notably that trapping was done without feedback. Briefly, motors were attached with monoclonal anti-GFP antibody (Chemicon, MAB3580) to nitrocellulose-coated cover slips. Actin dumbbells were formed between two trapping beads and pulled taught. Positional data was collected at 10 kHz and trap stiffness ranged from 0.006–0.012 pN nm−1. Binding events were determined by eye using a drop in the positional variance of the beads and the bead-to-bead . Transitions between processive steps were also scored by eye.
Supplementary Material
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
Acknowledgments
We thank A. Dunn, Z. Bryant and N. Geething of Stanford University for technical help with protein purification and motility assays, critical discussions and manuscript review; H.L. Sweeney of the University of Pennsylvania for plasmids; K. Holmes of the Max Planck Institute for Medical Research, Heidelberg, for the acto-myosin PDB model; T. Fenn of Stanford University for technical help with MALS analysis and graphical presentation; T. Purcell of the University of California, San Fransisco, for help with model building; S. Seifert of the Advanced Photon Source; R. Fenn of Stanford University for help with SAXS data collection; and S. Patel of Stanford University for MALDI analysis. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. B.J.S. is partially supported by grant T32 GM008294; S.D. is supported by grant PO1 GM066275; and J.A.S. is supported by grant GM33289, all from the US National Institutes of Health.
Footnotes
Published online at http://www.nature.com/nsmb/
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/
References
- 1.Rock RS, et al. Myosin VI is a processive motor with a large step size. Proc Natl Acad Sci USA. 2001;98:13655–13659. doi: 10.1073/pnas.191512398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishikawa S, et al. Class VI myosin moves processively along actin filaments backward with large steps. Biochem Biophys Res Commun. 2002;290:311–317. doi: 10.1006/bbrc.2001.6142. [DOI] [PubMed] [Google Scholar]
- 3.Altman D, Sweeney HL, Spudich JA. The mechanism of myosin VI translocation and its load-induced anchoring. Cell. 2004;116:737–749. doi: 10.1016/s0092-8674(04)00211-9. [DOI] [PubMed] [Google Scholar]
- 4.Wells AL, et al. Myosin VI is an actin-based motor that moves backwards. Nature. 1999;401:505–508. doi: 10.1038/46835. [DOI] [PubMed] [Google Scholar]
- 5.Menetrey J, et al. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature. 2005;435:779–785. doi: 10.1038/nature03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bryant Z, Altman D, Spudich JA. The power stroke of myosin VI and the basis of reverse directionality. Proc Natl Acad Sci USA. 2007;104:772–777. doi: 10.1073/pnas.0610144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park H, et al. The unique insert at the end of the myosin VI motor is the sole determinant of directionality. Proc Natl Acad Sci USA. 2007;104:778–783. doi: 10.1073/pnas.0610066104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okten Z, Churchman LS, Rock RS, Spudich JA. Myosin VI walks hand-over-hand along actin. Nat Struct Mol Biol. 2004;11:884–887. doi: 10.1038/nsmb815. [DOI] [PubMed] [Google Scholar]
- 9.Yildiz A, et al. Myosin VI steps via a hand-over-hand mechanism with its lever arm undergoing fluctuations when attached to actin. J Biol Chem. 2004;279:37223–37226. doi: 10.1074/jbc.C400252200. [DOI] [PubMed] [Google Scholar]
- 10.Balci H, Ha T, Sweeney HL, Selvin PR. Interhead distance measurements in myosin VI via SHRImP support a simplified hand-over-hand model. Biophys J. 2005;89:413–417. doi: 10.1529/biophysj.105.060608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ali MY, et al. Unconstrained steps of myosin VI appear longest among known molecular motors. Biophys J. 2004;86:3804–3810. doi: 10.1529/biophysj.103.037416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mehta AD, et al. Myosin-V is a processive actin-based motor. Nature. 1999;400:590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- 13.Purcell TJ, Morris C, Spudich JA, Sweeney HL. Role of the lever arm in the processive stepping of myosin V. Proc Natl Acad Sci USA. 2002;99:14159–14164. doi: 10.1073/pnas.182539599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veigel C, Wang F, Bartoo ML, Sellers JR, Molloy JE. The gated gait of the processive molecular motor, myosin V. Nat Cell Biol. 2002;4:59–65. doi: 10.1038/ncb732. [DOI] [PubMed] [Google Scholar]
- 15.Bahloul A, et al. The unique insert in myosin VI is a structural calcium-calmodulin binding site. Proc Natl Acad Sci USA. 2004;101:4787–4792. doi: 10.1073/pnas.0306892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spudich JA. The myosin swinging cross-bridge model. Nat Rev Mol Cell Biol. 2001;2:387–392. doi: 10.1038/35073086. [DOI] [PubMed] [Google Scholar]
- 17.Shih WM, Gryczynski Z, Lakowicz JR, Spudich JA. A FRET-based sensor reveals large ATP hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell. 2000;102:683–694. doi: 10.1016/s0092-8674(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 18.Forkey JN, Quinlan ME, Shaw MA, Corrie JE, Goldman YE. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature. 2003;422:399–404. doi: 10.1038/nature01529. [DOI] [PubMed] [Google Scholar]
- 19.Menetrey J, Llinas P, Mukherjea M, Sweeney HL, Houdusse A. The structural basis for the large powerstroke of myosin VI. Cell. 2007;131:300–308. doi: 10.1016/j.cell.2007.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Rock RS, et al. A flexible domain is essential for the large step size and processivity of myosin VI. Mol Cell. 2005;17:603–609. doi: 10.1016/j.molcel.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 21.Knight PJ, et al. The predicted coiled-coil domain of myosin 10 forms a novel elongated domain that lengthens the head. J Biol Chem. 2005;280:34702–34708. doi: 10.1074/jbc.M504887200. [DOI] [PubMed] [Google Scholar]
- 22.Lister I, et al. A monomeric myosin VI with a large working stroke. EMBO J. 2004;23:1729–1738. doi: 10.1038/sj.emboj.7600180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altman D, Goswami D, Hasson T, Spudich JA, Mayor S. Precise positioning of myosin VI on endocytic vesicles in vivo. PLoS Biol. 2007;5:e210. doi: 10.1371/journal.pbio.0050210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spudich G, et al. Myosin VI targeting to clathrin-coated structures and dimerization is mediated by binding to Disabled-2 and PtdIns(4,5)P2. Nat Cell Biol. 2007;9:176–183. doi: 10.1038/ncb1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park H, et al. Full-length myosin VI dimerizes and moves processively along actin filaments upon monomer clustering. Mol Cell. 2006;21:331–336. doi: 10.1016/j.molcel.2005.12.015. [DOI] [PubMed] [Google Scholar]
- 26.Berger B, et al. Predicting coiled coils by use of pairwise residue correlations. Proc Natl Acad Sci USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11:739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bonneau R, et al. De novo prediction of three-dimensional structures for major protein families. J Mol Biol. 2002;322:65–78. doi: 10.1016/s0022-2836(02)00698-8. [DOI] [PubMed] [Google Scholar]
- 29.Marqusee S, Baldwin RL. Helix stabilization by Glu-…Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci USA. 1987;84:8898–8902. doi: 10.1073/pnas.84.24.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Shea EK, Rutkowski R, Kim PS. Evidence that the leucine zipper is a coiled coil. Science. 1989;243:538–542. doi: 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- 31.Zaman MH, Berry RS, Sosnick TR. Entropic benefit of a cross-link in protein association. Proteins. 2002;48:341–351. doi: 10.1002/prot.10165. [DOI] [PubMed] [Google Scholar]
- 32.Rock RS, Rief M, Mehta AD, Spudich JA. In vitro assays of processive myosin motors. Methods. 2000;22:373–381. doi: 10.1006/meth.2000.1089. [DOI] [PubMed] [Google Scholar]
- 33.Lin HP, et al. Cell adhesion molecule Echinoid associates with unconventional myosin VI/Jaguar motor to regulate cell morphology during dorsal closure in Drosophila. Dev Biol. 2007;311:423–433. doi: 10.1016/j.ydbio.2007.08.043. [DOI] [PubMed] [Google Scholar]
- 34.Wang E, Wang CL. (i, i + 4) Ion pairs stabilize helical peptides derived from smooth muscle caldesmon. Arch Biochem Biophys. 1996;329:156–162. doi: 10.1006/abbi.1996.0204. [DOI] [PubMed] [Google Scholar]
- 35.Kuhlman B, Yang HY, Boice JA, Fairman R, Raleigh DP. An exceptionally stable helix from the ribosomal protein L9: implications for protein folding and stability. J Mol Biol. 1997;270:640–647. doi: 10.1006/jmbi.1997.1146. [DOI] [PubMed] [Google Scholar]
- 36.Howard J. Mechanics of Motor Proteins and the Cytoskeleton. xvi. Sinauer Associates; Sunderland: 2001. p. 367. [Google Scholar]
- 37.Idiris A, Alam MT, Ikai A. Spring mechanics of α-helical polypeptide. Protein Eng. 2000;13:763–770. doi: 10.1093/protein/13.11.763. [DOI] [PubMed] [Google Scholar]
- 38.Zagrovic B, Jayachandran G, Millett IS, Doniach S, Pande VS. How large is an α-helix? Studies of the radii of gyration of helical peptides by small-angle X-ray scattering and molecular dynamics. J Mol Biol. 2005;353:232–241. doi: 10.1016/j.jmb.2005.08.053. [DOI] [PubMed] [Google Scholar]
- 39.Sun Y, et al. Myosin VI walks “wiggly” on actin with large and variable tilting. Mol Cell. 2007;28:954–964. doi: 10.1016/j.molcel.2007.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De La Cruz EM, Ostap EM, Sweeney HL. Kinetic mechanism and regulation of myosin VI. J Biol Chem. 2001;276:32373–32381. doi: 10.1074/jbc.M104136200. [DOI] [PubMed] [Google Scholar]
- 41.Chen YH, Yang JT, Chau KH. Determination of the helix and β form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- 42.Lipfert J, Millett IS, Seifert S, Doniach S. Sample holder for small-angle X-ray scattering static and flow cell measurements. Rev Sci Instrum. 2006;77:046108. [Google Scholar]
- 43.Beno M, et al. Basic energy sciences synchrotron radiation center undulator sector at the advanced photon source. Nucl Instrum Methods Phys Res A. 2001;467–468:690–693. [Google Scholar]
- 44.Seifert S, Winans RE, Tiede DM, Thiyagarajan P. Design and performance of a ASAXS instrument at the Advanced Photon Source. J Appl Cryst. 2000;33:782–784. [Google Scholar]
- 45.Guinier A. La diffraction des rayons X aux tres petits angles: Application á l’etude de phenomenes ultramicroscopiques. Ann Phys (Paris) 1939;12:161–237. [Google Scholar]
- 46.Svergun D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr. 1992;25:495–503. [Google Scholar]
- 47.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Svergun DI, Petoukhov MV, Koch MH. Determination of domain structure of proteins from X-ray solution scattering. Biophys J. 2001;80:2946–2953. doi: 10.1016/S0006-3495(01)76260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kozin MB, Svergun DI. Automated matching of high- and low-resolution structural models. J Appl Crystallogr. 2001;34:33–41. [Google Scholar]
- 50.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wriggers W, Chacon P. Using Situs for the registration of protein structures with low-resolution bead models from X-ray solution scattering. J Appl Crystallogr. 2001;34:773–776. [Google Scholar]
- 52.Churchman LS, Okten Z, Rock RS, Dawson JF, Spudich JA. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proc Natl Acad Sci USA. 2005;102:1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rice SE, Purcell TJ, Spudich JA. Building and using optical traps to study properties of molecular motors. Methods Enzymol. 2003;361:112–133. doi: 10.1016/s0076-6879(03)61008-6. [DOI] [PubMed] [Google Scholar]
- 54.Holmes KC, Angert I, Kull FJ, Jahn W, Schroder RR. Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide. Nature. 2003;425:423–427. doi: 10.1038/nature02005. [DOI] [PubMed] [Google Scholar]
- 55.Pettersen EF, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.