

Background: Human γ-glutamyltranspeptidase 1 (hGGT1) is a key enzyme in cysteine metabolism and several diseases.
Results: We obtained the high resolution crystal structure of hGGT1.
Conclusion: The structure reveals the molecular basis for differences between the human and bacterial enzymes in autoprocessing and catalytic activity.
Significance: The structure provides a template for the structure-based design of therapeutic inhibitors of hGGT1.
Keywords: Enzyme Structure, Glycoprotein, Hydrolases, Oxidative Stress, Protease, gamma-glutamyltranspeptidase, gamma-Glutamylpeptidyltransferase, gamma-Glutamyltransferase, Anomalous Diffraction, Threonine Peptidase
Abstract
The enzyme γ-glutamyltranspeptidase 1 (GGT1) is a conserved member of the N-terminal nucleophile hydrolase family that cleaves the γ-glutamyl bond of glutathione and other γ-glutamyl compounds. In animals, GGT1 is expressed on the surface of the cell and has critical roles in maintaining cysteine levels in the body and regulating intracellular redox status. Expression of GGT1 has been implicated as a potentiator of asthma, cardiovascular disease, and cancer. The rational design of effective inhibitors of human GGT1 (hGGT1) has been delayed by the lack of a reliable structural model. The available crystal structures of several bacterial GGTs have been of limited use due to differences in the catalytic behavior of bacterial and mammalian GGTs. We report the high resolution (1.67 Å) crystal structure of glutamate-bound hGGT1, the first of any eukaryotic GGT. Comparisons of the active site architecture of hGGT1 with those of its bacterial orthologs highlight key differences in the residues responsible for substrate binding, including a bimodal switch in the orientation of the catalytic nucleophile (Thr-381) that is unique to the human enzyme. Compared with several bacterial counterparts, the lid loop in the crystal structure of hGGT1 adopts an open conformation that allows greater access to the active site. The hGGT1 structure also revealed tightly bound chlorides near the catalytic residue that may contribute to catalytic activity. These are absent in the bacterial GGTs. These differences between bacterial and mammalian GGTs and the new structural data will accelerate the development of new therapies for GGT1-dependent diseases.
Introduction
γ-Glutamyltranspeptidase 1 (GGT12; EC 2.3.2.2) is expressed in bacteria, plants, and animals. In humans, GGT1 (hGGT1, P19440) is a cell surface enzyme that hydrolyzes extracellular γ-glutamyl compounds, including glutathione (γ-Glu-Cys-Gly) and leukotriene C4. hGGT1 contributes to cysteine homeostasis, intracellular redox status, inflammation, and the renal mercapturic acid pathway (1–5). hGGT1 has been implicated as a potentiator of asthma, Parkinson disease, and cardiovascular disease. It is induced in many cancers, contributing to their inherent resistance to alkylating agents and other classes of clinical chemotherapeutic agents (6–10). Elevated hGGT1 activity in the serum is a common diagnostic marker of several diseases, including liver cancer, alcoholic hepatitis, disrupted bile formation, and pancreatic cancer. Despite its clinical importance, a complete characterization of hGGT1 and the development of therapeutic inhibitors have been hindered by the lack of a crystal structure for hGGT1 or any eukaryotic GGT (11).
Currently, the most potent inhibitors of the GGT family of enzymes are broad specificity glutamine substrate mimetics (competitive inhibitors), which have historically proven to be too toxic for use in humans (12–14). Rational improvements on this design have been hampered by unresolved differences in the binding mechanism between human and bacterial GGTs that are not readily discernable from their primary structures. We have recently identified a novel class of uncompetitive species-specific hGGT1 inhibitors that are several orders of magnitude less toxic than the canonical substrate analog, acivicin (15–17). Strategic refinements of the structure of the founding member of this class of hGGT1 inhibitors, OU749, have also been impeded by the lack of an accurate structural model for the human enzyme (or any eukaryotic GGT).
hGGT1 is a member of the N-terminal nucleophile hydrolase superfamily of enzymes (18). Like other members of this superfamily of enzymes, hGGT1 requires autocatalytic activation to become a mature amidase (19). As such, hGGT1 is expressed as a 569-amino acid propeptide that autocleaves into a large subunit (42 kDa, residues 1–380) and a small subunit (20 kDa, residues 381–569). The heterodimer localizes to the cell surface and is tethered to the plasma membrane by a single-pass transmembrane domain (residues 5–26) located at the N terminus of the large subunit. The same residue (Thr-381) that autocleaves the propeptide also serves as the catalytic nucleophile within the active site of the mature enzyme (20, 21).
Under physiological conditions, the primary reaction catalyzed by mammalian hGGT1 is the hydrolysis of γ-glutamyl amide bonds (2, 22, 23). hGGT1 cleaves the γ-glutamyl amide bond of all γ-glutamyl compounds that have been tested (5). The reaction is initiated by a nucleophilic attack by the side chain hydroxyl oxygen of Thr-381 on the γ-glutamyl amide bond. An acyl bond forms between the glutamate portion of the substrate (the δ-carbon of glutamate) and the γ-oxygen of the side chain of Thr-381 as the remainder of the substrate is released. The subsequent hydrolysis of the acyl bond releases glutamate and is the rate-limiting step of the reaction. hGGT1 can also catalyze a transpeptidation reaction in solutions with high concentrations of dipeptides and some amino acids (24). In the transpeptidation reaction, the γ-glutamyl group bound to Thr-381 is transferred to the α-nitrogen of a dipeptide acceptor, thereby forming a new γ-glutamyl compound. The hydrolysis and transpeptidation reactions proceed by a modified ping-pong mechanism (25, 26).
Crystal structures have been reported for one archaeal (Thermoplasma acidophilum; PDB code 2i3o) and four bacterial (Escherichia coli, Helicobacter pylori, Bacillus subtilis, and Bacillus halodurans) GGTs (27–29) (PDB codes 2dbx, 2qm6, 3A75, and 2nlz, respectively). Among these structures, GGT from E. coli (EcGGT; P18956) has the highest amino acid sequence homology to hGGT1 (P19440), with 32% (172 of 533 residues) identity, 53% (280 of 533 residues) similarity, and 4% (20 of 533 residues) gaps (Fig. 1). Despite these similarities, hGGT1 differs from bacterial GGTs in several ways. For example, hGGT1 catalyzes both a hydrolysis and a transpeptidation reaction (16, 17), whereas the characterized bacterial GGTs exhibit comparatively poorer transpeptidation activity (30–33). This suggests that the binding pocket surrounding the active site of hGGT1 differs from the binding pockets in the bacterial GGTs in a manner that is not readily discernible from sequence alignments between the primary structures of these orthologs. Furthermore, hGGT1 is a type II membrane protein tethered to the cell surface by a single-pass transmembrane domain, whereas GGTs in bacteria are untethered and localize to the periplasmic space (30, 34). Finally, hGGT1 has different requirements for autocleavage compared with bacterial GGTs. hGGT1 has seven N-glycosylation sites, and proper folding and autocleavage of the propeptide requires co-translational N-glycosylation (35, 36). The bacterial GGTs with crystal structures lack N-glycosylation, yet their propeptides are properly folded and undergo autocleavage (27–29, 37). Two cysteines (Cys-50 and Cys-74) are conserved in the large subunit of all eukaryotic GGT1s and are required for efficient autocleavage of hGGT1 (38, 39). Both of these sites are alanines in the bacterial GGTs. These differences in protein maturation and function reflect distinct structural features of hGGT1.
FIGURE 1.

Sequence alignment of crystallized GGT orthologs from humans and bacteria. Sequence numbering and secondary structural elements correspond to the human enzyme and are shown above the aligned residues. Identical residues are shaded in black, and similar residues are boxed. Additional shading highlights the positions of the conserved catalytic nucleophile (red), human GGT1 N-glycosylation sites (green), and cysteines that form intramolecular disulfide bonds in the human enzyme (yellow), using the amino acid sequences from human GGT1 (human, NCBI accession number NM_005265.2), E. coli GGT (NP_417904), H. pylori GGT (NP_207909), and B. subtilis GGT (NP_389723).
To improve our understanding of the structure of hGGT1, we have innovatively crystallized the soluble ectodomain (residues 28–569) of deglycosylated hGGT1 and determined its x-ray crystal structure to a resolution of 1.67 Å. Analysis of this active form of the enzyme has provided new insights into the folding, autocleavage, and kinetic activity of hGGT1 and revealed many key structural differences between the human and bacterial GGTs. These structural differences (a) underscore the differences in the catalytic behavior of these orthologous enzymes and (b) explain the difficulties in drug design based upon homology models of hGGT1 derived from the bacterial crystal structures. This new information will serve as an essential launching point for the structure-based design of hGGT1 inhibitors with enhanced therapeutic efficacy.
EXPERIMENTAL PROCEDURES
Expression and Purification of Deglycosylated Human GGT1
We developed a yeast recombinant system for the expression of hGGT1. To express the soluble ectodomain of the enzyme, the transmembrane domain (residues 1–27) was omitted and replaced with a tobacco etch virus (TEV) protease-cleavable polyhistidine tag. The N-terminal His6-tagged hGGT1 (P19440; amino acids 28–569) open reading frame was integrated into the genome of the Pichia pastoris strain X-33 (15). Two 40-ml volumes of BMGY medium (Invitrogen) containing 100 μg ml−1 of Zeocin were inoculated with the transformed X-33 strain and cultured at 30 °C to high density overnight at 250 rpm. These cultures were used to inoculate two 1.5-liter volumes of BMGY medium and were then propagated without an antibiotic at 30 °C until a final A600 of 15 was achieved. The cells were harvested by centrifugation at 1500 × g for 10 min, and the cell pellets were resuspended in two 1.5-liter volumes of pH 7.4 BMMY medium (Invitrogen) supplemented with 1% casamino acids and 1% methanol to induce expression of the recombinant hGGT1 enzyme. The cells were induced for 112 h with shaking (250 rpm) at 30 °C and the addition of 1% methanol at 24 and 72 h to sustain induction. The cells were harvested at 1500 × g for 10 min. The resulting supernatants contained the secreted hGGT1. The supernatants were pooled and filtered through 0.22-μm polyethersulfone bottle top filters (Corning), and the 3 liters of filtrate was concentrated 10-fold. The buffer was changed to binding buffer (25 mm HEPES, pH 8.0, 300 mm NaCl) by tangential flow filtration using a Pellicon PLCTK cassette with a size exclusion cut-off of 30 kDa (Millipore).
The resulting 300 ml of diafiltered medium was supplemented with imidazole to 10 mm and loaded onto Ni-NTA resin (PerfectPro; 1 ml of resin/liter of culture). The column was washed with 10 bed volumes of wash buffer (10 mm imidazole, 25 mm HEPES, pH 8.0, 300 mm NaCl), and the tagged protein was eluted with elution buffer (25 mm HEPES, pH 8.0, 100 mm NaCl, 250 mm imidazole). Eluate fractions were assayed for GGT activity as described previously (15). The active fractions (∼20 mg of total protein) were combined and dialyzed against 4 liters of 25 mm HEPES, pH 7.0, and 30 mm NaCl. The dialysate was then incubated with 300 mega units of Endo Hf (New England Biolabs) at 37 °C for 18 h to remove all but the basal N-acetylglucosamine (GlcNAc) residue from the enzyme. The deglycosylated hGGT1 pool was dialyzed against 4 liters of 25 mm HEPES (pH 8.0) and 100 mm NaCl buffer at 4 °C, and the dialysate was subjected to Ni-NTA purification as described above to remove the cleaved glycans and Endo Hf. The hGGT1-containing fractions were dialyzed against 4 liters of TEV cleavage buffer (25 mm Tris, pH 8.0, 1 mm DTT, 0.5 mm EDTA) at 4 °C. 150 μg of His6-tagged TEV protease was added at 4 °C for 20 h to cleave the hexahistidine tag. TEV-cleaved hGGT1 was separated from the hexahistidine tag and the His-tagged TEV protease by passage over a third Ni-NTA column. The untagged, deglycosylated hGGT1 was then dialyzed at 4 °C against 4 liters of 50 mm HEPES, pH 8.0, and concentrated at 4 °C in a Centriprep concentrator (YM-30, Amicon) to a final concentration of ∼20 mg ml−1. The concentrated protein stock was stored at 4 °C until used for crystallization.
Crystal Structure Studies
Crystals were grown by vapor diffusion with the hanging drop method. The protein stock solution was diluted to 5 mg ml−1 with 50 mm Na-HEPES, pH 8.0, 0.02% (w/v) sodium azide, and 0.5 mm Na-EDTA, pH 8.0. The crystallization drops were assembled by mixing 2 μl of protein solution with 2.0 μl of double-distilled H2O and 1.5 μl of reservoir solution. The crystals appeared in 1–4 weeks at room temperature over reservoir solutions containing 15–25% PEG 3350, 200 mm ammonium chloride, 0.5 mm l-glutamate, and 200 mm sodium cacodylate, pH 6.0, or Na-HEPES, pH 7.5. Similar crystals appeared in either sitting or hanging drops. The crystals were enlarged by adding to the reservoir 10 μl of 100% PEG 400 once and 10 μl of 50% glycerol five times during 2 weeks. The best diffracting crystals (500 × 100 × 30 μm) were isolated from clusters of thin plates. Crystals were cryoprotected by rapid passage through drops made by mixing 1.3 μl of 50% (w/v) PEG 1550 and 0.7 μl of reservoir solution supplemented with glutamate to a final concentration of 10 mm in the cryo drop.
X-ray (λ = 1.1 Å) diffraction data were collected at 100 K at beam line X25 of the National Synchrotron Light Source. The x-ray diffraction data were indexed, integrated, and scaled with XDS (40). The structure was determined by molecular replacement using MOLREP (41) and the crystal structure of the E. coli GGT (PDB code 2DBU) as a search model with twinned data at 2.9 Å and then 2.2 Å resolution. The structure was redetermined with MOLREP, and newly available data from an untwinned crystal (RS values for top solution/next solution were 0.410/0.644 with 47–1.98 Å data). The structure was refined against 1.67 Å x-ray data (Iobs > 0·σ(Iobs)) with PHENIX (42) between rounds of model rebuilding with COOT (43). The final R value was 0.145, and the final Rfree value was 0.174. The anomalous diffraction difference maps were calculated with ANODE (44). The refinement statistics and PDB accession codes are listed in Table 1. Sequence alignments were prepared with ClustalW (45) and ESPRIPT (46). DSSP was used to identify the secondary structure elements (47). Molecular images were made with PyMOL (Schrödinger, LLC). The buried molecular surface areas between the subunits and at intermolecular contacts were determined with the PISA server (48).
TABLE 1.
Data collection and refinement statistics
Values in parentheses refer to the highest resolution shell. All x-ray data were collected with 1.1-Å radiation. 5% of the reflections were randomly assigned to the test set and were excluded from refinement.
| Parameters | hGGT1-Glu | hGGT1-iodide |
|---|---|---|
| PDB code | 4DGX | 4GG2 |
| Data collection | ||
| Space group | C2221 | C2221 |
| Unit cell lengths a, b, c (Å) | 105.5, 125.2, 104.5 | 105.7, 126.7, 104.6 |
| Resolution range (Å) | 1.67–47.1 | 2.20–47.2 |
| High resolution bin (Å) | (1.67–1.76) | (2.20–2.26) |
| No. of unique reflections (test set) | 78,680 (3970) | 34,297 1864) |
| Completeness (%) | 98.3 (90.2) | 95.3 (73.2) |
| Multiplicity | 6.3 (4.6) | 18.5 (10.6) |
| Rmerge (%)a | 7.6 (74.0) | 10.8 (54.7) |
| Rmeas(%)b | 8.2 (83.0) | 11.1 (57.4) |
| Rp.i.m.(%)c | 3.2 (13.0) | 2.5 (16.3) |
| Mean I/σ(I) | 16.2 (2.1) | 26.2 3.3) |
| B factor from Wilson plot (Å2) | 17.2 | 26.9 |
| Refinement | ||
| Resolution range (Å) | 1.67–47.1 | 2.21–44.0 |
| R factor (%) | 14.5 | 14.0 |
| Free R factor (%) | 17.4 | 18.3 |
| Protein atomsd | 4538 | 4216 |
| Ligand atoms | 5 | 5 |
| Anions | 2 | 10 |
| Water molecules | 712 | 387 |
| Mean B (Å2), subunit A, B | 25.6, 21.0 | 30.5, 28.0 |
| Mean B (Å2), ligand, anions, water | 26.3, 13.7, 33.7 | 28.9, 44.6, 36.5 |
| All-atoms clashscoree | 1.6 | 4.91 |
| Ramachandran plot: favored/outliers (%) | 97.7, 0.19 | 97.4, 0.19 |
| No. of Cβ deviationse | 0 | 0 |
| Rotamer outliers (%) | 0.19 | 2.62 |
| Root mean square deviation from ideal geometry | ||
| Bond lengths (Å) | 0.010 | 0.012 |
| Bond angles (degrees) | 1.261 | 1.343 |
| Maximum likelihood coordinate error (Å) | 0.15 | 0.23 |
a Rmerge (merging R factor) = (ΣhΣl|Ihl − 〈Ih〉|)/(ΣhΣl 〈Ih〉).
b Rmeas (redundancy-independent R factor) = Σh(nh/(nh − 1)) (Σl|Ihl − 〈Ih〉|)/(ΣhΣl 〈Ih〉).
c Rp.i.m. (precision-indicating R factor) = Σh(1/(nh − 1))(Σl|Ihl − 〈Ih〉|)/(ΣhΣl 〈Ih〉).
d This number includes the covalently attached glycans but not the protein hydrogen atoms.
e Detected with MOLPROBITY (57).
Site-directed Mutagenesis
Mutagenesis at site 545 in hGGT1 was done with the QuikChange kit from Stratagene (La Jolla, CA). Full-length wild-type human GGT1 (EC 2.3.2.2) cDNA in a pcDNA3.1(+) plasmid served as the template for PCR mutagenesis (36). HEK293T cells were transiently transfected with either wild type hGGT1 or Q545K hGGT1 (36). The expression levels of hGGT1 were evaluated by Western blot. Equivalent amounts of wild-type or mutant hGGT1 protein were assayed for hydrolysis activity with d-γ-glutamyl-p-nitroanalide (3 mm) as the substrate (36).
RESULTS AND DISCUSSION
Structural Overview
The milligram quantities of hGGT1 required for crystallization made it impractical to purify the enzyme from human tissue, so we expressed a soluble form of hGGT1 in a yeast recombinant system. We showed previously that hGGT1 expressed in yeast is N-glycosylated, autocleaves into the expected large and small subunits, and has the same catalytic activity as the enzyme expressed in human tissues (15). To reduce structural heterogeneity, we cleaved the N-glycans on hGGT1 to leave one GlcNAc at each site. The purified enzyme (Fig. 2) was active (specific activity of 262 units/mg) and formed diffraction quality crystals within 10 days at room temperature in the presence of 0.25 mm l-glutamate. After cryoprotection, complete x-ray diffraction data were collected with synchrotron radiation from a single crystal for each structure (Table 1).
FIGURE 2.
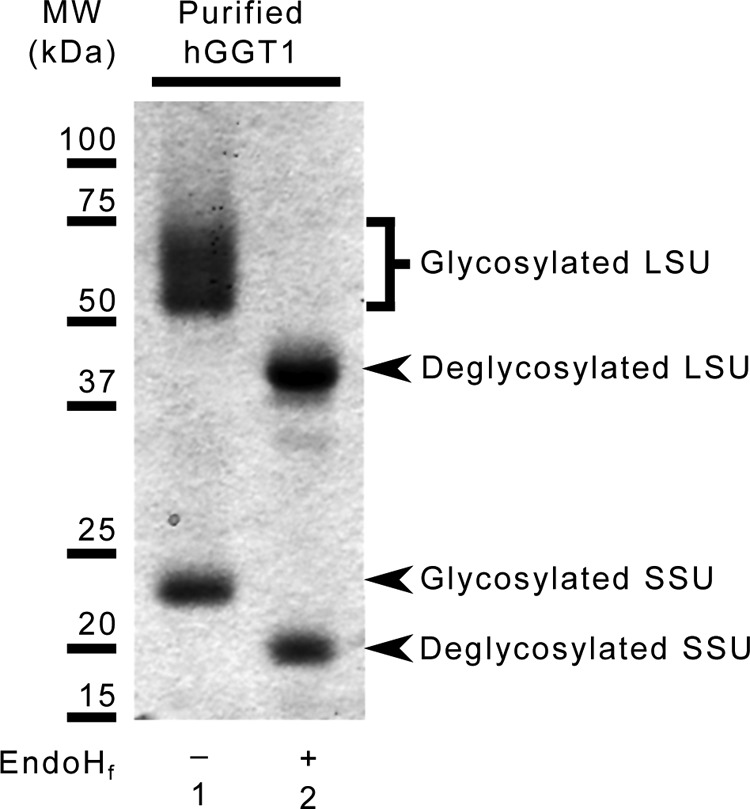
SDS-PAGE analysis of purified hGGT1 used for crystallography. 5 μg of untreated (left lane) and Endo Hf-treated (right lane) hGGT1 were subjected to SDS-PAGE and subsequently silver-stained to visualize the relative migration patterns of the glycosylated and deglycosylated large (LSU) and small (SSU) subunits.
The structure of hGGT1 was determined by molecular replacement with the crystal structure of the EcGGT (PDB code 2DBU) as a search model. The structure was refined with diffraction data to a resolution of 1.67 Å (Table 1). This resolution limit is within the range of resolution limits achieved for previous crystal structures of the bacterial enzymes (27–29).
The hGGT1 crystals contained one heterodimer per asymmetric unit. Other investigators have reported that the bacterial GGTs crystallized with two heterodimers per asymmetric unit in three space groups (17–19). The paired bacterial GGT heterodimers are related by an approximately 2-fold rotation and form two contact surfaces (27–29). In hGGT1, these two contacts were made with two different heterodimers rather than with one heterodimer and buried less molecular surface area (566 Å2) than the contacts between paired heterodimers of EcGGT (PDB code 2DBX, 932 Å2) or H. pylori GGT (HpGGT, PDB code 2NQO, 866 Å2) (27, 28). Dynamic light scattering and gel filtration studies of HpGGT have demonstrated that HpGGT exists as a heterotetramer in solution (33), so the paired heterodimers of the bacterial GGT may be biologically significant. Bacterial GGTs are nonglycosylated enzymes localized to the periplasmic space, whereas hGGT1 is both heavily N-glycosylated and tethered to the plasma membrane. These two structural features of hGGT1 probably underlie the monomeric state of the human enzyme.
Our structure revealed that hGGT1 formed a heterodimer (large subunit, residues 28–380; small subunit, residues 381–569) with a stacked α-β-β-α core structure reminiscent of the bacterial GGTs and other members of the N-terminal nucleophile hydrolase superfamily (Figs. 1 and 3, A, C, and D) (18, 27–29, 33, 49). As observed previously in the crystal structures of the bacterial GGT heterodimers (27–29), our structure showed a pattern of disordered termini (residues 28–32 and 375–380 omitted) in the large subunit and ordered termini (Thr-381 and Tyr-569) in the small subunit. The large subunit of hGGT1 surrounded three sides of the small subunit (Fig. 3, E and F) like a hand wrapped around a ball. The buried interface between the subunits was 6076 Å2; in comparison, the molecular surface area of the heterodimer was 19,329 Å2. The substrate channel formed a longitudinal crevice in the exposed surface of the small subunit (Fig. 3E). The entrance of the substrate channel (∼20% of the total crevice area) was open to the solvent, and the active site nucleophile, Thr-381, sat in the deepest part of the channel (Fig. 3E).
FIGURE 3.

Crystal structure of human GGT1. A, ribbon representation of the hGGT1 heterodimer. The large subunit (chain A) is colored blue, and the small subunit (chain B) is colored green. The active site Thr-381 is colored red. The orange oval outlines the active site cleft. B, view of the two disulfide bonds (Cys-50/Cys-74, Cys-192/Cys-196) in the large subunit of hGGT1. Ribbon representations of large (C) and small (D) subunits of hGGT1 are shown. E, the van der Waals surface of hGGT1 with the active site cleft facing the viewer. Thr-381 is colored red. The large subunit is colored dark gray, and the small subunit is white. The six GlcNAcs on the surface are represented as dark orange van der Waals spheres and are labeled by the residue number of the asparagine to which they are anchored (black and yellow numbers). A green sphere represents the anion-binding site. F, backside of view in E.
The N terminus of the large subunit of hGGT1 was located on the bottom of the enzyme below the active site cleft (Fig. 3A). The first ordered residue, Pro-33, projected outward at a right angle from the bottom of the heterodimer, suggesting that the N-terminal residues (1–32), which include the transmembrane helical domain in the full-length enzyme, extend away from the ectodomain and transverse the plasma membrane. All of the N-glycosylation sites point in the opposite direction toward the top of the figure (Fig. 3, E and F). To reflect the most likely orientation of hGGT1 on the plasma membrane in vivo, the structure in Fig. 3 is rotated 180° about an axis normal to the page relative to the published figures of crystal structures from the bacterial GGTs (27–29, 50). The top surface of hGGT1 in Fig. 3E probably faces the extracellular milieu.
If the six disordered C-terminal residues of the large subunit extended across the molecular surface of the protein, the distance between the termini in the full-length protein would be ∼27 Å (Fig. 3A). The large distance (29.9 Å through the protein rather than across the surface) between the C terminus of the large subunit and the N terminus of the small subunit suggests that the subunits rotate relative to each other after autocleavage of the propeptide. A similar rotation between the subunits was described in the comparison of the crystal structures of the mature heterodimeric enzyme of EcGGT with its uncleaved propeptide (50).
Co-translational Modifications of hGGT1 Provide Insight into hGGT1 Autocleavage
The hGGT1 crystal structure showed intramolecular disulfide bonds between Cys-50 and Cys-74 and between Cys-192 and Cys-196. The Cys-50/Cys-74 disulfide bond linked helices α-1 and -2 and was inaccessible to the solvent because it was buried 10 Å beneath the surface of the large subunit (Fig. 3B). The Cys-192/Cys-196 disulfide bond joined two small supersecondary structures by linking helix α-8 and strand β-6 (Fig. 3B). This bond was partly exposed to the solvent at the molecular surface. It was fully intact in the 4GG2 model of hGGT1, but it was a mix of intact and broken bonds in the 4DGX model, perhaps attributable to x-ray radiation damage. Site-directed mutagenesis studies with rat and human GGT1 demonstrated that both disulfide bonds are critical for optimal autocleavage of mammalian GGT1 into active heterodimers (36, 38, 39). The formation of the Cys-50/Cys-74 and Cys-192/Cys-196 disulfide bridges may contribute to the proper folding of hGGT1 and, thereby, facilitate autocleavage into the activated heterodimer. The bacterial GGTs contain a pair of conserved alanines at the corresponding positions (e.g. Ala-58 and Ala-82 in EcGGT; Fig. 1) but, nonetheless, cleave into active heterodimers. The lack of the disulfide bonds at these sites in the bacterial enzymes suggests that the human and bacterial enzymes move differently during the autocleavage reaction.
hGGT1 also requires co-translational N-glycosylation during its maturation into an active heterodimer, although the glycans can be removed from the mature enzyme without the loss of catalytic activity (36). Previous studies on the N-glycosylation pattern of hGGT1 isolated from human tissues identified seven N-glycosylation sites (35, 36). Because we trimmed the glycans to the proximal glycan with Endo Hf before crystallization, the N-glycosylation sites were marked by one GlcNAc residue. Our crystal structure of hGGT1 expressed in yeast showed glycosylation at six of the seven previously described N-glycosylation sites (Asn-95, Asn-120, Asn-230, Asn-266, and Asn-344) in the large subunit and Asn-511 in the small subunit (Fig. 3, E and F). N-glycosylation at Asn-297 occurred in hGGT1 isolated from human tissues but was absent in the crystal structure of the yeast-expressed hGGT1 (35).
Although mutagenesis studies have shown that the elimination of single N-glycosylation sites on hGGT1 is tolerated, mutating Asn-95 to prohibit N-glycosylation at that site decreased the cleavage efficiency of the propeptide (36). In our structural analysis, the GlcNAc at Asn-95 formed hydrogen bonds with His-148 and Arg-150 at the C-terminal end of helix α-3, thereby linking the ends of this helix with the Thr-97/Thr-98 turn that forms the crevice that contains Asn-95 (Fig. 4). The glycan on Asn-95 might chaperone proper folding of the hGGT1 propeptide into a conformation that induces autocatalytic cleavage into the mature heterodimeric enzyme. The presence of disulfide bond formation and co-translational N-glycosylation suggests that the hGGT1 has a more coordinated maturation process than its prokaryotic counterparts.
FIGURE 4.
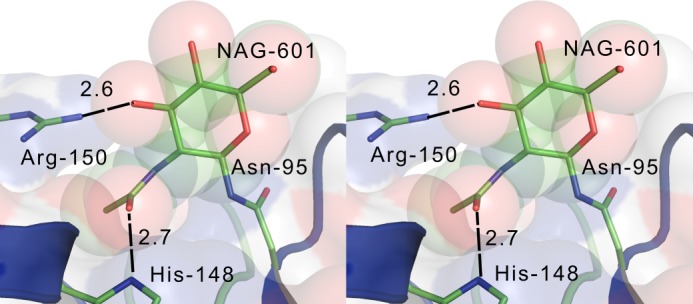
Stereo view of site 95. The GlcNAc residue (NAG-601) is represented by van der Waals spheres.
Lid Loop
Based on sequence alignments of hGGT1 with EcGGT and HpGGT, it has been proposed that residues Pro-427 through Ser-438 in hGGT1 form a loop that might regulate access to the active site (27–29). Both EcGGT and HpGGT have a “lid loop” (Pro-438 to Gly-449 in EcGGT and Pro-427 to Gly-438 in HpGGT) that covers most of the γ-glutamyl binding part of the active site and that leaves only the γ-carboxylate oxygen atoms of the bound glutamate exposed to solvent (Fig. 5, A and B) (27, 28). The analogous residues and loop are absent in BsGGT, which has a more open substrate channel (Fig. 5C) (29). Whereas the crystal structure of hGGT1 revealed that the anchoring termini of the lid loop were in comparable positions relative to those in the EcGGT and HpGGT, the loop itself was rotated away from the active site, leaving the substrate channel open (Fig. 5D).
FIGURE 5.

Comparison of the molecular surfaces and lid loop over the active site cleft in the crystal structures of four GGTs. The lid loop is colored black, the catalytic threonine is shown as black sticks, and the glutamate is shown as spheres. A, EcGGT; B, HpGGT; C, BsGGT; D, hGGT1. In the top row of panels, the large subunit is colored dark gray, and the small unit is colored light gray. In the bottom row, the lid loop is colored black, and both subunits are colored light gray.
In EcGGT and HpGGT, the lid loops in both the apoenzyme and the glutamate-bound structures adopt a closed conformation and are held in place by hydrogen bonds (27, 28). The side chain of an exposed tyrosine residue (Tyr-444 in EcGGT and Tyr-433 in HpGGT) at the apex of the bacterial lid loop gates the active site by hydrogen-bonding to a conserved asparagine residue (Asn-411 in EcGGT and Asn-400 in HpGGT) adjacent to the catalytic threonine (Fig. 6C) (27, 28). Phe-433 is the corresponding gating residue in hGGT1, but its side chain cannot hydrogen-bond with Asn-401 (Fig. 7). In EcGGT, the distance between the CZ carbon atom of Tyr-444 and the OD1 atom of Asn-411 is 3.5 Å. In HpGGT, the corresponding distance between Tyr-433 and Asn-400 is also 3.5 Å. (In hGGT1, the distance between the corresponding Phe-433 and Asn-401 is about 7 times longer, 24.7 Å, which also conflicts with the homology models of hGGT1 built from the crystal structures of bacterial GGT (51).) In both EcGGT and HpGGT, the tyrosine side chain is within 4.5 Å of the bound substrate (Fig. 5, A and B). Their lid loops may block or limit the binding of acceptor molecules and thereby limit the catalytic efficiency of the transpeptidation reaction within the active sites, an observation that has previously been documented for EcGGT and HpGGT (30, 33).
FIGURE 6.
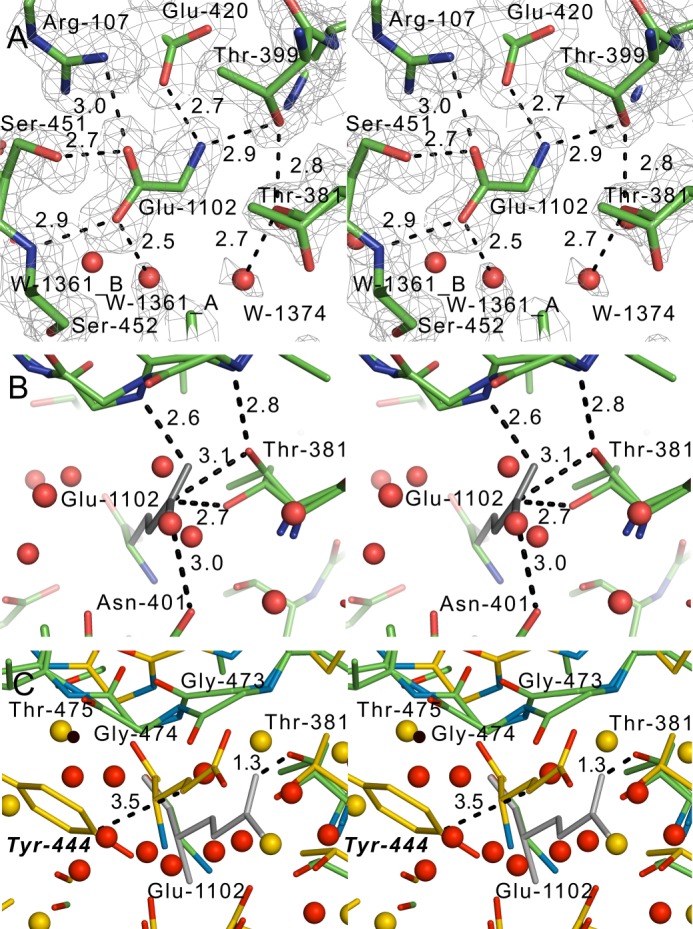
Stereo views of the active site. A, a 2mFo − DFc simulated annealing omit map contoured at the 1 σ level, where Glu-1102 was omitted. Dashed lines, hydrogen bonds; red spheres, water molecules. B, modeled side chain of Glu-1102 (colored gray). C, stereo view of the crystal structure of EcGGT (2DBX; yellow carbon atoms) superposed on hGGT1 (green carbon atoms). The yellow-colored glutamate is from EcGGT. The gray sticks represent the modeled side chain atoms of the glutamate in the structure of hGGT1. The residue numbers in boldface italic type are for EcGGT. The dashed lines represent interatomic distances, not hydrogen bonds. The yellow spheres are water molecules in the EcGGT structure.
FIGURE 7.

The open lid loop of hGGT1. A, stereo view of the 2mFo − DFc simulated annealing omit map of the hGGT1 lid loop with a 1-σ contour interval. The catalytic Thr-381 is about 25 Å to the left of the ring of Phe-433, which projects away from the surface of the protein. B, stereo view of the pocket into which Phe-433 and part of the lid loop packs on the surface of a symmetry-related protein.
The hGGT1 lid loop has higher B factors on average than the rest of the structure; therefore, our structure suggests that the lid loop of hGGT1 has greater flexibility than the lid loop of HpGGT or EcGGT. A more flexible lid loop would allow hGGT1 to accommodate a wider range of substrates, achieve faster substrate turnover, or both. This idea is supported by the 33- and 100-fold greater transpeptidation rates of BsGGT and hGGT1, respectively, relative to rates of EcGGT or HpGGT, whereas the hydrolysis rates are similar for all four GGTs (30–33).
Binding of the γ-Glutamyl Group in the Active Site
hGGT1 was co-crystallized with glutamate, the common product of the hydrolysis of γ-glutamyl bonds. The α-carboxyl and α-amino groups of the bound glutamate showed significant electron density in maps made with the glutamate omitted from the calculation (Glu-1102; Fig. 6A), but no significant electron density was observed for the side chain atoms. Nonetheless, the pattern of hydrogen bonding and salt bridges between α-carboxyl and α-amino groups of glutamate and neighboring residues within the hGGT1 active site allowed for the characterization of the intermolecular contacts that secure the γ-glutamyl moiety of substrates within the active site (Fig. 8A) (52). The α-carboxyl group of l-glutamate formed hydrogen bonds with Arg-107 (NH1), Ser-451 (OG), and Ser-452 (N) (Fig. 6A). These three amino acids are conserved among eukaryotic and prokaryotic GGTs (Fig. 1), and the same interactions have been observed in the bacterial GGTs crystallized with glutamate (27–29). In addition, the α-amino group of the glutamate formed a salt bridge with Glu-420 (OE1) of hGGT1. A similar bond has been documented in the bacterial GGTs (Gln-433 in EcGGT, Glu-419 in HpGGT, and Glu-442 in BsGGT; Fig. 1) (27–29); therefore, several of the substrate-enzyme interactions present in bacterial GGTs are found in hGGT1.
FIGURE 8.
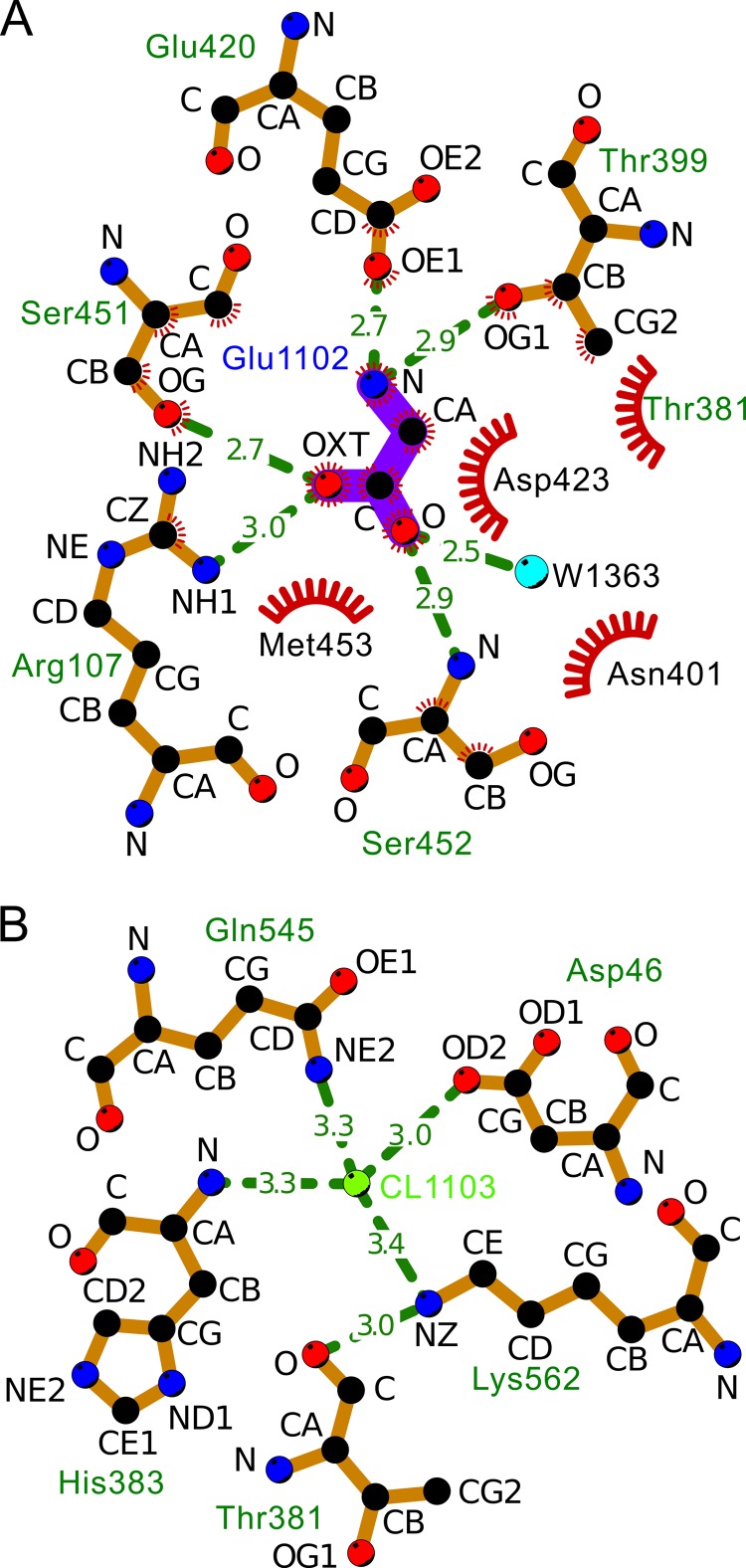
LIGPLOT diagrams. A, interactions between the backbone of hGGT1 and the α-nitrogen and α-carboxylate groups of glutamate (Glu-1102) in the substrate binding site of the enzyme (bond lengths not to scale); B, interactions between CL-1103 and the surrounding atoms, including the backbone of Thr-381. The ligand bonds are colored purple. The crescents with the bristles represent hydrophobic interactions. The hydrophobic atoms are colored black and are labeled with black letters. The polar atoms are in red (oxygen) or blue (nitrogen). The dashed lines represent hydrogen bonds.
However, substrate binding differed between hGGT1 and the bacterial GGTs. For example, our crystal structure revealed a hydrogen bond between Thr-399 of hGGT1 and the α-amino group of the bound glutamate (Fig. 6A). Thr-399 is conserved between human and bacterial GGT, but this interaction is absent in the bacterial GGTs. It is unlikely to occur in bacterial GGTs because the distance between the OG atom of Thr-399 and the amine of the glutamate is greater in the bacterial GGTs than in hGGT1 (e.g. greater by 2.3 Å for EcGGT; 2DBX). In contrast, the distance between the side chain of Asn-401 in hGGT1 and the α-amino group of the l-glutamate was too large to favor the formation of the hydrogen bond that is formed in each of the bacterial GGTs (Asn-411 in EcGGT, Asn-400 in HpGGT, and Glu-423 in BsGGT). Finally, the functional group of the conserved residue Asp-423 in hGGT1 is translated 1.7 Å away from its position in EcGGT. As a result, the cumulative distance between Asp-423 and the backbone amine of the bound glutamate in hGGT1 is 3.6 Å, a distance that is too great for hydrogen bond formation. Whereas Asp-423 in bacterial GGTs forms a hydrogen bond with the α-amino group of glutamate, the same residue in hGGT1 forms a novel salt bridge with Arg-107. This salt bridge can stabilize the hydrogen-bonding interaction of Arg-107 with the glutamate (Fig. 6A). Although several of the substrate-enzyme interactions were shared between the active site of hGGT1 and its bacterial orthologs, the crystal structure of hGGT1 had several key differences that can be exploited to develop novel inhibitors.
Active Site Geometry
Unlike the well ordered active sites of the crystallized bacterial GGTs, our crystal structure revealed structural disorder (as indicated by poorly defined electron density and high temperature factors) at three key regions within the active site: (a) the glutamate side chain, (b) the side chain of the catalytic nucleophile (Thr-381), and (c) the tripeptide (Gly-473, Gly-474, and Thr-475) that forms the oxyanion hole (Fig. 6, A and B). Because the side chain of the bound glutamate that co-crystallized with hGGT1 was absent in the electron density map, a complete glutamate was modeled within the active site to examine whether the side chain in a likely conformation would collide with the surrounding atoms (Fig. 6B). To this end, the backbone atoms of the glutamate were superimposed on their observed positions in the crystal structure, and the side chain was placed in the most frequent of 27 rotamers for glutamate in the 2010 Dunbrack backbone-independent rotamer library (19.1% frequency, χ1 = −67.7°, χ2 = −177.8°, and χ3 = −2.2°) (53). The OE1 oxygen atom of the modeled glutamate was 1.1 Å from water 1373, suggesting that the OE1 oxygen atom was in an appropriate orientation, so we included this modeled glutamate in our analysis of the geometry of the hGGT1 active site.
The tripeptide that forms the oxyanion hole was modeled in two conformations to better fit the ill defined electron density. Within the hGGT1 active site, two conformations for the side chain of the catalytic nucleophile, Thr-381, were observed; conformer A had a χ1 torsion angle of 41.9°, and conformer B had a χ1 torsion angle of −52.7°. The OG1 hydroxyl oxygen of Thr-381 in conformer A was 2.1 Å from the OE1 oxygen atom of the modeled glutamate and was oriented to form an acyl bond between the δ-carbon of the γ-glutamyl substrate and the hydroxyl (γ) oxygen on the side chain of Thr-381. Conversely, the side chain oxygen atom of conformer B was 2.4 Å from the OE1 oxygen atom and lacked a favorable alignment for covalent bond formation. This suggests that the formation of the acyl bond that creates the enzyme-substrate intermediate (the F-form of the enzyme) occurs when Thr-381 is in conformer A. Conformer B may reflect the position of Thr-381 after hydrolysis of the acyl bond as the glutamate is released.
To better fit the electron density, the tripeptide (Gly-473 through Thr-475) was modeled in two conformations. In conformers A and B, the backbone nitrogen of Gly-474 was within 3.1 and 2.6 Å, respectively, of the OE1 oxygen atom of the modeled glutamate and aligned favorably for hydrogen bond formation. Based on its position, Gly-473 was predicted to form a hydrogen bond with the side chain of Thr-381 in conformer A. By stabilizing conformer A of Thr-381, this hydrogen bond would promote acyl bond formation between the substrate and enzyme. In bacterial GGTs, Gly-473 and Gly-474 cooperate to stabilize the side chain carboxylate of the bound glutamate within a rigid oxyanion hole that includes Gly-473 through Thr-475 (27–29). The nitrogen atoms of Gly-473 and Gly-474 in hGGT1 are close enough to the carboxylate in the modeled glutamate to stabilize it by forming a similar oxyanion hole despite their disorder.
Chloride Ions near the Catalytic Residue
We found both weakly and strongly bound chloride anions near the catalytic Thr-381 at the positively charged N terminus of the small subunit (Figs. 9 and 10). (We also found a sodium cation contributing to structural stability (Fig. 10B).) The weak anomalous signal from chlorides in the native diffraction data for 4DGX was used to make an anomalous diffraction electron density map (not shown). Significant difference peaks suggested chloride bound to two sites: (a) near Pro-211 at the N terminus of helix α-9 and 18.6 Å from the positively charged backbone nitrogen of Thr-381 and (b) near the side chain amine of Lys-562. The amine of Lys-562, in turn, binds to the backbone carbonyl of Thr-381 (Fig. 8B). The latter chloride is 6.9 Å from the backbone nitrogen of Thr-381. The crystals of hGGT1-Glu were soaked with sodium iodide to verify the putative chloride binding sites by anomalous difference electron density maps and (Foiodide − Fonative) exp(−iσnative) isomorphous difference maps (Fig. 9). One soaked crystal gave 2.2 Å x-ray data with 1.1-Å wavelength x-rays. The imaginary component of the anomalous scattering from iodide is significant at this wavelength (f″ = 3.9 e−). The anomalous signal (i.e. the Bijvoet ratio (〈|ΔF±|〉/〈|F|〉)) for the hGGT1-iodide data set (PDB code 4GG2) was 0.048. There have been no reports of chlorides bound in the active site of any bacterial GGT, but two iodide sites in hGGT1 were assigned as legitimate chloride binding sites based on the geometry and distances to the surrounding protein ligand atoms (PDB code 4GG2). The first site was positioned adjacent to the free and positively charged backbone nitrogen of Thr-381, facilitating the burial of the N terminus of the small subunit (IOD 1104:A in Fig. 10A). This site was partially occupied by the iodide, as expected based on the different sizes of this site in conformer A (2.4 Å) and conformer B (3.6 Å). Because the van der Waals radius of chloride is 3.2 Å, only conformer B could accommodate chloride at this position. This site was missing in the native anomalous map, probably due to partial occupancy at this site as well as the weak nature of the anomalous signal (0.38 electrons) from the chlorides at the wavelength used to collect the diffraction data. Only conformer B of Thr-381 would accommodate an iodide anion at this site (distance 3.6 Å). The second part of this iodide was bound by the side chain amine of Lys-562 (Fig. 10A).
FIGURE 9.

Difference electron density maps with peaks corresponding to anion sites. Shown are a Bijvoet difference map showing the sites of the bound iodides in the crystal structure of hGGT1-iodide (PDB code 4GG2) (A) and an hGGT1-iodide minus hGGT1-Glu (PDB code 4DGX) isomorphorous difference map (B). The large subunit is represented by a C-α trace that is colored black. The small subunit is represented by a C-α trace that is colored white. B, the iodide sites were >15 σ in height in the (Foiodide − Fonative) exp(−iαnative) isomorphous difference map. In comparison, the difference peaks due to non-isomorphism between the protein structures were less than 5 σ. This second difference map provided independent evidence of the presence of the iodides. Only one (residue 606) of the eight iodides were assigned as additional chlorides (giving a total of two chlorides) in the crystal structure of hGGT1-Glu (PDB code 4GDX). The other iodide binding sites are probably not well occupied by chlorides because the chlorides were too small to interact as well with the protein.
FIGURE 10.
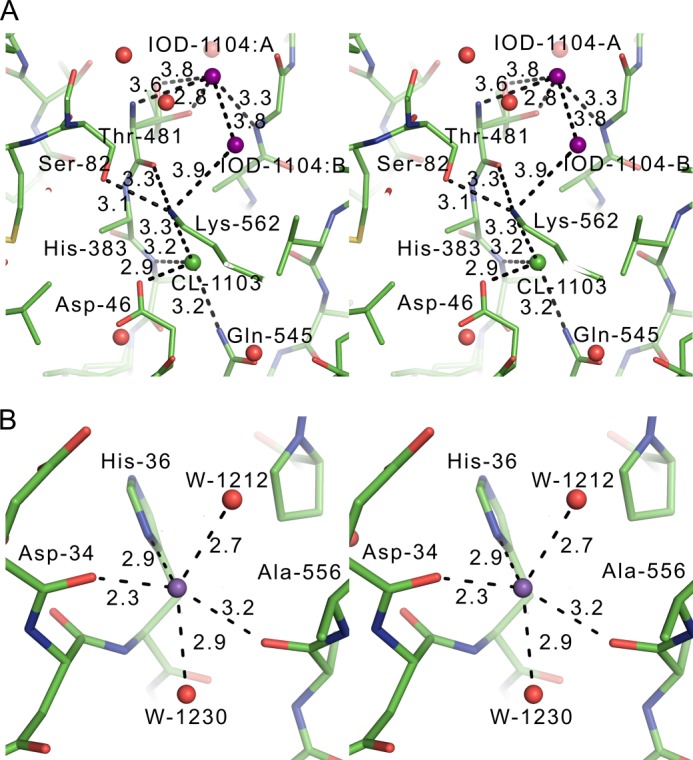
Anions and cations that stabilized the folded state of hGGT1. The dashed lines represent distances and not necessarily hydrogen bonds. A, stereo view of a split iodide anion (colored purple) and a chloride anion (colored green) near the catalytic Thr-381 in the crystal structure of iodide soak hGGT1-Glu (PDB code 4GG2). The top half of the iodide interacts directly with Thr-381 by binding the free and positively charged backbone nitrogen of Thr-381, which is the N terminus of the small subunit. This interaction stabilizes the burial of the N terminus of the small subunit. Part B of IOD-1004 partly stabilized Lys-562, which in turn bound to the backbone carbonyl of Thr-381. The completely buried chloride at the bottom (CL-1103) was not displaced by iodide. CL-1103 also stabilized Lys-562. B, sodium binding site near the N terminus of the large subunit of hGGT1-Glu. Shown is a stereo view of an apparent sodium cation (colored purple) identified by interatomic distances shown in angstroms. The sodium cation stabilized the extended loop near the N terminus and orients the first visible residue, Pro-33. The transmembrane helix extends away from Pro-33.
Interestingly, the iodides failed to displace the chloride (Cl-1103) bound to the opposite side of this lysine amine because the available space was too small for iodide to bind. The chloride-to-nitrogen and chloride-to-oxygen distances were 3.21–3.36 Å at this site. In contrast, iodide-to-oxygen distances are usually 3.5 Å. This chloride bound to the buried side chain of Asp-46. To allow this direct interaction with the chloride, Asp-46 was probably protonated to avoid the very high energetic penalty for burying a negatively charged side chain. These observations suggest that the buried chloride ion (Cl-1103) near Lys-562 is an integral part of the protein structure and should be included in future molecular simulations of this enzyme. Furthermore, the positioning of the tandem chloride ions around Thr-381 implies that there is excess positive charge around the catalytic residue in the presence of the glutamate ligand. This finding is in contrast to the hypothesis that the catalytic residue is neutral in N-terminal nucleophile hydrolases before acyl bond formation (19), but it is consistent with the suggestion that the catalytic residue is zwitterionic when it participates in catalysis (54). The presence or absence of the chlorides in the absence of glutamate can be tested in the eventual crystal structures of the apo-hGGT1. In any event, protein electrostatics are undoubtedly important in stabilizing the structure of the active site, binding of the substrate, and fine tuning of the catalytic efficiency of the active site nucleophile.
Insights into hGGT1 Mutants with Altered Enzymatic Activity
The crystal structure of hGGT1 provided insights into the role of residues that have been shown by mutagenesis studies to contribute to the catalytic activity of hGGT1. Mutagenesis of His-383 to an alanine resulted in an 86% loss of hydrolysis activity, which was proposed to affect substrate release (55). The crystal structure revealed a bond between the backbone amide of His-383 and the buried chloride (Cl-1103) (Fig. 8B). The side chain of His-383 is buried in this structure, so the replacement of the bulky side chain of the histidine with the small side chain of the alanine would create a cavity that the alanine would try to fill by moving away from the chloride. Movement of the alanine's backbone of even a few tenths of an angstrom would be enough to greatly reduce the affinity of this site for the chloride. The loss of the chloride would destabilize the burial of the positively charged Lys-562 and upset the hydrogen bond between its side chain amine group and the backbone carbonyl of Thr-381. Single-site mutagenesis of Arg-107, Ser-451, or Ser-452 resulted in a complete or nearly complete loss of enzymatic activity (31, 56). Each of these residues forms a hydrogen bond with the substrate glutamate, suggesting that these residues stabilize the substrate in the active site (Figs. 6A and 8A). Similarly, mutagenesis of Asp-423 resulted in a 1000-fold increase in the Km for d-γ-glutamyl-p-nitroanalide (31). Asp-423 forms a salt bridge with Arg-107 and makes van der Waals contacts (Fig. 8A) with the glutamate portion of the substrate, so the disruption of these interactions by amino acid substitutions at site 423 probably accounts for the loss of substrate affinity (Fig. 8A).
In addition, the crystal structure provides insight into our studies with a Q545K mutant of hGGT1. Our data showed that the Q545K hGGT1 was expressed and autocleaved into the heterodimer. Equal amounts of the wild-type and Q545K hGGT1 protein (based on Western analysis) were assayed for hydrolysis activity with d-γ-glutamyl-p-nitroanalide as the substrate. The Q545K hGGT1 had only 3% of the activity of the wild-type protein. Fig. 8B shows that Gln-545, Asp-46, Lys-562, and His-383 all bond to Cl-1103. The Q545K mutant would have destabilized the structure by burying a positive charge and introducing a number of steric clashes. This destabilization of the structure would explain the loss in activity.
CONCLUSION
The hGGT1 crystal structure has provided new insights into the maturation and catalytic activity of the enzyme. The validation of post-translational changes that are unique to hGGT1, such as disulfide bond formation and multiple N-glycosylation sites, supports previous site-directed mutagenesis studies and provides a conceptual framework to better understand how the eukaryotic enzyme differs in its maturation from bacterial GGTs. The new insights into the function of hGGT1 included the discovery that it adopts an open active site, has several new substrate interactions, attracts anions to bind near the catalytic residue, has a buried chloride ion that is an integral part of the protein structure, and exhibits substrate-dependent conformational changes around the catalytic nucleophile. These unique features of the active site can be exploited in the structure-based design of new classes of inhibitors that specifically target this enzyme.
Acknowledgments
We thank J. Donald Capra for a critical review of the manuscript. X-ray diffraction data were collected at the National Synchrotron Light Source, for which financial support comes principally from the United States Department of Energy Offices of Biological and Environmental Research and of Basic Energy Sciences and from National Institutes of Health Grants P41RR012408 and P41GM103473.
This work was supported, in whole or in part, by National Institutes of Health Grants P20GM103640 (an Institutional Development Award (IDeA), R01 AI088011 (to B. H. M. M.), R56 CA57530 (to M. H. H.), and F32 CA128338 (to M. B. W.). This work was also supported by University of Oklahoma Health Sciences Center Department of Biochemistry and Molecular Biology Startup Funds (to B. H. M. M.) and a Seed Grant from the Stephenson Oklahoma Cancer Center.
The atomic coordinates and structure factors (codes 4GDX and 4GG2) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GGT1
- γ-glutamyltranspeptidase 1
- hGGT1
- human GGT1
- EcGGT, HpGGT, and BsGGT
- E. coli, H. pylori, and B. subtilis γ-glutamyltranspeptidase, respectively
- TEV
- tobacco etch virus
- Ni-NTA
- nickel-nitrilotriacetic acid
- PDB
- Protein Data Bank
- GlcNAc
- N-acetylglucosamine.
REFERENCES
- 1. Lieberman M. W., Wiseman A. L., Shi Z. Z., Carter B. Z., Barrios R., Ou C. N., Chévez-Barrios P., Wang Y., Habib G. M., Goodman J. C., Huang S. L., Lebovitz R. M., Matzuk M. M. (1996) Growth retardation and cysteine deficiency in γ-glutamyl transpeptidase-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 93, 7923–7926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanigan M. H., Ricketts W. A. (1993) Extracellular glutathione is a source of cysteine for cells that express γ-glutamyl transpeptidase. Biochemistry 32, 6302–6306 [DOI] [PubMed] [Google Scholar]
- 3. Cooper A. J., Hanigan M. H. (2010) Enzymes involved in processing glutathione conjugates. in Comprehensive Toxicology: Biotransformation (Guengerich F. P., ed) pp. 323–365, Elsevier, New York [Google Scholar]
- 4. Rojas E., Valverde M., Kala S. V., Kala G., Lieberman M. W. (2000) Accumulation of DNA damage in the organs of mice deficient in γ-glutamyltranspeptidase. Mutat. Res. 447, 305–316 [DOI] [PubMed] [Google Scholar]
- 5. Wickham S., West M. B., Cook P. F., Hanigan M. H. (2011) γ-Glutamyl compounds. Substrate specificity of γ-glutamyl transpeptidase enzymes. Anal. Biochem. 414, 208–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lowry M. H., McAllister B. P., Jean J. C., Brown L. A., Hughey R. P., Cruikshank W. W., Amar S., Lucey E. C., Braun K., Johnson P., Wight T. N., Joyce-Brady M. (2008) Lung lining fluid glutathione attenuates IL-13-induced asthma. Am. J. Respir. Cell Mol. Biol. 38, 509–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sian J., Dexter D. T., Lees A. J., Daniel S., Jenner P., Marsden C. D. (1994) Glutathione-related enzymes in brain in Parkinson's disease. Ann. Neurol. 36, 356–361 [DOI] [PubMed] [Google Scholar]
- 8. Emdin M., Pompella A., Paolicchi A. (2005) γ-glutamyltransferase, atherosclerosis, and cardiovascular disease. Triggering oxidative stress within the plaque. Circulation 112, 2078–2080 [DOI] [PubMed] [Google Scholar]
- 9. Diergaarde B., Brand R., Lamb J., Cheong S. Y., Stello K., Barmada M. M., Feingold E., Whitcomb D. C. (2010) Pooling-based genome-wide association study implicates γ-glutamyltransferase 1 (GGT1) gene in pancreatic carcinogenesis. Pancreatology 10, 194–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hanigan M. H., Gallagher B. C., Townsend D. M., Gabarra V. (1999) γ-Glutamyl transpeptidase accelerates tumor growth and increases the resistance of tumors to cisplatin in vivo. Carcinogenesis 20, 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castellano I., Merlino A. (2012) γ-Glutamyltranspeptidases. Sequence, structure, biochemical properties, and biotechnological applications. Cell. Mol. Life Sci. 69, 3381–3394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Earhart R. H., Koeller J. M., Davis T. E., Borden E. C., McGovren J. P., Davis H. L., Tormey D. C. (1983) Phase I trial and pharmacokinetics of acivicin administered by 72-hour infusion. Cancer Treat. Rep. 67, 683–692 [PubMed] [Google Scholar]
- 13. Fleishman G., Yap H. Y., Murphy W. K., Bodey G. (1983) Phase II trial of acivicin in advanced metastatic breast cancer. Cancer Treat. Rep. 67, 843–844 [PubMed] [Google Scholar]
- 14. Hidalgo M., Rodriguez G., Kuhn J. G., Brown T., Weiss G., MacGovren J. P., Von Hoff D. D., Rowinsky E. K. (1998) A Phase I and pharmacological study of the glutamine antagonist acivicin with the amino acid solution aminosyn in patients with advanced solid malignancies. Clin. Cancer Res. 4, 2763–2770 [PubMed] [Google Scholar]
- 15. King J. B., West M. B., Cook P. F., Hanigan M. H. (2009) A novel, species-specific class of uncompetitive inhibitors of γ-glutamyl transpeptidase. J. Biol. Chem. 284, 9059–9065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wickham S., Regan N., West M. B., Kumar V. P., Thai J., Li P. K., Cook P. F., Hanigan M. H. (2012) Divergent effects of compounds on the hydrolysis and transpeptidation reactions of γ-glutamyl transpeptidase. J. Enzyme Inhib. Med. Chem. 27, 476–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wickham S., Regan N., West M. B., Thai J., Cook P. F., Terzyan S. S., Li P. K., Hanigan M. H. (2013) Inhibition of human γ-glutamyl transpeptidase. Development of more potent, physiologically relevant, uncompetitive inhibitors. Biochem. J. 450, 547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duggleby H. J., Tolley S. P., Hill C. P., Dodson E. J., Dodson G., Moody P. C. (1995) Penicillin acylase has a single-amino-acid catalytic centre. Nature 373, 264–268 [DOI] [PubMed] [Google Scholar]
- 19. Oinonen C., Rouvinen J. (2000) Structural comparison of Ntn-hydrolases. Protein Sci. 9, 2329–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castonguay R., Halim D., Morin M., Furtos A., Lherbet C., Bonneil E., Thibault P., Keillor J. W. (2007) Kinetic characterization and identification of the acylation and glycosylation sites of recombinant human γ-glutamyltranspeptidase. Biochemistry 46, 12253–12262 [DOI] [PubMed] [Google Scholar]
- 21. Ekici O. D., Paetzel M., Dalbey R. E. (2008) Unconventional serine proteases. Variations on the catalytic Ser/His/Asp triad configuration. Protein Sci. 17, 2023–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elce J. S., Broxmeyer B. (1976) γ-Glutamyltransferase of rat-kidney. simultaneous assay of hydrolysis and transfer-reactions with [glutamate-C-14]glutathione. Biochem. J. 153, 223–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Curthoys N. P., Hughey R. P. (1979) Characterization and physiological-function of rat renal γ-glutamyl-transpeptidase. Enzyme 24, 383–403 [DOI] [PubMed] [Google Scholar]
- 24. Thompson G. A., Meister A. (1976) Hydrolysis and transfer reactions catalyzed by γ-glutamyl transpeptidase. Evidence for separate substrate sites and for high affinity of l-cystine. Biochem. Biophys. Res. Commun. 71, 32–36 [DOI] [PubMed] [Google Scholar]
- 25. Allison R. D. (1985) γ-Glutamyl transpeptidase. Kinetics and mechanism. Methods Enzymol. 113, 419–437 [DOI] [PubMed] [Google Scholar]
- 26. Taniguchi N., Ikeda Y. (1998) γ-Glutamyl transpeptidase. Catalytic mechanism and gene expression. Adv. Enzymol. Relat. Areas Mol. Biol. 72, 239–278 [DOI] [PubMed] [Google Scholar]
- 27. Morrow A. L., Williams K., Sand A., Boanca G., Barycki J. J. (2007) Characterization of Helicobacter pylori γ-glutamyltranspeptidase reveals the molecular basis for substrate specificity and a critical role for the tyrosine 433-containing loop in catalysis. Biochemistry 46, 13407–13414 [DOI] [PubMed] [Google Scholar]
- 28. Okada T., Suzuki H., Wada K., Kumagai H., Fukuyama K. (2006) Crystal structures of γ-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Proc. Natl. Acad. Sci. U.S.A. 103, 6471–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wada K., Irie M., Suzuki H., Fukuyama K. (2010) Crystal structure of the halotolerant γ-glutamyltranspeptidase from Bacillus subtilis in complex with glutamate reveals a unique architecture of the solvent-exposed catalytic pocket. FEBS J. 277, 1000–1009 [DOI] [PubMed] [Google Scholar]
- 30. Suzuki H., Kumagai H., Tochikura T. (1986) γ-Glutamyl-transpeptidase from Escherichia coli K-12. Purification and properties. J. Bacteriol. 168, 1325–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ikeda Y., Fujii J., Anderson M. E., Taniguchi N., Meister A. (1995) Involvement of Ser-451 and Ser-452 in the catalysis of human γ-glutamyl transpeptidase. J. Biol. Chem. 270, 22223–22228 [DOI] [PubMed] [Google Scholar]
- 32. Minami H., Suzuki H., Kumagai H. (2003) Salt-tolerant γ-glutamyltranspeptidase from Bacillus subtilis 168 with glutaminase activity. Enzyme Microb. Technol. 32, 431–438, http://dx.doi.org/10.1016/S0141-0229(02)00314-9 [Google Scholar]
- 33. Boanca G., Sand A., Barycki J. J. (2006) Uncoupling the enzymatic and autoprocessing activities of Helicobacter pylori γ-glutamyltranspeptidase. J. Biol. Chem. 281, 19029–19037 [DOI] [PubMed] [Google Scholar]
- 34. Rajpert-De Meyts E., Heisterkamp N., Groffen J. (1988) Cloning and nucleotide sequence of human γ-glutamyl transpeptidase. Proc. Natl. Acad. Sci. U.S.A. 85, 8840–8844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. West M. B., Segu Z. M., Feasley C. L., Kang P., Klouckova I., Li C., Novotny M. V., West C. M., Mechref Y., Hanigan M. H. (2010) Analysis of site-specific glycosylation of renal and hepatic γ-glutamyl transpeptidase from normal human tissue. J. Biol. Chem. 285, 29511–29524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. West M. B., Wickham S., Quinalty L. M., Pavlovicz R. E., Li C., Hanigan M. H. (2011) Autocatalytic cleavage of human γ-glutamyl transpeptidase is highly dependent on N-glycosylation at asparagine 95. J. Biol. Chem. 286, 28876–28888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Suzuki H., Kumagai H. (2002) Autocatalytic processing of γ-glutamyltranspeptidase. J. Biol. Chem. 277, 43536–43543 [DOI] [PubMed] [Google Scholar]
- 38. Kinlough C. L., Poland P. A., Bruns J. B., Hughey R. P. (2005) γ-Glutamyltranspeptidase. Disulfide bridges, propeptide cleavage, and activation in the endoplasmic reticulum. Methods Enzymol. 401, 426–449 [DOI] [PubMed] [Google Scholar]
- 39. West M. B., Wickham S., Parks E. E., Sherry D. M., Hanigan M. H. (2013) Human GGT2 does not autocleave into a functional enzyme. A cautionary tale for interpretation of microarray data on redox signaling. Antioxid. Redox Signal. 10.1089/ars.2012.4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vagin A., Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 42. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thorn A., Sheldrick G. M. (2011) ANODE. Anomalous and heavy-atom density calculation. J. Appl. Crystallogr. 44, 1285–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thompson J. D., Higgins D. G., Gibson T. J. (1994) CLUSTAL W. Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript. Analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
- 47. Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 48. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 49. Boanca G., Sand A., Okada T., Suzuki H., Kumagai H., Fukuyama K., Barycki J. J. (2007) Autoprocessing of Helicobacter pylori γ-glutamyltranspeptidase leads to the formation of a threonine-threonine catalytic dyad. J. Biol. Chem. 282, 534–541 [DOI] [PubMed] [Google Scholar]
- 50. Okada T., Suzuki H., Wada K., Kumagai H., Fukuyama K. (2007) Crystal structure of the γ-glutamyltranspeptidase precursor protein from Escherichia coli. Structural changes upon autocatalytic processing and implications for the maturation mechanism. J. Biol. Chem. 282, 2433–2439 [DOI] [PubMed] [Google Scholar]
- 51. Hu X., Legler P. M., Khavrutskii I., Scorpio A., Compton J. R., Robertson K. L., Friedlander A. M., Wallqvist A. (2012) Probing the donor and acceptor substrate specificity of the γ-glutamyl transpeptidase. Biochemistry 51, 1199–1212 [DOI] [PubMed] [Google Scholar]
- 52. Laskowski R. A., Swindells M. B. (2011) LigPlot+. Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 [DOI] [PubMed] [Google Scholar]
- 53. Shapovalov M. V., Dunbrack R. L., Jr. (2011) A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lodola A., Branduardi D., De Vivo M., Capoferri L., Mor M., Piomelli D., Cavalli A. (2012) A catalytic mechanism for cysteine N-terminal nucleophile hydrolases, as revealed by free energy simulations. PLoS One 7, e32397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ikeda Y., Fujii J., Taniguchi N. (1996) Effects of substitutions of the conserved histidine residues in human γ-glutamyl transpeptidase. J. Biochem. 119, 1166–1170 [DOI] [PubMed] [Google Scholar]
- 56. Ikeda Y., Fujii J., Taniguchi N. (1993) Significance of Arg-107 and Glu-108 in the catalytic mechanism of human γ-glutamyl transpeptidase. Identification by site-directed mutagenesis. J. Biol. Chem. 268, 3980–3985 [PubMed] [Google Scholar]
- 57. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity. All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]