

Abstract
Promyelocytic leukemia protein (PML) is emerging as an important tumor suppressor. Its expression is lost during the progression of several types of cancer, including lung cancer. The EGF receptor (EGFR), a membrane-bound receptor tyrosine kinase, transduces intracellular signals responsible for cell proliferation, differentiation and migration. EGFR activity is frequently abnormally upregulated in lung adenocarcinoma (LAC) and thus is considered to be a driving oncogene for LAC. EGFR translocates into the nucleus and transcriptionally activates genes, such as CCND1, that promote cell growth. Recently, we demonstrated that PML interacted with nuclear EGFR (nEGFR) and suppressed the nEGFR-mediated transcriptional activation of CCND1 in lung cancer cells, thereby restraining cell growth. When we further investigated the interplay between PML and EGFR in lung cancer metastasis, we found that the matrix metalloprotease-2 gene (MMP2) was a novel nEGFR target gene and was repressed by PML. We provide evidence that nEGFR bound to the AT-rich sequence (ATRS) in the MMP2 promoter and enhanced its transcriptional activity. In addition, we demonstrated that PML repressed nEGFR-induced MMP2 transcription and reduced cell invasion. PML was recruited by nEGFR to the MMP2 promoter where it reduced histone acetylation, leading to the transcriptional repression of MMP2. Finally, we demonstrated that PML upregulation by interferon-β (IFNβ) in lung cancer cells decreased MMP2 expression and cell invasion. Together, our results suggested that IFNβ induced PML to inhibit lung cancer metastasis by repressing the nEGFR-mediated transcriptional activation of MMP2.
Keywords: ATRS, Interferon-β, lung adenocarcinoma, MMP2, nuclear EGFR, PML, STAT3
Abbreviations
- PML
promyelocytic leukemia protein
- EGFR
EGF receptor
- LAC
lung adenocarcinoma
- nEGFR
nuclear EGFR
- MMP2
matrix metalloprotease-2
- ATRS
AT-rich sequence
- IFNβ
interferon-β
- INM
inner nuclear membrane
Introduction
Lung cancer is the leading cause of cancer-related death worldwide1 Because most lung cancer-associated deaths are caused by metastases, understanding the regulation of lung cancer metastasis is critically important.
Cancer metastasis is a complicated multi-step process involving intravasation, survival in the circulation, extravasation and colonization.2 During the intravasation and extravasation steps, secreted proteases degrade the extracellular matrix to facilitate cancer cell invasion through the basement membrane, which otherwise hinders this process. Matrix metalloprotease-2 (MMP2) is a type IV collagenase that efficiently cleaves collagen IV, the major component of the basement membrane. The expression level of MMP2 is positively correlated with the invasiveness of cancer cells and with poor prognosis for lung cancer patients.3,4
PML has been suggested to be a tumor suppressor because its expression is lost in human cancers from multiple histological origins.5,6 Its roles in regulating cell proliferation and apoptosis contribute to its tumor suppressive function.7,8 In addition, it represses tumor neoangiogenesis through translational control of HIF1α.9 Recently, it has been discovered that PML also plays a role in regulating cell migration through the transcriptional control of integrin β1 and the translational control of HIF1α.10,11 In addition, PML plays important roles in regulating cancer stem cells.12 Nevertheless, the role of PML in the process of lung cancer metastasis remains unclear.
EGFR is a cell surface receptor tyrosine kinase that transduces signals to stimulate cell proliferation, migration and invasion. Recently, EGFR has been shown to translocate into the nucleus to regulate multiple cellular functions, including cell proliferation,13 DNA damage repair,14-16 drug and radiation resistance16-20 and transcription.21-24 The transcriptional activity of nuclear EGFR (nEGFR) has been attributed to its ability to bind to AT-rich sequences (ATRSs) in promoter regions and to its intrinsic transcriptional activation domain.21,24 Clinical studies have indicated that increased nEGFR expression is associated with poor prognosis.25,26
The nuclear translocation of EGFR is initiated by ligand-induced endocytosis.27 Endosome-associated EGFR is recognized by importin β in the cytoplasm and is then transported to the inner nuclear membrane (INM) via the INTERNET (integral trafficking from the ER to the nuclear envelope transport) mechanism.28,29 Finally, Sec61β in the INM releases membrane-bound EGFR into the nucleoplasm.30 In addition, EGFR phosphorylation by AKT and Src family kinases promotes the nuclear translocation of EGFR18,31-33; the syntaxin 6-mediated transportation of EGFR to the Golgi body is also important for its transport into the nucleus.34
Recently, we reported that PML suppressed EGFR-mediated lung cancer cell proliferation. In this study, we examined the effect of PML on lung cancer cell invasion. We found that PML repressed EGF-induced lung cancer cell invasion by decreasing the expression of MMP2, which is important for cancer metastasis.3 We also identified MMP2 as a novel nEGFR target gene and characterized the recruitment of nEGFR to the ATRS in the MMP2 promoter. We determined that PML was essential for the IFNβ-mediated repression of MMP2 expression in lung cancer cells. We report here that PML suppressed EGFR-mediated lung cancer cell invasion by inhibiting the nEGFR-mediated transcriptional activation of MMP2.
Results
PML suppressed EGF-mediated lung cancer cell invasion
We performed a tail vein metastasis assay to explore the role of PML in regulating lung cancer metastasis. When an equal number of control or PML-knockdown luciferase-expressing A549 cells were injected into NOD/SCID mice, higher luciferase activity was detected in the lungs of mice injected with PML-knockdown cells (Fig. 1A). These mice also died earlier than the control mice (Fig. 1B). These results suggested that PML- knockdown cells colonized the lungs more efficiently than control cells and implied that PML might suppress lung cancer metastasis. We further examined the effect of PML on lung cancer cell invasion, which is important for intravasation and extravasation during metastasis. In the invasion assay, PML significantly regulated the invasion of H1975 cells but not of A549 cells (Fig. 1C, left and Fig. S1). Because H1975 cells harbor a mutation that constitutively actives EGFR,35 we determined the effect of PML on the invasive activity of A549 cells treated with EGF. Under this condition, PML knockdown increased A549 cell invasion (Fig. 1C, right). Similarly, PML overexpression significantly reduced the invasion activity of EGF-treated A549 and CL1-5 cells (Figs. 1D and 1E). These results suggested that PML suppressed lung cancer invasion in the presence of activated EGFR.
Figure 1.
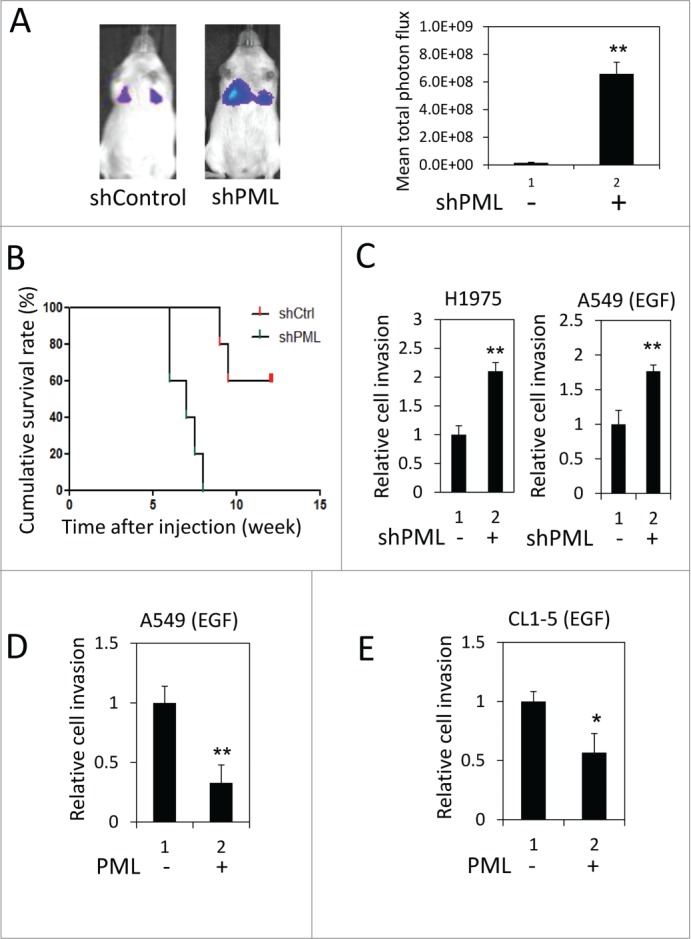
PML suppressed lung cancer cell invasion when EGFR was activated. (A) Left, Luciferase-expressing A549-shControl or shPML cells were injected via the tail vein into immunocompromised NOD/SCID mice (n = 5 for each group). The mice were monitored for colonization of the injected cells in the lungs as described in the Materials and Methods. Representative images from week 6 post-injection are shown. Right, The mean total photon flux measured by IVIS at week 6 is shown. The data are presented as the mean ± SEM. (B) The cumulative survival rate of the same mice described in Figure 1A was monitored weekly until 13th week after cell injection. (C) Left, H1975-shControl and H1975-shPML cell invasion was assayed as described in the Materials and Methods. Right, A549-shControl and A549-shPML cells were cultured overnight in low-serum starvation conditions (0.5% FBS). These cells were then subjected to the cell invasion assay in the low serum-medium with 50 ng/ml EGF. (D) A549-Control and A549-PML cells were cultured overnight in low-serum starvation conditions (0.5% FBS). These cells were then subjected to the cell invasion assay in the low serum-medium with 50 ng/ml EGF. (E) CL1-5-Control and CL1-5-PML cell invasion was assayed as described in (D).
PML suppressed nEGFR-induced MMP2 expression
We used 2 shRNA sequences (shPML and shPML-2) to knockdown endogenous PML expression. In H1975 cells, shPML decreased PML mRNA expression by more than 80%, and shPML-2 reduced PML mRNA expression by 50% (Fig. 2, gray bar). We then examined the effect of PML on the expression of several genes important for cancer metastasis, including MMP2, MMP9, ITGB1 and MMP14.36-39 We found that the mRNA expression of MMP2, but not of the other 3 genes, was significantly upregulated in response to PML knockdown (Fig. 2A and Fig. S2, left). MMP2 protein expression was also upregulated by PML knockdown (Fig. S2, right). Consistent with these data, introducing shRNA-resistant PML (rPML) to H1975-shPML cells decreased MMP2 mRNA expression (Fig. 2B). These results suggested that PML suppressed MMP2 expression in H1975 cells. Because we recently reported that PML repressed gene expression by decreasing the transcriptional activity of nEGFR,40 we determined the effects of nEGFR and PML on MMP2 expression. The experiments were performed in 293T cells because of its high transfection efficiency and low nEGFR level (data not shown). In a luciferase reporter gene assay, EGFR overexpression slightly increased MMP2 promoter activity. However, tethering an exogenous nuclear localization sequence (NLS) to EGFR significantly enhanced its ability to activate the MMP2 promoter (Fig. 2C). These results suggested that nEGFR was involved in regulating MMP2 promoter activity. We further investigated the interplay between nEGFR and PML in regulating MMP2 promoter activity. In a reporter gene assay, PML significantly reduced EGF-induced MMP2 promoter activity (Fig. 2D). More importantly, PML repressed nEGFR-induced MMP2 promoter activity and mRNA expression in a dose-dependent manner (Figs. 2E and 2F). These results suggested that MMP2 was a novel nEGFR target gene and that PML repressed the nEGFR-mediated activation of MMP2.
Figure 2.
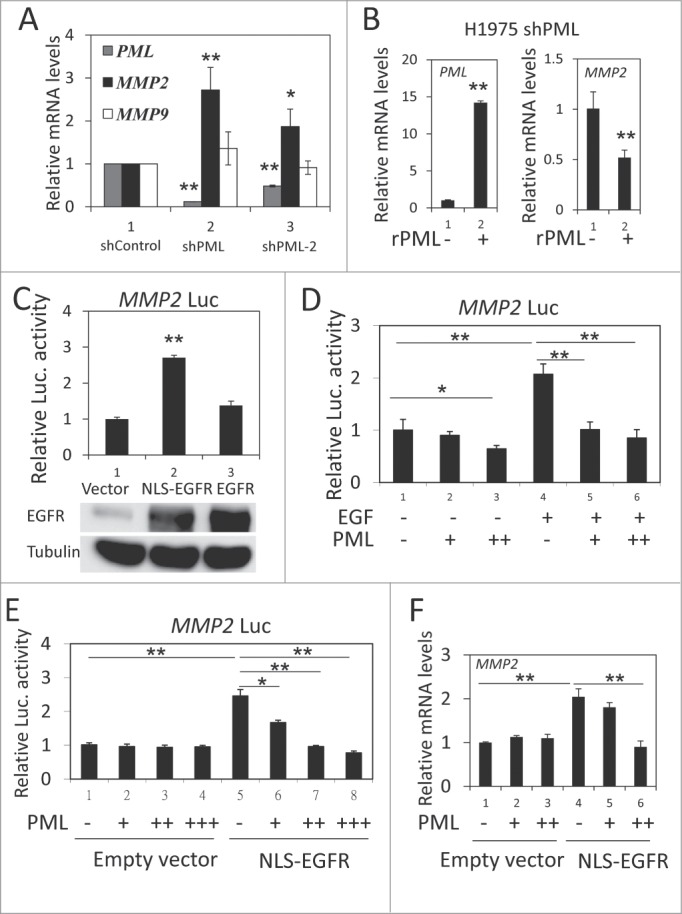
PML repressed nEGFR-induced MMP2 expression. (A) RT-qPCR was performed to determine the relative mRNA expression of PML, MMP2 and MMP9 in H1975-shControl, H1975-shPML and H1975-shPML-2 cells as described in the Materials and Methods. (B) H1975 shPML cells with or without overexpression of shRNA-resistant PML (rPML) were analyzed by RT-qPCR to determine the relative mRNA expression of PML and MMP2. (C) 293T cells transfected with pMMP2 Luc and 100 ng of pCMV-EGFR NLS or pCMV-EGFR were analyzed using the luciferase reporter gene assay as described in the Materials and Methods. (D) A549 cells were transfected with pMMP2 Luc and increasing amounts of the pHA-PML expression plasmid. Twenty-four hours after transfection, the cells were treated with or without 50 ng/ml EGF for 18 h and then subjected to a luciferase reporter gene assay. (E) 293T cells were transfected with pMMP2 Luc, 100 ng of pCMV-EGFR NLS or empty vector and increasing amounts of PML expression plasmid as indicated. The cells were subjected to a luciferase reporter gene assay 48 h after transfection. (F) 293T cells were transfected in 6-well plate with 1 μg of pCMV-EGFR NLS or empty vector, and increasing amounts of pHA-PML as indicated. Forty-eight hours after transfection, the cells were analyzed by RT-qPCR to detect MMP2 mRNA expression.
nEGFR bound to the MMP2 promoter
We next identified the region of the MMP2 promoter that was responsible for the nEGFR-induced activation. In the reporter gene assay, the activities of the long and medium fragments of the MMP2 promoter were enhanced by nEGFR, whereas the short form did not respond to nEGFR (Fig. 3A). These results indicated that the -666 to -134 region of the MMP2 promoter was required for nEGFR-mediated induction. Using ChIP, we demonstrated that nEGFR specifically bound to a DNA fragment encompassing this region (Fig. 3B). An inspection of the nucleotide sequence of this region revealed a putative ATRS. Mutating this putative ATRS in the MMP2 promoter from TATTT to either TGGTT or GATTT abolished the nEGFR-induced activation in the reporter gene assay and nEGFR binding in the ChIP assay (Figs. 3C and 3D). Furthermore, nEGFR significantly enhanced the histone acetylation of the wild-type, but not the mutant, ATRS in the MMP2 promoter (Fig. 3E). These results suggested that the MMP2 promoter contained an ATRS that was bound by nEGFR and was responsible for nEGFR-mediated activation.
Figure 3.
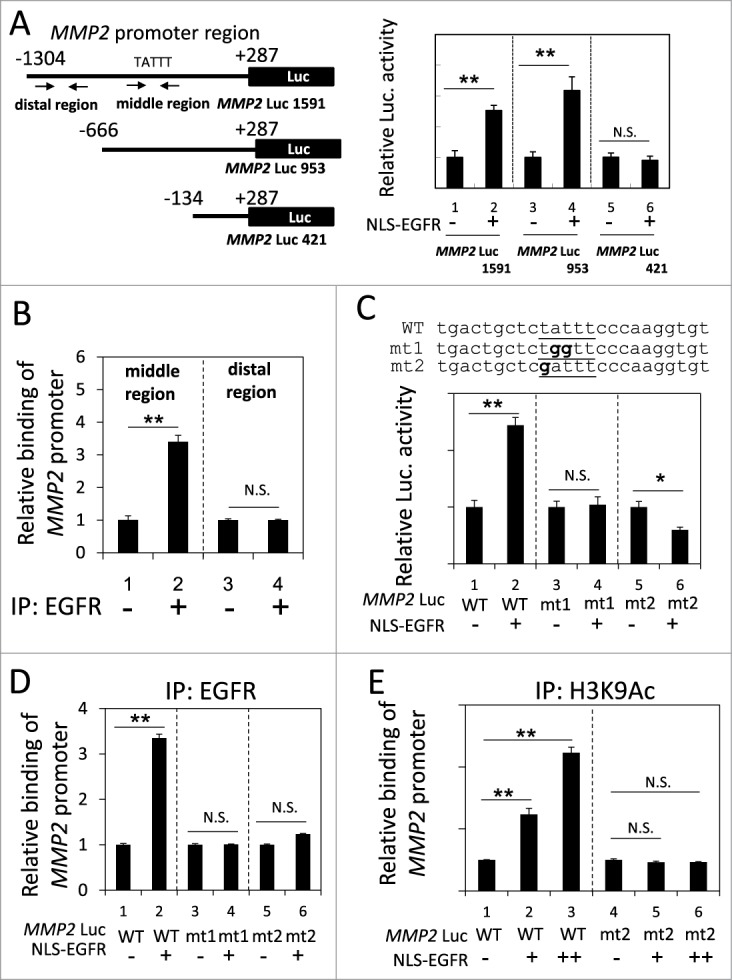
Identification of an ATRS in the MMP2 promoter. (A) Left, A list of the MMP2 promoter luciferase reporter constructs. The promoter region is indicated relative to the transcription start site. The arrows indicate the 2 primer pairs used for PCR amplification in the ChIP assay. The putative ATRS in the middle region of the MMP2 promoter is indicated. Right, 293T cells transfected with the 3 pMMP2 Luc constructs with or without pCMV-NLS-EGFR, were subjected to a luciferase reporter gene assay. (B) H1975 cells were analyzed by ChIP using preimmune IgG or anti-EGFR antibody as described in the Materials and Methods. The relative amount of immunoprecipitated DNA from the middle or distal regions of the MMP2 promoter was quantified by RT-qPCR with specific primers. (C) Upper, The sequences of the wild-type and mutant pMMP2 Luc near the ATRS are provided. The wild-type and mutated ATRS sequences are underlined with the mutated nucleotides in boldface. Lower, 293T cells transfected with wild-type or mutant ATRS pMMP2 Luc with or without pCMV-NLS-EGFR were subjected to a luciferase reporter gene assay. (D) 293T cells transfected with pMMP2 Luc with a wild-type or a mutant ATRS and pCMV-NLS-EGFR were subjected to ChIP with an anti-EGFR antibody. The relative level of immunoprecipitated wild-type or mutant MMP2 ATRS were Figure 3 (See previous page). quantified by RT-qPCR with specific primers. (E) 293T cells transfected with pMMP2 Luc with a wild-type or a mutated ATRS and increasing amounts of pCMV-NLS-EGFR were subjected to ChIP with an anti-H3K9Ac antibody. The relative amount of ATRS derived from the immunoprecipitated MMP2 promoter was quantified by RT-qPCR with specific primers.
nEGFR cooperated with STAT3 to activate the MMP2 promoter
Recently, a STAT3 binding site was identified near the ATRS in the MMP2 promoter (Fig. 4A).41 Because nEGFR interacts with STAT3 in the promoters of their target genes,22 we investigated the role of STAT3 in the nEGFR-mediated activation of the MMP2 promoter. In H1975 cells, STAT3 knockdown by 2 different shRNA sequences significantly reduced MMP2 mRNA expression (Fig. 4B). Stattic, a STAT3 inhibitor, also decreased MMP2 mRNA expression (Fig. S3). Notably, STAT3 knockdown abolished nEGFR-regulated MMP2 promoter activity (Fig. 4C). Mutating the STAT3 binding site in the MMP2 promoter also abolished nEGFR-mediated MMP2 transcriptional activation (Fig. 4D). In addition, STAT3 overexpression further enhanced nEGFR-induced MMP2 promoter activity (Fig. 4E). Both nEGFR and STAT3 bound to the ATRS in the MMP2 promoter in the ChIP assay (Fig. 4F, left). After sequential immunoprecipitation with EGFR and STAT3 antibodies in the re-ChIP assay, more MMP2 ATRS was recovered in the immunoprecipitated complex than in the negative control sample, indicating that these 2 proteins associated with the same MMP2 ATRS. These results suggested that STAT3 and nEGFR interacted and bound together to the ATRS in the MMP2 promoter and that STAT3 was essential for nEGFR to activate the MMP2 promoter.
Figure 4.
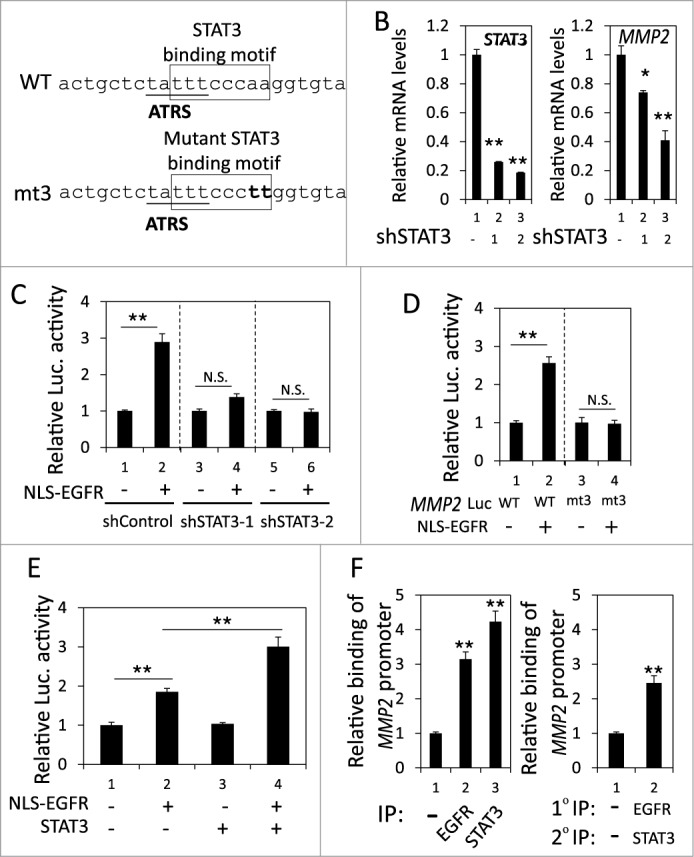
nEGFR and STAT3 cooperatively activated the MMP2 promoter. (A) The sequence near the ATRS in MMP2 promoter is illustrated. The ATRS is underlined. The wild-type and mutant STAT3 binding sites are specified in the open box. The mutated nucleotides in the STAT3 binding motif are capitalized. (B) shControl, shSTAT3-1 and shSTAT3-2 H1975 cells were subjected to RT-qPCR to analyze the MMP2 and STAT3 mRNA expression. (C) 293T cells transfected with pMMP2 Luc, pCMV-NLS-EGFR and the STAT3 shRNA-expression plasmid were subjected to a luciferase reporter gene assay. (D) 293T cells transfected with wild-type or mutant pMMP2 Luc and pCMV-NLS-EGFR were subjected to a luciferase reporter gene assay. (E) 293T cells transfected with pMMP2 Luc, pCMV-NLS-EGFR and/or a STAT3-expressing plasmid were subjected to a luciferase reporter gene assay. (F) Left, H1975 cells were subjected to ChIP with preimmune IgG or an anti-EGFR or anti-STAT3 antibody. Right, H1975 cells were subjected to ChIP with sequential immunoprecipitation with preimmune IgG or an anti-EGFR or anti-STAT3 antibody. The relative amount of the immunoprecipitated MMP2 ATRS DNA fragment was quantified by RT-qPCR with specific primers.
PML repressed the STAT3- and nEGFR-mediated induction of the MMP2 promoter by decreasing histone acetylation
We ascertained the effect of PML on the occupancy of STAT3 and nEGFR at the MMP2 promoter. In the reporter assay, PML overexpression reduced the MMP2 promoter activity induced by co-transfecting STAT3- and nEGFR-expressing plasmids, indicating that PML repressed STAT3/nEGFR-mediated MMP2 transcriptional activation (Fig. 5A). To determine the effect of PML on the binding of STAT3 and/or nEGFR to the MMP2 ATRS, we performed ChIP and found that the binding of STAT3 and nEGFR to the MMP2 promoter was not affected by PML overexpression or knockdown; these results indicated that PML did not affect the association of STAT3 and nEGFR with the MMP2 promoter (Fig. 5B and Fig. S4A). However, EGF increased PML binding to the MMP2 ATRS (Fig. S4B). In accordance with this observation, PML overexpression increased its association with the MMP2 promoter, and PML knockdown decreased its association with the MMP2 promoter, indicating that PML bound to the MMP2 promoter (Fig. 5C and Fig. S4C). Furthermore, the histone acetylation of the MMP2 promoter was significantly reduced by PML overexpression (Fig. 5D). Moreover, we performed ChIP and demonstrated that knockdown of EGFR significantly reduced the association of STAT3 and PML with the MMP2 ATRS; and knockdown of STAT3 significantly reduced the association of EGFR and PML with the MMP2 ATRS, indicating that both nEGFR and STAT3 were important for PML binding to the MMP2 ATRS (Fig. 5E, Fig. S4D, and S4E). Together, these results suggested that PML repressed STAT3/nEGFR-mediated MMP2 promoter activation by reducing the histone acetylation of this promoter.
Figure 5.
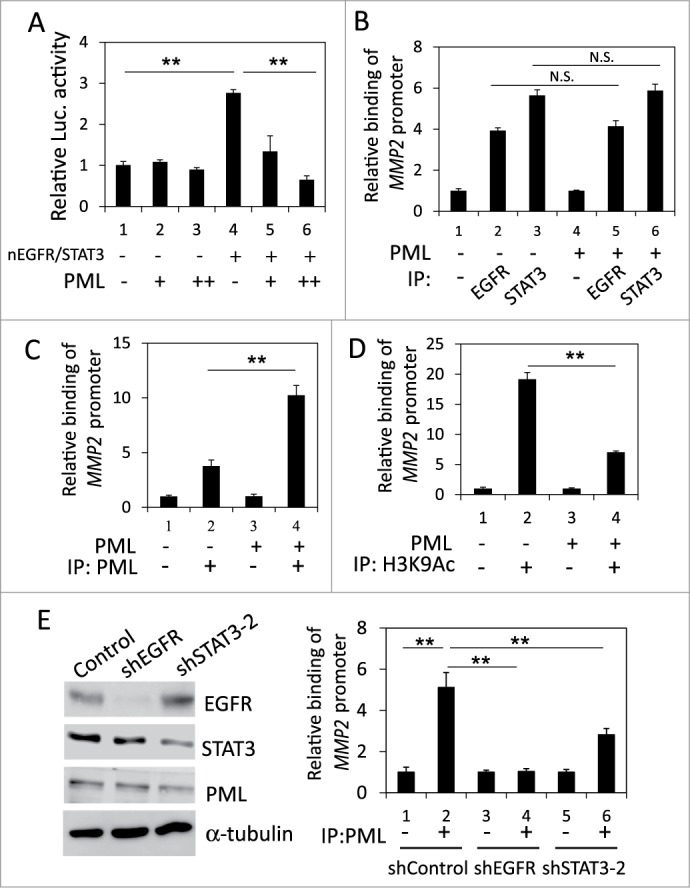
PML repressed the coactivation of the MMP2 promoter by nEGFR and STAT3. (A) 293T cells transfected with pMMP2 Luc, pCMV-NLS-EGFR, the STAT3 expression vector and increasing amounts of the PML expression plasmid were subjected to a luciferase reporter assay. (B) PML-knockdown H1975 cells with or without shRNA-resistant PML expression were subjected to ChIP with preimmune IgG or an anti-EGFR or anti-STAT3 antibody. The relative amount of the immunoprecipitated MMP2 ATRS was quantified by RT-qPCR. (C and D) PML-knockdown H1975 cells with or without shRNA-resistant PML expression were subjected to ChIP with preimmune IgG or an anti-PML or anti-H3K9Ac antibody. The relative amount of the immunoprecipitated MMP2 ATRS was quantified by RT-qPCR. (E) Left, H1975 cells infected with control, shEGFR or shSTAT3-2 lentiviral vectors were subjected to Western blotting with indicated antibodies 2 d after infection. Right, The same stable cell lines as in the left panel were subjected to ChIP with preimmune IgG or an anti-PML antibody. The amount of immunoprecipitated MMP2 ATRS was quantified by RT-qPCR.
PML was essential for the IFNβ-mediated repression of lung cancer cell invasion
It has been reported that PML expression is induced by numerous cytokines, including IFNβ.42 We examined whether IFNβ induced PML expression in lung cancer cells when EGFR was activated. Using real-time qPCR and Western blotting, we determined that IFNβ significantly enhanced PML mRNA and protein expression in H1975 cells and in EGF-treated A549 cells; therefore, IFNβ induced PML expression in lung cancer cells when EGFR was activated (Figs. 6A and 6B). We then investigated the effect of IFNβ on MMP2 expression in the above cells. IFNβ treatment significantly reduced MMP2 mRNA expression in H1975 cells and in EGF-treated A549 cells (Figs. S5 and 6C). However, the IFNβ-mediated suppression of MMP2 expression was attenuated by PML knockdown (Fig. 6D). Consistently, the invasion of EGF-treated A549 cells was significantly reduced by IFNβ (Fig. 6E, lane 3 versus lane 4). Together, these results suggested that IFNβ suppressed the invasion of EGFR-activated lung cancer cells by upregulating PML.
Figure 6.
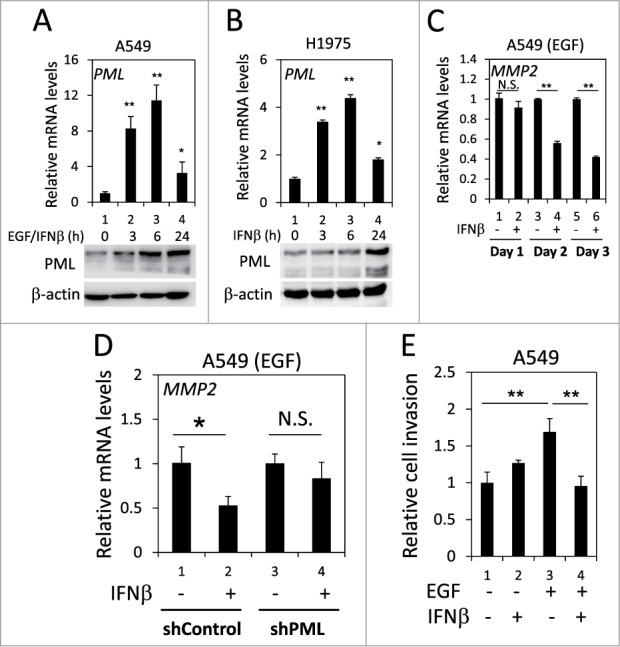
PML upregulation was involved in the IFNβ-mediated suppression of lung cancer cell invasion. (A) After overnight serum starvation, A549 cells were treated with EGF and IFNβ for the indicated periods of time. The cells were then subjected to RT-qPCR and Western blotting for PML mRNA and protein expression, respectively. (B) After overnight serum starvation, H1975 cells were treated with IFNβ for the indicated periods of time. The cells were analyzed as in (A). (C) After overnight serum starvation, A549 cells were treated with EGF and IFNβ for the indicated periods of time. The relative MMP2 mRNA expression was quantified by RT-qPCR with specific primers. (D) Control and PML-knockdown A549 cells were treated with EGF or IFNβ for 3 d and then analyzed by RT-qPCR to determine the relative MMP2 mRNA expression. (E) A549 cells treated with EGF and/or IFNβ for 24 h were subjected to a cell invasion assay as described in the Materials and Methods.
Discussion
Elevated expression of nEGFR predicts poor prognosis in many types of cancer, including lung cancer.43 nEGFR binds to the promoters of target genes, such as CCND1 and iNOS, and activates their transcription, leading to enhanced cancer cell proliferation and survival. Here we provide evidence that MMP2 is a novel nEGFR target gene that increases lung cancer cell invasion. The results presented here, along with previous findings, suggest that nEGFR promotes tumor progression by enhancing cancer cell proliferation, survival and invasion.
We demonstrated that the ATRS in the MMP2 promoter partially overlaps with a known STAT3 binding site (Fig. 4A).44 This observation suggested that nEGFR and STAT3 compete for binding to the MMP2 promoter. However, our ChIP and reporter gene assay results suggested that nEGFR and STAT3 bound cooperatively to the MMP2 ATRS and activated MMP2 gene transcription (Figs. 4E and 4F). Whether this novel binding motif is specific for the binding of the nEGFR/STAT3 complex to the MMP2 ATRS remains to be determined.
It has been reported that PML interacts with STAT3 to inhibit its binding to target DNA sequences and to repress STAT3 transcriptional activity.45 However, the ChIP results suggested that PML did not affect the association of STAT3 with the MMP2 promoter (Fig. 5B and Fig. S4A). This discrepancy may be due to the colocalization of nEGFR and STAT3 at the MMP2 promoter (Fig. 4F). In addition, knockdown of STAT3 led to reduced EGFR association with the MMP2 promoter, suggesting that STAT3 may facilitate the binding of EGFR to the MMP2 promoter.
In Figure 5E, knockdown of EGFR or STAT3 partially repressed the PML protein expression. This may lead to the decrease of PML binding to the MMP2 promoter. However, EGFR knockdown completely abolished the PML binding to MMP2 promoter while STAT3 knockdown only decreased half of that. Therefore, EGFR might be more important than STAT3 for the recruitment of PML to the MMP2 promoter.
Consistent with our result that knockdown of STAT3 reduced PML expression (Fig. 5E, left), it has been reported that STAT3 can bind to the IRSE of the PML promoter and activate its transcription.46 We also showed that knockdown of EGFR decreased STAT3 expression, although the underlying mechanism remains unclear (Fig. 5E, left).47 However, this result implied that the decrease of PML expression mediated by EGFR knockdown could be due to the downregulation of STAT3.
PML expression is induced by numerous cytokines, such as IFNγ, IL-6 and IFNβ. Among these cytokines, IFNβ is used clinically to treat patients with multiple sclerosis (MS).48 It has been proposed that the IFNβ-mediated downregulation of MMP2 expression in monocytes contributes to the beneficial effect of IFNβ in MS.49 Treating tumor cells with IFNβ decreases MMP2 expression, which suppresses tumor metastasis.50 De novo protein synthesis is required for optimal interferon-mediated inhibition of MMP2 expression.51 We determined that PML induction was essential for IFNβ to repress MMP2 expression in lung cancer cells. In many types of cancer, PML expression is post-translationally downregulated.6,52 Therefore, our results suggested that cancer cells in which PML is downregulated may escape regulation by IFNβ in the tumor microenvironment and by IFNβ-based gene or cell therapies.53
In conclusion, we demonstrated that PML repressed MMP2 expression, which decreased lung cancer cell invasion when EGFR was activated. Furthermore, we determined that MMP2 was a novel nEGFR target gene and that PML repressed the nEGFR-induced MMP2 promoter activity. We also showed that the induction of PML was essential for the IFNβ-mediated downregulation of MMP2 expression in lung cancer cells. Our findings suggested that lung cancer cells in which PML is downregulated may escape regulation by cytokines, such as IFNβ in the tumor microenvironment.
Materials and Methods
Cell culture and transfection
A549, H1975 and 293T cells were obtained from American Type Culture Collection. The CL1-5 cell line was established in our laboratories.54 The lung cancer and 293T cell lines were grown in RPMI-1640 and DMEM medium, respectively, with 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin in a humidified incubator with 5% CO2 at 37°C. The cells were transfected with DNA by using Lipofectamine 2000 (Invitrogen, 11668-019, http://www.lifetechnologies.com/order/catalog/product/11668019) according to the manufacturer's instructions.
Growth factors, cytokines and antibodies
Recombinant EGF (AF-100-15, http://www.peprotech.com/en-US/Pages/Product/Recombinant-Proteins/Growth-Factors-Cytokines/Recombinant_Human_EGF/AF-100-15) and IFNβ (AK8262-0002, http://www.akronbiotech.com/products/index.cfm?flist=1&famDetail=1&famDetailID=81&familyID=81) were purchased from PeproTech and Akron Biotech, respectively. The EGFR antibodies for Western blotting (D38B1, http://www.cellsignal.com/products/primary-antibodies/4267) and ChIP (MA5-13697, http://www.pierce-antibodies.com/EGFR-antibody-clone-528–19912-Mo noclonal–MA513697.html) were purchased from Cell Signaling Technology, and Thermo Scientific, respectively. The PML antibody was a gift from Dr. Kun-Sang Chang. The histone H3K9Ac antibody (GTX61103, http://www.genetex.com/Histone-H3K9ac-acetyl-Lys9-antibody-Y28-GTX61103.html) was purchased from GeneTex. The STAT3 antibody (D3Z2G, http://www.cellsignal.com/products/primary-antibodies/12640) was purchased from Cell Signaling Technology. The MMP2 antibody (AB19167, http://www.millipore.com/catalog/item/ab19167) was purchased from Millipore.
Plasmids
pLKO-shPML, pLKOneo-gD-EYFP and pLKOneo-gD-EYFP-rPML (shPML-resistant) were gifts from Dr. Roger Everette (MRC-University of Glasgow, Scotland, UK). pLKO-shPML-2, pLKO-shSTAT3-1, pLKO-shSTAT3-2 and pLKO-shEGFR were obtained from the RNAi Core Facility, Academia Sinica (Taipei, Taiwan). pLKO-shControl and pCMV-EGFR NLS have been previously described.40,55 The pMMP2 Luc plasmid was constructed by inserting MMP2 promoter fragments into the pGL3-Basic vector using the KpnI and XhoI restriction sites. The primer sequences used to generate the MMP2 promoter fragments are available in the supplementary information (Table S3). The pMMP2 Luc ATRS mutants were generated by site-directed mutagenesis with specific primers (sequence available in Table S4).
Invasion assay
A FluoroBlok 24-Multiwell Insert System with an 8-μm pore size polyethylene terephthalate membrane (BD Falcon, 35-1152) was used for the cell invasion assays. Before the cells were seeded, the insert chamber was coated with diluted Matrigel to mimic the in vivo basement membrane. Each well was filled with 700 μl medium, and 2 × 104 cells (4 × 104 cells for the experiments in Fig. 6E) were seeded into the insert chamber in 300 μl medium. After 18 h, the medium was removed, and the chamber was washed with 1X PBS and fixed in 100% methanol overnight at −20°C. After staining the cells with propidium iodide for 30 min, the migratory cells were visualized using an inverted fluorescence microscope. The cell number was quantitated using the ImageJ software. The data are presented as the mean ± SD of 3 independent experiments.
Luciferase reporter assay
Five × 104 A549 cells or 2 × 105 293T cells per well were seeded in 24-well plates the day before transfection. The cells were transfected with a total of 500 ng per well, including 200 ng pMMP2 Luc, various amount of plasmids as indicated in the figures and 100 ng pTK-RL to normalize for transfection efficiency. Firefly and Renilla luciferase activity were assayed sequentially using the Dual-Glo® Luciferase Assay System (Promega, E2940) 48 h after transfection. The data are presented as the mean ± SD of 4 independent experiments.
RNA extraction, RT-PCR and real time quantitative PCR. RNA was extracted from cells using TriPure Isolation Reagent (Roche, 11-667-165-001) according to the manufacturer's protocol. Extracted total RNA was reverse transcribed into cDNA using random hexamer primers and Transcriptor First Strand cDNA Synthesis Kit (Roche, 04-897-030-001). Each cDNA reaction was equally diluted for subsequent PCR amplification with the KAPA SYBR FAST ABI Prism 2X qPCR Master Mix (KAPA Biosystems, KK4602) using the StepOne Plus Real-Time PCR system (Applied Biosystems). The sequences of the primers designed to detect specific genes are available in Supplementary Table S1. The relative gene expression was normalized to GAPDH and was calculated using the 2−ΔΔCt methods. The data are presented as the mean ± SD of 3 independent experiments.
Chromatin immunoprecipitation
The cells were fixed with 1% formaldehyde for 15 min at room temperature and then 125 mM glycine was added. After nuclear fractionation, the chromatin was sheared, pre-cleared and immunoprecipitated with 3 μg of the indicated antibodies. Bound DNA-protein complexes were washed and then eluted. The cross-linked complexes were removed by incubation in 250 mM NaCl at 65°C for 4 h followed by precipitation and proteinase K digestion. The resulting ChIP DNA samples were purified by using a QIAquick PCR purification kit (Qiagen) and were eluted in 50 μl of elution buffer (10 mM Tris pH 8.0, and 1 mM EDTA). Five percent of the ChIP product or 1% of the input chromatin (control) was used in each PCR reaction. Specific ATRS sequences in the MMP2 promoter in the ChIP samples were detected by RT-qPCR with specific primers (the sequences are available in Supplementary Table S2). The percentage of the input recovered in the immunoprecipitated samples was normalized to the percentage of the input in the control IgG sample to obtain the relative binding. The data are presented as the mean ± SD of 3 independent experiments.
Virus generation, cell infection and stable cell line selection
In total, 2.2 × 106 293T cells were seeded in a 100-mm culture dish the day before transfection. The cells were co-transfected with the pLKO.1-based lentiviral vector, the Δ8.9 plasmid, and a vesicular stomatitis virus G protein plasmid. The medium was replaced with fresh culture medium after 24 h, and the lentivirus-containing medium was collected after another 24 h. The lentiviral infections were performed by adding the virus containing medium to the cells at the desired multiplicity of infection in the presence of 8 ng/ml polybrene. To generate PML-knockdown and the corresponding control cells (A549-shPML, A549-shControl, H1975-shPML, H1975-shPML-2 and H1975-shControl), the parental cells were infected with lentivirus expressing shPML or shControl; 24 h after infection, the cells were selected with puromycin (2 μg/ml) for 2 d To generate PML-overexpressing and the corresponding control cells (A549-PML, A549-Control, CL1–5-PML and CL1–5-Control), the parental cells were infected with PML or EYFP expressing lentivirus; 24 h after infection, the cells were selected with G418 (800μg/ml) for 7 d To generate PML add-back and the corresponding control H1975-shPML cells (H1975-shPML-rPML and H1975-shPML-Control), the H1975-shPML cells were further infected with rPML or EYFP expressing lentivirus; 24 h after infection, the cells were selected with G418 and puromycin for 7 d.
Tail vein metastasis assay
The animal procedures were performed in accordance with the Institutional Animal Care and Use Committee. A549 cells were engineered to stably express luciferase and shRNA using lentivirus particles. For the tail vein injection, 1 × 106 luciferase-expressing A549-shControl or A549-shPML cells in a volume of 0.1 ml were injected into 8-week-old male NOD/SCID mice via the tail vein (BioLasco Taiwan Co.). Pulmonary metastasis was monitored weekly via bioluminescence using the IVIS 200 system (PerkinElmer). Briefly, 150 mg/kg D-Luciferin (PerkinElmer, 122796, http://www.perkinelmer.com/Catalog/Product/ID/122796 ) was administrated approximately 5 to 10 minutes before imaging via an intraperitoneal injection, and the images were acquired at 1-minute intervals.
Statistical analysis
All the statistical analyses were performed using a 2-tailed Student's t-test. P values less than 0.05 were considered statistically significant (*P < 0.05, **P < 0.01).
Supplementary Material
Funding Statement
This research was supported by the Institute of Biomedical Sciences, Academia Sinica, the National Yang-Ming University (103AC-P902), the Ministry of Health and Welfare (MOHW103-TD-B-111-02), and the Ministry of Science and Technology (NSC102-2325-B-010-010; NSC102-2321-B-010-010), Taiwan.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Roger D. Everett, Dr. Kun-Sang Chang for providing reagents. We are very grateful to Miss Hsin-Yi Hsieh (National Defense Medical Center, Taiwan) and Mr. Pei-Hsiang Hou (Institute of Biomedical Sciences, Academia Sinica) for helping metastatic models. We also thank the core facility of the Taiwan Mouse Clinic.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69-90; PMID:21296855; http://dx.doi.org/ 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 2. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 2006; 12:895-904; PMID: 16892035; http://dx.doi.org/ 10.1038/nm1469 [DOI] [PubMed] [Google Scholar]
- 3. Guo CB, Wang S, Deng C, Zhang DL, Wang FL, Jin XQ. Relationship between matrix metalloproteinase 2 and lung cancer progression. Mol Diagn Ther 2007; 11:183-92; PMID:17570740; http://dx.doi.org/ 10.1007/BF03256240 [DOI] [PubMed] [Google Scholar]
- 4. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev 2006; 25:9-34; PMID:16680569; http://dx.doi.org/ 10.1007/s10555-006-7886-9 [DOI] [PubMed] [Google Scholar]
- 5. Reineke EL, Kao HY. PML: an emerging tumor suppressor and a target with therapeutic potential. Cancer Ther 2009; 7:219-26; PMID:19756257 [PMC free article] [PubMed] [Google Scholar]
- 6. Gurrieri C, Capodieci P, Bernardi R, Scaglioni PP, Nafa K, Rush LJ, Verbel DA, Cordon-Cardo C, Pandolfi PP. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J Natl Cancer Inst 2004; 96:269-79; PMID:14970276; http://dx.doi.org/ 10.1093/jnci/djh043 [DOI] [PubMed] [Google Scholar]
- 7. Li Q, He Y, Wei L, Wu X, Wu D, Lin S, Wang Z, Ye Z, Lin SC. AXIN is an essential co-activator for the promyelocytic leukemia protein in p53 activation. Oncogene 2011; 30:1194-204; PMID:21057547; http://dx.doi.org/ 10.1038/onc.2010.499 [DOI] [PubMed] [Google Scholar]
- 8. Vannucchi S, Percario ZA, Chiantore MV, Matarrese P, Chelbi-Alix MK, Fagioli M, Pelicci PG, Malorni W, Fiorucci G, Romeo G, et al. Interferon-beta induces S phase slowing via up-regulated expression of PML in squamous carcinoma cells. Oncogene 2000; 19:5041-53; PMID:11042692; http://dx.doi.org/ 10.1038/sj.onc.1203883 [DOI] [PubMed] [Google Scholar]
- 9. Bernardi R, Guernah I, Jin D, Grisendi S, Alimonti A, Teruya-Feldstein J, Cordon-Cardo C, Simon MC, Rafii S, Pandolfi PP. PML inhibits HIF-1alpha translation and neoangiogenesis through repression of mTOR. Nature 2006; 442:779-85; PMID:16915281; http://dx.doi.org/ 10.1038/nature05029 [DOI] [PubMed] [Google Scholar]
- 10. Reineke EL, Liu Y, Kao HY. Promyelocytic leukemia protein controls cell migration in response to hydrogen peroxide and insulin-like growth factor-1. J Biol Chem 2010; 285:9485-92; PMID:20100838; http://dx.doi.org/ 10.1074/jbc.M109.063362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan WC, Lee YR, Huang SF, Lin YM, Chen TY, Chung HC, Tsai CH, Chen HY, Chiang CT, Lai CK, et al. A Cullin3-KLHL20 Ubiquitin ligase-dependent pathway targets PML to potentiate HIF-1 signaling and prostate cancer progression. Cancer Cell 2011; 20:214-28; PMID:21840486; http://dx.doi.org/ 10.1016/j.ccr.2011.07.008 [DOI] [PubMed] [Google Scholar]
- 12. Zhou W, Bao S. PML-mediated signaling and its role in cancer stem cells. Oncogene 2013; 33:1475-84; PMID:23563177; http://dx.doi.org/ 10.1038/onc.2013.111 [DOI] [PubMed] [Google Scholar]
- 13. Wang SC, Nakajima Y, Yu YL, Xia W, Chen CT, Yang CC, McIntush EW, Li LY, Hawke DH, Kobayashi R, et al. Tyrosine phosphorylation controls PCNA function through protein stability. Nat Cell Biol 2006; 8:1359-68; PMID:17115032 [DOI] [PubMed] [Google Scholar]
- 14. Liccardi G, Hartley JA, Hochhauser D. EGFR nuclear translocation modulates DNA repair following cisplatin and ionizing radiation treatment. Cancer Res 2011; 71:1103-14; PMID:21266349; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-2384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dittmann K, Mayer C, Fehrenbacher B, Schaller M, Kehlbach R, Rodemann HP. Nuclear EGFR shuttling induced by ionizing radiation is regulated by phosphorylation at residue Thr654. FEBS Lett 2010; 584:3878-84; PMID:20692258; http://dx.doi.org/ 10.1016/j.febslet.2010.08.005 [DOI] [PubMed] [Google Scholar]
- 16. Hsu SC, Miller SA, Wang Y, Hung MC. Nuclear EGFR is required for cisplatin resistance and DNA repair. Am J Transl Res 2009; 1:249-58; PMID:19956435 [PMC free article] [PubMed] [Google Scholar]
- 17. Li C, Iida M, Dunn EF, Ghia AJ, Wheeler DL. Nuclear EGFR contributes to acquired resistance to cetuximab. Oncogene 2009; 28:3801-13; PMID:19684613; http://dx.doi.org/ 10.1038/onc.2009.234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang WC, Chen YJ, Li LY, Wei YL, Hsu SC, Tsai SL, Chiu PC, Huang WP, Wang YN, Chen CH, et al. Nuclear translocation of epidermal growth factor receptor by Akt-dependent phosphorylation enhances breast cancer-resistant protein expression in gefitinib-resistant cells. J Biol Chem 2011; 286:20558-68; PMID:21487020; http://dx.doi.org/ 10.1074/jbc.M111.240796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee PC, Lee HJ, Kakadiya R, Sanjiv K, Su TL, Lee TC. Multidrug-resistant cells overexpressing P-glycoprotein are susceptible to DNA crosslinking agents due to attenuated Src/nuclear EGFR cascade-activated DNA repair activity. Oncogene 2012; 32:1144-54; PMID:22525278; http://dx.doi.org/ 10.1038/onc.2012.133 [DOI] [PubMed] [Google Scholar]
- 20. Yu YL, Chou RH, Wu CH, Wang YN, Chang WJ, Tseng YJ, Chang WC, Lai CC, Lee HJ, Huo L, et al. Nuclear EGFR suppresses ribonuclease activity of polynucleotide phosphorylase through DNAPK-mediated phosphorylation at serine 776. J Biol Chem 2012; 287:31015-26; PMID:22815474; http://dx.doi.org/ 10.1074/jbc.M112.358077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lin SY, Makino K, Xia W, Matin A, Wen Y, Kwong KY, Bourguignon L, Hung MC. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat Cell Biol 2001; 3:802-8; PMID:11533659 [DOI] [PubMed] [Google Scholar]
- 22. Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell 2005; 7:575-89; PMID:15950906; http://dx.doi.org/ 10.1016/j.ccr.2005.05.007 [DOI] [PubMed] [Google Scholar]
- 23. Hanada N, Lo HW, Day CP, Pan Y, Nakajima Y, Hung MC. Co-regulation of B-Myb expression by E2F1 and EGF receptor. Mol Carcinog 2006; 45:10-7; PMID:16299810; http://dx.doi.org/ 10.1002/mc.20147 [DOI] [PubMed] [Google Scholar]
- 24. Huo L, Wang YN, Xia W, Hsu SC, Lai CC, Li LY, Chang WC, Wang Y, Hsu MC, Yu YL, et al. RNA helicase A is a DNA-binding partner for EGFR-mediated transcriptional activation in the nucleus. Proc Natl Acad Sci U S A 2010; 107:16125-30; PMID:20802156; http://dx.doi.org/ 10.1073/pnas.1000743107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xia W, Wei Y, Du Y, Liu J, Chang B, Yu YL, Huo LF, Miller S, Hung MC. Nuclear expression of epidermal growth factor receptor is a novel prognostic value in patients with ovarian cancer. Mol Carcinog 2009; 48:610-7; PMID:19058255; http://dx.doi.org/ 10.1002/mc.20504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hadzisejdic I, Mustac E, Jonjic N, Petkovic M, Grahovac B. Nuclear EGFR in ductal invasive breast cancer: correlation with cyclin-D1 and prognosis. Mod Pathol 2010; 23:392-403; PMID:20062009; http://dx.doi.org/ 10.1038/modpathol.2009.166 [DOI] [PubMed] [Google Scholar]
- 27. Wang YN, Wang H, Yamaguchi H, Lee HJ, Lee HH, Hung MC. COPI-mediated retrograde trafficking from the Golgi to the ER regulates EGFR nuclear transport. Biochem Biophys Res Commun 2010; 399:498-504; PMID:20674546; http://dx.doi.org/ 10.1016/j.bbrc.2010.07.096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem 2007; 282:10432-40; PMID:17283074; http://dx.doi.org/ 10.1074/jbc.M610014200 [DOI] [PubMed] [Google Scholar]
- 29. Wang YN, Yamaguchi H, Hsu JM, Hung MC. Nuclear trafficking of the epidermal growth factor receptor family membrane proteins. Oncogene 2010; 29:3997-4006; PMID:20473332; http://dx.doi.org/ 10.1038/onc.2010.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang YN, Yamaguchi H, Huo L, Du Y, Lee HJ, Lee HH, Wang H, Hsu JM, Hung MC. The translocon Sec61beta localized in the inner nuclear membrane transports membrane-embedded EGF receptor to the nucleus. J Biol Chem 2010; 285:38720-9; PMID:20937808; http://dx.doi.org/ 10.1074/jbc.M110.158659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Iida M, Brand TM, Campbell DA, Li C, Wheeler DL. Yes and Lyn play a role in nuclear translocation of the epidermal growth factor receptor. Oncogene 2012; 32:759-67; PMID:22430206; http://dx.doi.org/ 10.1038/onc.2012.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li C, Iida M, Dunn EF, Wheeler DL. Dasatinib blocks cetuximab- and radiation-induced nuclear translocation of the epidermal growth factor receptor in head and neck squamous cell carcinoma. Radiother Oncol 2010; 97:330-7; PMID:20667610; http://dx.doi.org/ 10.1016/j.radonc.2010.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brand TM, Iida M, Dunn EF, Luthar N, Kostopoulos KT, Corrigan KL, Wleklinski MJ, Yang D, Wisinski KB, Salgia R, et al. Nuclear epidermal growth factor receptor is a functional molecular target in triple-negative breast cancer. Mol Cancer Ther 2014; 13:1356-1368. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 34. Du Y, Shen J, Hsu JL, Han Z, Hsu MC, Yang CC, Kuo HP, Wang YN, Yamaguchi H, Miller SA, et al. Syntaxin 6-mediated Golgi translocation plays an important role in nuclear functions of EGFR through microtubule-dependent trafficking. Oncogene 2014; 33:756-70; PMID:23376851; http://dx.doi.org/ 10.1038/onc.2013.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004; 305:1163-7; PMID:15284455; http://dx.doi.org/ 10.1126/science.1101637 [DOI] [PubMed] [Google Scholar]
- 36. Leinonen T, Pirinen R, Bohm J, Johansson R, Kosma VM. Increased expression of matrix metalloproteinase-2 (MMP-2) predicts tumour recurrence and unfavourable outcome in non-small cell lung cancer. Histol Histopathol 2008; 23:693-700; PMID:18366007 [DOI] [PubMed] [Google Scholar]
- 37. Xu M, Wang YZ. miR133a suppresses cell proliferation, migration and invasion in human lung cancer by targeting MMP14. Oncol Rep 2013; 30:1398-404; PMID:23783274 [DOI] [PubMed] [Google Scholar]
- 38. Zheng S, Chang Y, Hodges KB, Sun Y, Ma X, Xue Y, Williamson SR, Lopez-Beltran A, Montironi R, Cheng L. Expression of KISS1 and MMP-9 in non-small cell lung cancer and their relations to metastasis and survival. Anticancer Res 2010; 30:713-8; PMID:20392988 [PubMed] [Google Scholar]
- 39. Wang XM, Li J, Yan MX, Liu L, Jia DS, Geng Q, Lin HC, He XH, Li JJ, Yao M. Integrative analyses identify osteopontin, LAMB3 and ITGB1 as critical pro-metastatic genes for lung cancer. PLoS One 2013; 8:e55714; PMID:23441154; http://dx.doi.org/ 10.1371/journal.pone.0055714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuo HY, Chen YC, Chang HY, Jeng JC, Lin EH, Pan CM, Chang YW, Wang ML, Chou YT, Shih HM, et al. The PML isoform IV is a negative regulator of nuclear EGFR's transcriptional activity in lung cancer. Carcinogenesis 2013; 34:1708-16; PMID:23563092; http://dx.doi.org/ 10.1093/carcin/bgt109 [DOI] [PubMed] [Google Scholar]
- 41. Xie TX, Wei D, Liu M, Gao AC, Ali-Osman F, Sawaya R, Huang S. Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 2004; 23:3550-60; PMID:15116091; http://dx.doi.org/ 10.1038/sj.onc.1207383 [DOI] [PubMed] [Google Scholar]
- 42. Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saib A, Quignon F, Pelicano L, Guillemin MC, Schindler C, et al. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 1995; 11:2565-73; PMID:8545113 [PubMed] [Google Scholar]
- 43. Traynor AM, Weigel TL, Oettel KR, Yang DT, Zhang C, Kim K, Salgia R, Iida M, Brand TM, Hoang T, et al. Nuclear EGFR protein expression predicts poor survival in early stage non-small cell lung cancer. Lung Cancer 2013; 81:138-41; PMID:23628526; http://dx.doi.org/ 10.1016/j.lungcan.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang W, Yu LF, Zhong J, Wu W, Zhu JY, Jiang FX, Wu YL. Stat3 is involved in angiotensin II-induced expression of MMP2 in gastric cancer cells. Dig Dis Sci 2009; 54:2056-62; PMID:19082717; http://dx.doi.org/ 10.1007/s10620-008-0617-z [DOI] [PubMed] [Google Scholar]
- 45. Kawasaki A, Matsumura I, Kataoka Y, Takigawa E, Nakajima K, Kanakura Y. Opposing effects of PML and PML/RAR alpha on STAT3 activity. Blood 2003; 101:3668-73; PMID:12506013; http://dx.doi.org/ 10.1182/blood-2002-08-2474 [DOI] [PubMed] [Google Scholar]
- 46. Hubackova S, Krejcikova K, Bartek J, Hodny Z. Interleukin 6 signaling regulates promyelocytic leukemia protein gene expression in human normal and cancer cells. J Biol Chem 2012; 287:26702-14; PMID:22711534; http://dx.doi.org/ 10.1074/jbc.M111.316869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Viti J, Feathers A, Phillips J, Lillien L. Epidermal growth factor receptors control competence to interpret leukemia inhibitory factor as an astrocyte inducer in developing cortex. J Neurosci 2003; 23:3385-93; PMID:12716946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arnason BG. Immunologic therapy of multiple sclerosis. Annu Rev Med 1999; 50:291-302; PMID:10073279; http://dx.doi.org/ 10.1146/annurev.med.50.1.291 [DOI] [PubMed] [Google Scholar]
- 49. Galboiz Y, Shapiro S, Lahat N, Miller A. Modulation of monocytes matrix metalloproteinase-2, MT1-MMP and TIMP-2 by interferon-gamma and -beta: implications to multiple sclerosis. J Neuroimmunol 2002; 131:191-200; PMID:12458052; http://dx.doi.org/ 10.1016/S0165-5728(02)00266-7 [DOI] [PubMed] [Google Scholar]
- 50. Ling X, Marini F, Konopleva M, Schober W, Shi Y, Burks J, Clise-Dwyer K, Wang RY, Zhang W, Yuan X, et al. Mesenchymal stem cells overexpressing IFN-beta inhibit breast cancer growth and metastases through Stat3 signaling in a syngeneic Tumor model. Cancer Microenviron 2010; 3:83-95; PMID:21209776; http://dx.doi.org/ 10.1007/s12307-010-0041-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Qin H, Moellinger JD, Wells A, Windsor LJ, Sun Y, Benveniste EN. Transcriptional suppression of matrix metalloproteinase-2 gene expression in human astroglioma cells by TNF-alpha and IFN-gamma. J Immunol 1998; 161:6664-73; PMID:9862695 [PubMed] [Google Scholar]
- 52. Guan D, Factor D, Liu Y, Wang Z, Kao HY. The epigenetic regulator UHRF1 promotes ubiquitination-mediated degradation of the tumor-suppressor protein promyelocytic leukemia protein. Oncogene 2013; 32:3819-28; PMID:22945642; http://dx.doi.org/ 10.1038/onc.2012.406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Park MY, Kim DR, Jung HW, Yoon HI, Lee JH, Lee CT. Genetic immunotherapy of lung cancer using conditionally replicating adenovirus and adenovirus-interferon-beta. Cancer Gene Ther 2010; 17:356-64; PMID:19893592; http://dx.doi.org/ 10.1038/cgt.2009.78 [DOI] [PubMed] [Google Scholar]
- 54. Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R, Wu CW. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol 1997; 17:353-60; PMID:9308922; http://dx.doi.org/ 10.1165/ajrcmb.17.3.2837 [DOI] [PubMed] [Google Scholar]
- 55. Chou YT, Lin HH, Lien YC, Wang YH, Hong CF, Kao YR, Lin SC, Chang YC, Lin SY, Chen SJ, et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res 2010; 70:8822-31; PMID:20978205; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0638 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.