

Summary
Our objective was to determine the role of the inflammatory cytokine interleukin‐23 (IL‐23) in promoting neutrophil recruitment, inflammatory cytokine expression and intestinal histopathology in response to Clostridium difficile infection. Wild‐type (WT) and p19−/− (IL‐23KO) mice were pre‐treated with cefoperazone in their drinking water for 5 days, and after a 2‐day recovery period were challenged with spores from C. difficile strain VPI 10463. Interleukin‐23 deficiency was associated with significant defects in both the recruitment of CD11bHigh Ly6GH igh neutrophils to the colon and the expression of neutrophil chemoattractants and stabilization factors including Cxcl1, Cxcl2, Ccl3 and Csf3 within the colonic mucosa as compared with WT animals. Furthermore, the expression of inflammatory cytokines including Il33, Tnf and Il6 was significantly reduced in IL‐23‐deficient animals. There was also a trend towards less severe colonic histopathology in the absence of IL‐23. The induction of Il17a and Il22 was also significantly abrogated in IL‐23KO mice. Inflammatory cytokine expression and neutrophilic inflammation were not reduced in IL‐17a‐deficient mice or in mice treated with anti‐IL‐22 depleting monoclonal antibody. However, induction of RegIIIg was significantly reduced in animals treated with anti‐IL‐22 antibody. Taken together, these data indicate that IL‐23, but not IL‐17a or IL‐22, promotes neutrophil recruitment and inflammatory cytokine and chemokine expression in the colon in response to C. difficile infection.
Keywords: Clostridium difficile, colitis, innate inflammation, mucosal inflammation, neutrophils
Introduction
Marked neutrophil recruitment is one of the most prominent host responses to Clostridium difficile infection in antibiotic pre‐treated mice.1, 2, 3, 4, 5, 6 This neutrophil recruitment appears to be protective to the host, as numerous studies have demonstrated reduced survival during C. difficile infection following interventions that reduced neutrophil recruitment.1, 2, 3 Although recent studies have demonstrated roles for Myeloid Differentiation Primary Response 88,3 Apoptosis‐Associated Speck‐Like Protein Containing a CARD,1 Nucleotide Binding Oligomerization Domain 12 and interleukin‐22 (IL‐22) and CD1606 in driving neutrophil recruitment to the large bowel during C. difficile infection, our understanding of the host signals promoting neutrophil recruitment remains incomplete.
Neutrophil mobilization and recruitment are intimately associated with C. difficile infection in humans as well. Neutrophilic infiltration is prominent in large colon tissue from patients with pseudomembranous colitis,7 and increased neutrophilia in the bloodstream is common during C. difficile colitis.8 Furthermore, the mobilization of neutrophils may play a role in determining the outcome of disease, as a recent study has reported a strong association between increased neutrophil numbers in the blood and increased risk of mortality.9
Neutrophil recruitment is a rapid and common host response to insult at mucosal sites.2, 4, 5, 10, 11, 12, 13 Neutrophils are myeloid cells that are rapidly recruited to sites of inflammation,1, 2, 3, 4, 5, 6, 10 and are characterized by high levels of Ly6G and CD11b expression.1, 2, 3, 10, 14 The influx of neutrophils into peripheral tissues is often associated with expression or production of two potent neutrophil chemokines, CXCL1 and CXCL2, which activate neutrophils and promote their egress from the bone marrow.1, 3, 4, 5, 11, 14, 15, 16
Interleukin‐23 is a heterodimeric cytokine comprised of a p19 and a p40 subunit,17 which promotes innate inflammatory responses during mucosal inflammation at numerous sites.10, 11, 18, 19, 20 Neutrophil recruitment during both Pseudomonas aeruginosa 18, 19 and bleomycin‐mediated20 pulmonary inflammation is partially dependent upon IL‐23 signalling. Specific to intestinal inflammation, IL‐23 also promotes neutrophil recruitment in response to Salmonella typhimurium typhlocolitis11 and Dextran Sodium Sulphate‐induced colitis.10 Interleukin‐23 can also promote the development of severe intestinal histopathology in response to infectious insult.21 Recent studies have also reported IL‐23 secretion by bone marrow‐derived dendritic cells in response to stimulation with a combination of microbial pathogen‐associated molecular patterns and C. difficile toxins,22 and reduced mortality and morbidity following C. difficile infection of IL‐23‐deficient mice.23 However, the role of IL‐23 in driving neutrophil recruitment during C. difficile colitis has yet to be investigated.
Both IL‐17 and IL‐22 also promote neutrophil recruitment to mucosal sites, and are induced in response to mucosal inflammation. Interleukin‐17 is known to promote neutrophil recruitment and epithelial damage during inflammatory responses in both the lung and the intestine.12, 13, 24, 25, 26 Interleukin‐22 signalling can promote neutrophil recruitment during pulmonary inflammation24 as well as drive neutrophil‐attractive chemokine expression from colonic cells;27, 28 IL‐22 also protects against severe histopathology during infectious murine colitis.29 Furthermore, IL‐23 can drive the induction of both IL‐22 and IL‐17 in numerous models of mucosal inflammation.10, 11, 19, 21, 29
In the current study, our objective was to investigate the role of IL‐23 in driving neutrophil recruitment, inflammatory cytokine expression and intestinal histopathology during C. difficile colitis. If so, our objective was to investigate the relative contributions of IL‐17 and IL‐22, two cytokines whose induction at sites of mucosal inflammation is controlled by IL‐23, in driving neutrophil recruitment, colonic histopathology and inflammatory cytokine expression in the colon in response to C. difficile infection.
Materials and methods
Animals and housing
Male C57BL/6 mice aged 5–11 weeks, and male and female IL‐17a−/− (IL‐17KO) and p19−/− (IL‐23KO) mice aged 5–14 weeks were used in the current study. C57BL/6 mice were obtained from an in‐house colony founded by Jackson breeders, and IL‐17a−/− (IL‐17KO) and p19−/− (IL‐23KO) on a C57BL/6 background were likewise obtained from a breeding colony maintained at the University of Michigan. The breeding pairs of IL‐23−/− mice were a kind gift from Ben Segal at the University of Michigan. Mice were maintained under specific pathogen‐free conditions, and autoclaved food, water and bedding were provided ad libitum. All animal manipulations were carried out in a laminar flow hood. All experiments were conducted in accordance with a protocol approved by the University Committee on Use and Care of Animals at the University of Michigan.
Clostridium difficile spore preparation
VPI 10463 spore stocks were generated by plating an earlier spore preparation on taurocholate cefoxitin cycloserine fructose agar (TCCFA) plates anaerobically. Single colonies were isolated, and grown overnight in Columbia broth. Two millilitres of the overnight culture was inoculated into 40 ml of Clospore broth,30 and the culture was allowed to grow for 7 days. Spores were collected by centrifugation, and washed to remove vegetative cell debris. All spore stocks were stored in water at 4° until used.
Clostridium difficile infection
Mice were given a 5‐day course of cefoperazone (0·5 g/l) in their drinking water to permit C. difficile infection as described previously.31 After a 2‐day recovery period, mice were challenged via oral gavage with 5·70 ± 0·25 log10 C. difficile spores from strain VPI 10463. Animals were followed for an additional 2 days, and all samples were collected at 2 days post infection. Inoculum dosage was confirmed by serially diluting and plating an aliquot of the inoculum on TCCFA plates anaerobically. Animals were monitored following infection for signs of severe disease, including lethargy, hunched posture and > 20% weight loss. Any animals found moribund were humanely euthanized. Untreated animals did not receive antibiotics or C. difficile challenge.
Anti‐IL‐22 treatment
Animals were given two intraperitoneal injections of anti‐IL‐22 monoclonal antibody (mAb; clone 8E11). Each mouse received 150 μg antibody 1 day before and 1 day after infection.29 The anti‐IL‐22 mAb was a kind gift from Dr Wenjun Ouyang.
Histology
Colonic tissue was fixed in 10% formalin for at least 24 hr, and then transferred to 70% ethanol. Tissue was processed, paraffin embedded, sectioned and used to prepare haematoxylin & eosin stained slides by McClinchey Histology Labs Inc., Stockbridge, MI. Representative images were acquired using an Olympus BX40 light microscope (Olympus Corporation, Center Valley, PA) and a qimaging micropublisher RTV 5.0 5 megapixel camera. All images were acquired at a total magnification of 400 ×. Panels were assembled in adobe photoshop CS5, version 12.0. Image processing was restricted to global adjustments of brightness, contrast and image size.
Histological scoring
Light microscopic evaluation of haematoxylin & eosin stained colonic sections was performed by a board‐certified veterinary pathologist. The pathologist was blinded to experimental groupings at the time of the evaluation, and sections were scored using a previously established system,32, 33 Oedema: 0 no oedema #bib1 mild, focal or multifocal oedema with minimal submucosal expansion (< 2×), 2 moderate multifocal oedema with moderate submucosal expansion (2–3 ×), 3 severe multifocal to coalescing oedema with severe submucosal expansion (> 3 ×), 4 same as 3 with diffuse submucosal expansion. Inflammation: 0 no inflammation, 1 minimal, multifocal neutrophilic infiltration, 2 moderate, multifocal neutrophilic infiltration (greater submucosal involvement), 3 severe multifocal to coalescing neutrophilic infiltration (greater submucosal ± mural involvement), 4 same as 3 with abscesses or extensive transmural involvement. Epithelial damage: 0 no epithelial damage, 1 mild multifocal, superficial damage (vacuolation, increased apoptosis, villus tip attenuation/necrosis), 2 moderate, multifocal superficial damage (same qualitative changes as above), 3 severe multifocal to coalescing mucosal damage ± pseudomembrane formation (intraluminal aggregate of neutrophils and sloughed epithelium in a fibrinous matrix covering eroded or ulcerated mucosa), 4 same as 3 with extensive pseudomembrane or ulcer formation.
RNA isolation and expression analysis
Colonic tissue samples (~ 1 cm2) were collected from the centre of the colon and stored in RNAlater (Ambion, Austin, TX). RNA isolation and purification from colonic tissue was performed as described previously.4, 5, 6, 31 Tissue was homogenized in TRIzol reagent (Life Technologies, Carlsbad, CA) and the resulting RNA was purified using the RNeasy Mini kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The concentration of the purified RNA was determined using a Nanodrop instrument (Thermo Fisher, Waltham, MA). Synthesis of cDNA, using the purified RNA as a template, was performed using the RT2 First Strand kit (Qiagen), and colonic gene expression was assessed using RT2 Profiler PCR arrays (Qiagen). All reactions were run on a Roche Lightcycler 480. To correct for variation between RT2 Profiler PCR arrays, cross card normalization was performed as described previously.5, 34 ΔC t (dC t) values were calculated by subtracting the geometric mean of two internal control genes from the Ct value of the gene of interest.35 The 2−ddCt method was used to calculate fold change gene expression in treatment groups compared with untreated animals for all comparisons.36
Leucocyte isolation
Leucocytes were isolated from colonic tissue as described previously.4 Isolated colonic tissue was minced with serrated scissors to physically disrupt the tissue, and was subsequently incubated in 20 ml Hanks' balanced salt solution (HBSS) supplemented with 2·5% fetal bovine serum #bib5 mm EDTA and 1 mm dithiothreitol for 20 min at 37°. Tissue was then incubated in 20 ml of a digest solution consisting of HBSS supplemented with 2·5% fetal bovine serum #bib400 U/ml collagenase type 3 (Worthington Biochemical, Lakewood, NJ) and 0·5 mg/ml DNAse I (Roche, Basel, Switzerland) for 60 min at 37°. Samples were then resuspended in 20% Percoll (Sigma, St Louis, MO) in PBS, and centrifuged at 900 g for 30 min at room temperature without brake. The resulting single cell suspensions were stained for flow cytometric analysis.
Flow staining and analysis
Single‐cell suspensions were plated at a concentration of approximately 106 cells per well in a 96‐well plate. Cells were blocked with unlabelled FC RIII/II, and then stained with fluorescently labelled antibodies for 30 min. Cells were washed to remove excess antibody, and resuspended in stabilizing fixative (BD Biosciences, Franklin Lakes, CA). Data were collected on a three‐laser Canto II using FACSdiva software (BD Biosciences). All data analysis was performed in flowjo (Treestar, Ashland, OR). Isolated colonic cells were stained with the following antibodies: CD45 (clone 30‐F11), CD11b (clone M1/70) and Ly6G (clone IA8) as well as Fc RIII/II (clone 2·4G2). All antibodies were purchased from eBioscience (San Diego, CA), BD Pharmingen (Franklin Lakes, CA) and Biolegend (San Diego, CA).
Total number of neutrophils per colon was calculated by multiplying the frequency of CD45High CD11bHigh Ly6GHigh neutrophils as defined by flow cytometry by the total number of cells in the colon in question. For all animals, the entirety of the colon was taken and processed for leucocyte isolation and analysis by FACS.
Statistical analysis
Statistically significant differences in gene expression were determined using a one‐way analysis of variance with Tukey's post hoc test for multiple comparisons. For all quantitative PCR data, statistical tests were performed on normalized dC t values.4, 31 A one‐way analysis of variance with Tukey's post hoc test was also used to identify significant differences in the number of neutrophils per colon. Significant differences in histopathological scoring were determined using the Kruskal–Wallis test followed by Dunn's multiple comparisons test. For all analyses, significance was set at P ≤ 0·05.
Results
Effect of IL‐23 deficiency on colonic neutrophil recruitment
For these studies, WT and p19−/− (IL‐23KO) mice were given cefoperazone (0·5 g/l) in their drinking water for 5 days as described previously.6, 31 Following a 2‐day recovery period on regular water, mice were challenged with 5·70 ± 0·25log10 C. difficile spores (strain VPI 10463). Animals were followed for an additional 2 days, and all samples were collected at 2 days post‐infection. All infected groups had a mean C. difficile colonization level of ≥ 105 CFU/g host tissue (data now shown).
To determine the role of IL‐23 in driving neutrophil recruitment in response to C. difficile colitis, flow cytometry was used to identify recruited leucocytes. Analysis of colonic leucocytes isolated from WT animals revealed a drastic influx of CD11bHigh Ly6GHigh neutrophils following C. difficile infection (Fig. 1a). In contrast, the frequency of the CD11bHigh Ly6GHigh neutrophil population was markedly reduced in IL‐23KO animals (Fig. 1a). Further quantification of the total number of CD11bHigh Ly6GHigh neutrophils per colon revealed a statistically significant reduction in the total number of neutrophils recruited to the colons of IL‐23KO animals compared with WT (Fig. 1b). These data demonstrate a significant reduction in neutrophil recruitment to the colon in response to C. difficile colitis in the absence of IL‐23.
Figure 1.
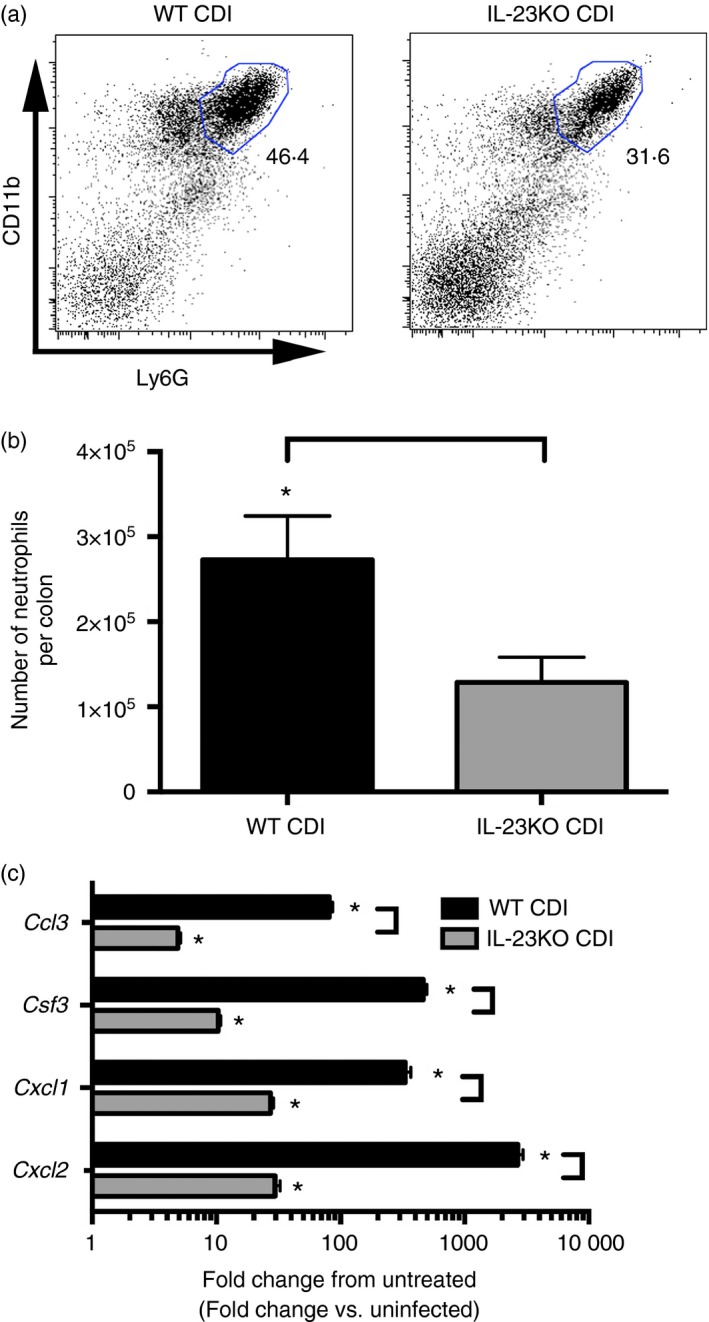
Colonic neutrophil recruitment and neutrophil chemokine expression in response to Clostridium difficile infection in the absence of interleukin‐23 (IL‐23) signalling. (a) Analysis of CD11b and Ly6G expression profiles of CD45+ colonic leucocytes. The number in bold type represents the percentage of total CD45+ leucocytes contained within the indicated gate. (b) Total number of recruited CD11bHigh Ly6GH igh neutrophils as defined in (a). Bars represent mean ± SEM number of recruited neutrophils for the indicated group. n = 8 per group. For all animals, the entire colon was taken for FACS analysis. (c) Colonic gene expression was assessed using quantitative PCR as outlined in the Materials and methods. n ≥ 6 per group. Data are shown as mean ± SEM fold change gene expression of wild‐type (WT) C. difficile‐infected (black bars) and IL‐23KO C. difficile‐infected (grey bars) animals compared with untreated WT mice. CDI = C. difficile infected. For all analyses, *P < 0·05 compared with untreated WT animals and brackets indicate P < 0·05 for the differences between indicated groups.
To investigate the effect of IL‐23 in driving chemokine and granulocyte colony‐stimulating factor (CSF3) expression during C. difficile colitis, quantitative RT‐PCR was used to examine colonic cytokine expression at 2 days post infection. Clostridium difficile infection was associated with increased expression of the neutrophil chemokines Cxcl1, Cxcl2 and Ccl3, as well as the neutrophil stabilization factor Csf3 within the colonic mucosa (Fig. 1c). Consistent with the reduced neutrophilic influx observed in response to C. difficile infection in IL‐23KO mice (Fig. 1a,b), Cxcl1, Cxcl2, Ccl3 and Csf3 expression levels were significantly reduced in IL‐23KO animals compared with WT (Fig. 1c). There was no defect in the expression of the eosinophil chemokine Ccl11, or the T‐cell chemokines Cxcl9 and Cxcl10 in IL‐23‐deficient animals (Fig. 2a). Furthermore, Ccl24 expression was significantly increased in the absence of IL‐23 (Fig. 2a). Taken together, these data demonstrate that IL‐23 deficiency is associated with significant defects in both the recruitment of neutrophils to the colon and the expression of neutrophil chemoattractants within the colonic mucosa during C. difficile colitis.
Figure 2.
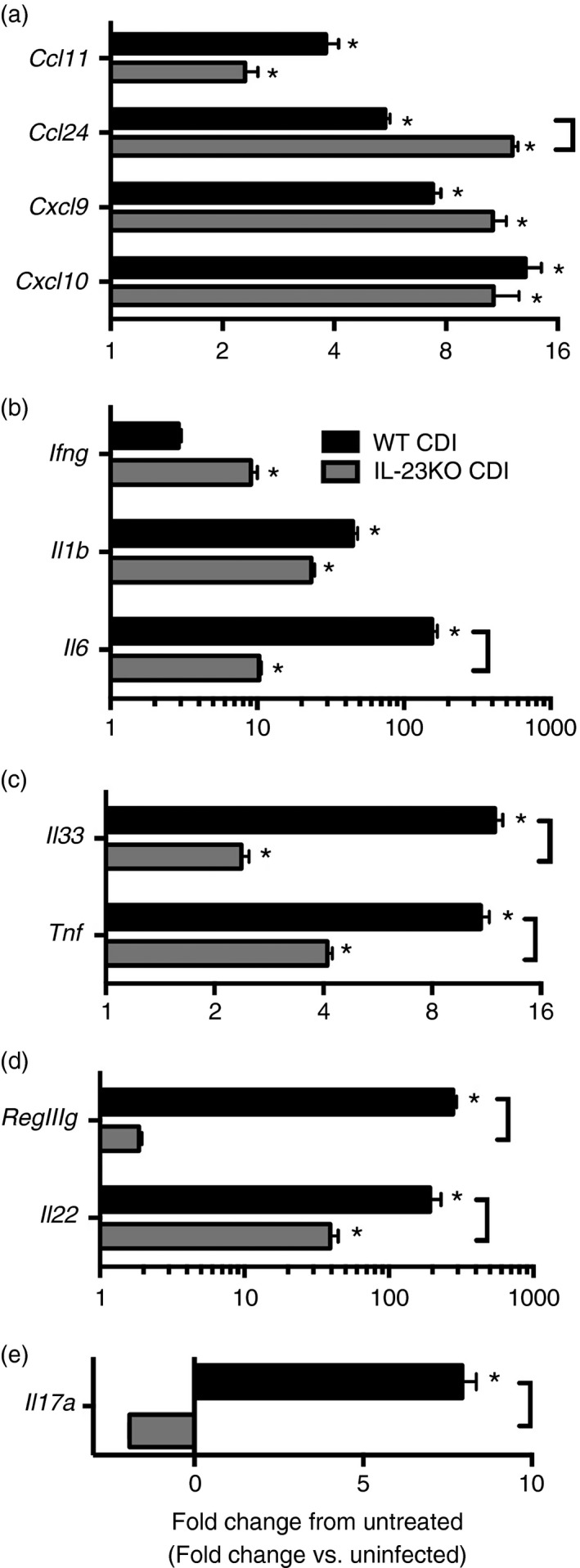
Effect of interleukin‐23 (IL‐23) deficiency on colonic (a) chemokine and (b–e) inflammatory cytokine expression during Clostridium difficile colitis. Host gene expression was measured as outlined in the methods. n ≥ 6 per group. Data are shown as mean ± SEM fold change gene expression of wild‐type (WT) C. difficile‐infected (black bars) and IL‐23KO C. difficile‐infected (grey bars) animals compared with untreated WT mice. CDI = C. difficile infected. *P < 0·05 compared with untreated WT animals. Brackets indicate P < 0·05 for the differences between indicated groups.
Effect of IL‐23 deficiency on colonic inflammatory cytokine expression
Analysis by RT‐PCR was also used to determine the role of IL‐23 in promoting inflammatory cytokine expression in response to C. difficile infection. Expression of the antimicrobial C‐type lectin RegIIIg was significantly increased in response to C. difficile infection, and this induction was significantly reduced in IL‐23KO mice (Fig. 2d). Interleukin‐23‐deficient animals displayed no reduction in Il1b or Ifng expression levels in response to C. difficile infection (Fig. 2b). However, expression levels of the inflammatory cytokines Il6, Il33 and Tnf were all significantly reduced in the absence of IL‐23 (Fig. 2b,c). Additionally, the increased expression of Il17a and Il22 seen in WT animals was completely abrogated in IL‐23KO mice (Fig. 2d,e). Hence, these data indicate that IL‐23 promotes the induction of numerous inflammatory cytokines including Il6, Il17a and Tnf, as well as the pleiotropic cytokine Il22, in response to C. difficile colitis.
To assess the contribution of IL‐23 signalling towards the development of intestinal inflammation and epithelial destruction during C. difficile colitis, sections of the colonic mucosa were examined for histopathological evidence of severe inflammation. In addition to significant neutrophilic influx (Fig. 1a,b), C. difficile infection was associated with significant epithelial damage and oedema, indicative of severe intestinal inflammation (Fig. 3). Interestingly, although the absence of IL‐23 had no impact on the development of colonic epithelial damage (Fig. 3c), there was a trend towards reduced oedema in IL‐23KO mice (Fig. 3b). Neutrophilic inflammation was also reduced in IL‐23KO mice (data not shown). These data suggest that IL‐23 also promotes the development of colonic oedema, but not epithelial damage, during C. difficile colitis.
Figure 3.
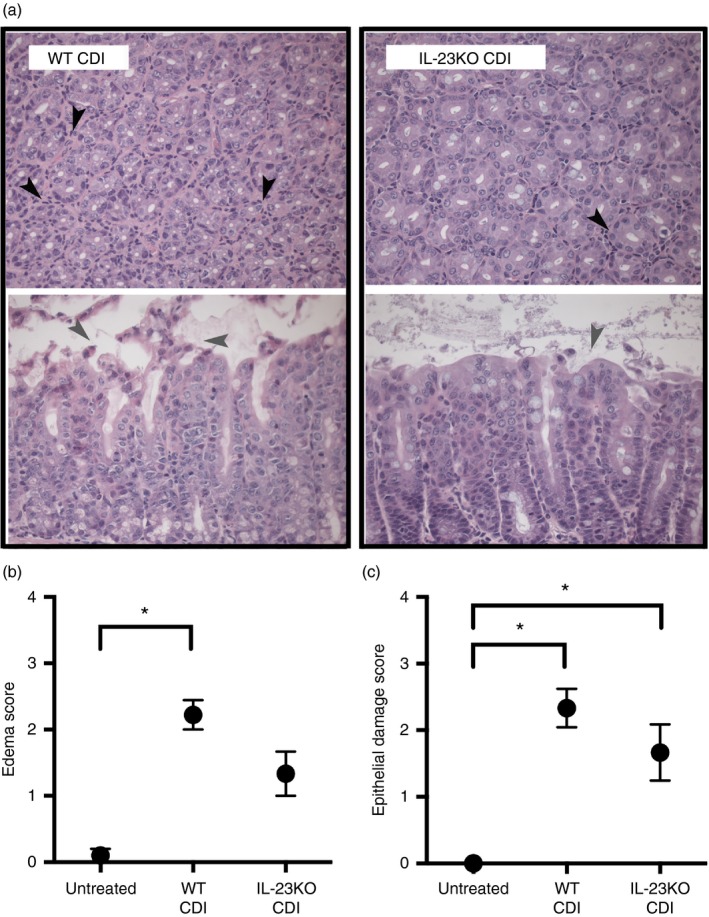
Colonic histopathology during Clostridium difficile infection in the absence of interleukin‐23 (IL‐23). (a) Representative photomicrographs of haematoxylin & eosin‐stained colonic sections from wild‐type (WT) C. difficile‐infected and IL‐23KO C. difficile‐infected animals. Cross‐sections of colonic crypts (upper images) and longitudinal sections of the epithelial–luminal interface (lower images) are shown for each genotype. Black arrowheads highlight cellular infiltrate, whereas grey arrowheads highlight epithelial damage. Total magnification for all images is 400×. (b, c) Histopathological scoring of colonic sections from Untreated, WT CDI, and IL‐23KO CDI mice. Slides were scored for oedema (b) and epithelial damage (c) as described in the Materials and methods. Data are shown as mean ± SEM. n ≥ 6 per group. CDI = C. difficile infected. Brackets indicate P < 0·05 for the differences between indicated groups.
The role of IL‐17 during C. difficile colitis
To investigate the contribution of IL‐17 in supporting neutrophil recruitment and mucosal inflammatory responses during C. difficile colitis, IL‐17a−/− (IL‐17KO) mice were infected with C. difficile. As in previous experiments, all samples were collected at 2 days post infection. Compared with C. difficile infection in WT animals, there was no reduction in expression of the neutrophil chemokines Cxcl1, Cxcl2 and Ccl3 within the colonic mucosa of IL‐17KO mice (Fig. 4c). Consistently, cellular infiltrates were apparent in colonic sections from IL‐17KO mice (Fig. 5a) and the levels of colonic neutrophilic inflammation were equivalent between IL‐17KO and WT animals infected with C. difficile (Fig. 5b). These data suggest that IL‐17 signalling is not required for the expression of neutrophil chemokines or the development of neutrophilic inflammation in response to C. difficile colitis.
Figure 4.
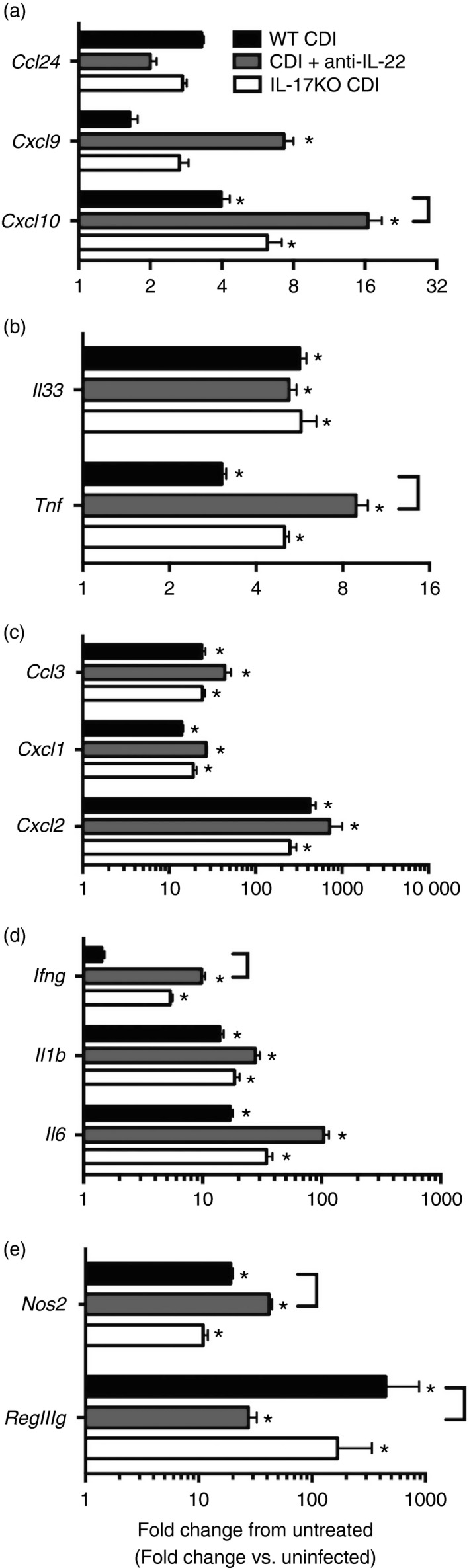
Effect of anti‐interleukin‐22 (IL‐22) treatment or IL‐17 deficiency on colonic (a, c) chemokine and (b, d, e) inflammatory cytokine expression during Clostridium difficile colitis. Colonic gene expression was assessed via quantitative PCR as outlined in the Materials and methods. n ≥ 6 per group. Data are shown as mean ± SEM fold change gene expression of wild‐type (WT) C. difficile‐infected (black bars), C. difficile‐infected and anti‐IL‐22‐treated (grey bars), and IL‐17KO C. difficile‐infected (white bars) animals compared with untreated WT mice. CDI = C. difficile infected. *P < 0·05 compared with untreated WT animals. Brackets indicate P < 0·05 for the differences in expression levels between indicated groups.
Figure 5.
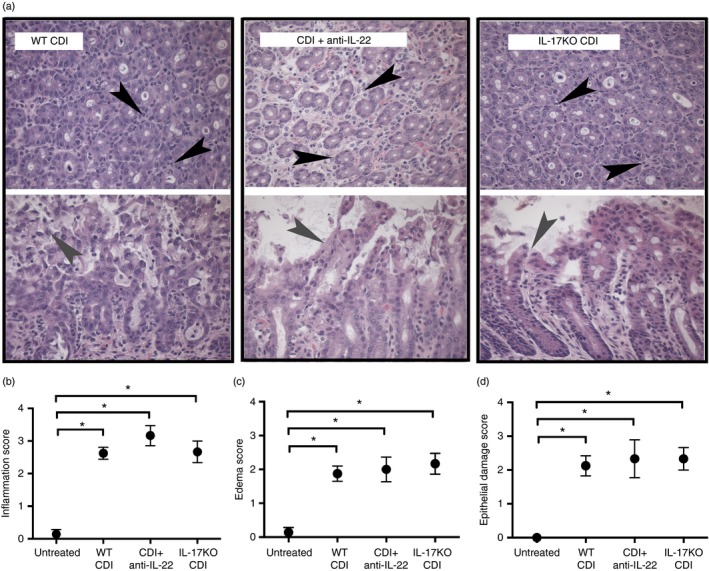
Colonic histopathology during Clostridium difficile infection in the absence of interleukin‐17 (IL‐17) or following anti‐IL‐22 treatment. Representative photomicrographs of haematoxylin & eosin‐stained colonic sections from wild‐type (WT) C. difficile‐infected, C. difficile‐infected and anti‐IL‐22‐treated, and IL‐17KO C. difficile‐infected animals. Cross‐sections of colonic crypts (upper images) and longitudinal sections of the epithelial–luminal interface (lower images) are shown for each genotype. Black arrowheads highlight leucocytic infiltrate, and grey arrowheads highlight areas of epithelial damage. Total magnification for all images is 400×. (b–d) Histopathological scoring of colonic sections from Untreated, WT CDI, CDI + anti‐IL‐22, and IL‐17KO CDI mice. Slides were scored for neutrophilic inflammation (b), oedema (c), and epithelial damage (d) as described in the Materials and methods. Data are shown as mean ± SEM. n ≥ 6 per group. CDI = C. difficile infected. Brackets indicate P < 0·05 for the differences between indicated groups.
Analysis by RT‐PCR was also used to investigate the role of IL‐17 in promoting inflammatory cytokine expression in response to C. difficile infection. Interestingly, the absence of IL‐17 was not associated with any reduction in inflammatory cytokines, including Ifng, Il1b, Il6, Il33 and Tnf (Fig. 4b,d). Additionally, Ccl24, Cxcl9 and Cxcl10 expression levels were unchanged in IL‐17KO mice (Fig. 4a). Consistent with the unaltered induction of inflammatory cytokines seen in these animals, IL‐17KO mice were not protected against the development of significant colonic epithelial damage and oedema during C. difficile infection (Fig. 5c,d). Taken together, these data support the hypothesis that neutrophil recruitment, inflammatory cytokine expression, and the development of colonic histopathology during C. difficile colitis are independent of IL‐17 signalling.
The role of IL‐22 during C. difficile colitis
To determine the role of IL‐22 in supporting mucosal inflammatory responses to C. difficile infection, mice were treated with an anti‐IL‐22 mAb (clone 8E11) 1 day before and 1 day after C. difficile infection. Animals were followed for 2 days post infection at which point all samples were collected. Colonic sections from anti‐IL‐22‐treated mice were examined for signs of marked histopathology. Anti‐IL‐22 treatment was associated with no reduction in epithelial damage or oedema compared with WT C. difficile‐infected animals (Fig. 5c,d). In agreement with these findings, colonic expression of numerous pro‐inflammatory cytokines including Il1b, Il6 and Il33 were unchanged in anti‐IL‐22‐treated mice (Fig. 4b,d). Interestingly, the expression levels of other inflammatory cytokines, most notably Tnf and Ifng as well as Cxcl10, were significantly increased following anti‐IL‐22 treatment (Fig. 4a,b,d,). RegIIIg expression was significantly reduced in anti‐IL‐22‐treated mice (Fig. 4e), indicating that the anti‐IL‐22 mAb treatment was sufficient to ablate IL‐22 signalling in vivo. Colonic sections from anti‐IL‐22‐treated mice were scored for neutrophilic inflammation. Anti‐IL‐22 treatment was not associated with any reduction in neutrophilic inflammation (Fig. 5a,b), and consistently, the expression levels of Cxcl1, Cxcl2 and Ccl3 were unchanged following anti‐IL‐22 treatment (Fig. 4c). Taken together, these data suggest that IL‐22 does promote the induction of RegIIIg, but does not promote neutrophil recruitment or affect histopathology during the response to C. difficile colitis.
Discussion
In the current study, we reported decreased neutrophil recruitment in IL‐23‐deficient animals in response to C. difficile colitis. This decrease in neutrophil recruitment was associated with decreases in Cxcl1 and Cxcl2 expression, as well as with reduced expression of Il‐17a and Il‐22. However, neither Cxcl1 and Cxcl2 expression nor neutrophilic inflammation was reduced in either IL‐17‐deficient mice or mice treated with a depleting anti‐IL‐22 mAb. Hence, our data strongly suggest that IL‐23, independent of IL‐17 or IL‐22, drives neutrophil recruitment and innate inflammatory responses during C. difficile colitis.
In the absence of IL‐23, neutrophil recruitment was significantly reduced in response to C. difficile colitis. Recent studies have demonstrated increased levels of IL‐23 in colonic biopsies from C. difficile‐infected patients,23 as well as increased levels of IL‐23 production from myeloid cells stimulated with C. difficile toxins in vitro.22 However, the role of IL‐23 in supporting innate inflammatory responses, including neutrophil recruitment, remains poorly understood. In the current study, we observed reduced neutrophil recruitment in association with decreased expression of neutrophil chemotactic factors in the absence of IL‐23. Previous studies have reported a role for IL‐23 in supporting neutrophil recruitment and neutrophil chemokine production in other models of mucosal inflammation.10, 11, 18, 19, 20, 37, 38 Interleukin‐23 is required for the full recruitment of neutrophils to the large intestine in response to both S. typhimurium typhlocolitis,11 as well as dextran sodium sulphate‐induced colitis.10 Additionally, neutrophil recruitment during pulmonary inflammation in response to both chemical20 and microbial18, 19, 38 challenges is supported by IL‐23. We observed a significant defect in the recruitment of CD11bHigh Ly6GHigh neutrophils, as well as the induction of the neutrophil chemokines Cxcl1 and Cxcl2, in IL‐23‐deficient mice infected with C. difficile. Taken together, these data strongly suggest that IL‐23 promotes neutrophil chemokine expression and neutrophil recruitment to the colon during C. difficile colitis.
Despite the robust increase in Il22 expression within the colonic mucosa during C. difficile colitis, we observed no decrease in inflammatory cytokine and chemokine expression following anti‐IL‐22 treatment. Interleukin‐22 promotes CXCL1 production from mouse tracheal epithelial cells,39 and also supports neutrophil recruitment in response to chemical pulmonary challenge.24 Additionally, IL‐22 is capable of stimulating neutrophil chemokine expression from both colonic epithelial cells28 and subepithelial myofibroblasts27 in vitro. However, in agreement with a recent study by Hasegawa et al.,14 we observed no decrease in expression of the neutrophil chemokines Cxcl1 or Cxcl2, or reduced expression of the inflammatory cytokines Il1b, Il6 or Il33 following anti‐IL‐22 treatment. Neutrophilic inflammation, oedema, and epithelial damage levels were also unchanged following the administration of anti‐IL‐22. Furthermore, anti‐IL‐22 treatment was associated with increased expression of Cxcl10, Ifng and Tnf. Hence, these data strongly suggest that IL‐22 is not a major driver of neutrophil chemokine or inflammatory cytokine expression in response to C. difficile colitis.
Likewise, we observed no significant change in the severity of intestinal histopathology following anti‐IL‐22 treatment. The role of IL‐22 during mucosal inflammation is pleiotropic: although IL‐22 is protective against severe colonic histopathology and mortality during Citrobacter rodentium infection,29 IL‐22 drives severe intestinal histopathology and necrosis during Toxoplasma gondii infection.21 Recent studies have reported no change in the severity of intestinal histopathology in IL‐22KO animals14 or following anti‐IL‐22 treatment6 during C. difficile infection. Consistently, we found that anti‐IL‐22 treatment was not associated with a significant increase or amelioration of neutrophilic inflammation, colonic epithelial damage or oedema during C. difficile colitis. Hence, the data presented here indicate that the development of colonic histopathology is independent of IL‐22.
The expression of the antimicrobial‐peptide RegIIIγ was significantly decreased in both IL‐23KO and anti‐IL‐22‐treated IL‐23‐sufficient mice. RegIIIg induction during Citrobacter rodentium colitis is induced by IL‐22 signalling, which is in turn induced by IL‐23.29 Marked induction of RegIIIg transcript has previously been reported in response to C. difficile infection of the large bowel.5, 6, 31 Additionally, a recent study from our laboratory has demonstrated a significant reduction in RegIIIg expression during C. difficile colitis following neutralization of IL‐22.6 In addition to reduced RegIIIg expression following anti‐IL‐22 treatment, in the current study, we also observed a concomitant reduction in both Il22 and RegIIIg expression in IL‐23‐deficient mice infected with C. difficile. Hence, these data strongly suggest that IL‐23‐dependent IL‐22 signalling is required for full induction of RegIIIg expression during C. difficile colitis.
Interleukin‐17 deficiency was not associated with any reduction in expression of inflammatory cytokines or neutrophil‐attracting chemokines, including Cxcl1 and Cxcl2 within the colonic mucosal following C. difficile infection. Interleukin‐17 has a well‐documented role supporting neutrophil recruitment during inflammatory responses at mucosal sites.12, 13, 24, 25, 26, 40 Specific to the gut, IL‐17 promotes CXCL1 expression in response to S. typhimurium typhlocolitis,25 as well as supporting neutrophil recruitment during both dextran sodium sulphate‐induced12 and 2 #bib4 #bib6‐trinitrobenzenesulphonic acid‐induced13 colitis. However, we observed no reduction in expression of the neutrophil chemoattractants Cxcl1, Cxcl2 or Ccl3 in the absence of IL‐17. In agreement with the unaltered levels of chemokine expression, as well as the unaltered expression of the inflammatory cytokines Il1b, Il6, Il33 and Tnf, IL‐17 deficiency was not associated with a reduction in the severity of colonic histopathology. Taken together, our data support the hypothesis that IL‐17 is dispensable for the recruitment of neutrophils, the induction of inflammatory cytokines, and the development of intestinal histopathology during C. difficile colitis.
In addition to decreased neutrophil recruitment, IL‐23 deficiency was associated with decreased expression of the inflammatory cytokines Il6, Il33 and Tnf as well as a trend towards decreased colonic oedema. Interleukin‐23 contributes to IL‐6 production in response to Pseudomonas aeruginosa pulmonary infection,18 and IL‐6 and IL‐1β expression in response to Toxoplasma gondii ileitis is partially dependent upon IL‐23.21 Furthermore, interference with IL‐23 signalling has been shown to reduce the severity of intestinal histopathology in both infectious21 and chemical10 models of gastrointestinal inflammation. In the current study, we report decreased colonic oedema in association with reduced inflammatory cytokine expression in IL‐23‐deficient animals. These data suggest that IL‐23 contributes to the development of severe intestinal histopathology and drives the induction of inflammatory cytokines including Il6 and Il33 during C. difficile colitis.
The data presented in the current study suggest a clear role for IL‐23 in supporting neutrophil recruitment, the induction of inflammatory cytokines, and the development of severe colonic histopathology during C. difficile infection. One possible model that could explain these phenomena is that neutrophils, recruited in part by IL‐23 signalling, contribute to colonic histopathology and severe disease outcomes. Indeed, a recent study has demonstrated reduced morbidity and mortality in IL‐23‐deficient mice infected with C. difficile.23 However, numerous previous studies have reported increased mortality during C. difficile infection following interventions that reduced neutrophilic influx,1, 2, 3 suggesting a protective role for neutrophil recruitment. Furthermore, a recent study from our laboratory found no reduction in the severity of colonic histopathology during C. difficile colitis following anti‐Gr‐1 treatment.4 Taken together these studies suggest that IL‐23 signalling may ultimately play a dual role during C. difficile colitis by both promoting neutrophil recruitment as well as other innate responses that contribute to morbidity and intestinal histopathology during infection.
Author contributions
AJM and GBH conceived, designed and interpreted the experiments. NRF, RAM and VBY contributed to their design and interpretation. AJM, RAM, NRF and CRP performed the experiments. AJM, RAM, NRF, CRP and GBH analysed the data. AJM and GBH wrote the manuscript, and all other authors provided commentary and advice on the manuscript.
Disclosures
The authors declare no conflicts of interest.
Acknowledgements
The authors would like to thank the staff of the University of Michigan Flow Cytometry Core for their assistance in performing the flow cytometry experiments. We thank the University of Michigan Unit for Laboratory Animal Medicine In‐Vivo Animal Core, and specifically Dr Ingrid Bergin, for scoring of histological sections. We thank Dr Wenjun Ouyang for his kind gift of anti‐IL‐22 mAb. We thank Dr Paul Carlson Jr for supplying C. difficile spores for this study. We thank Dr John Erb‐Downward for his discussions and contributions to the project. We thank Charles Frank for technical contributions to the project. We also thank Dr Merritt Gillilland III for his critical reading of this manuscript. This work was supported by National Institutes of Health grants U19 AI090871 (GBH and VBY) and P30 DK034933 (GBH and VBY). Support was also provided by the Host‐Microbiome Initiative (HMI) of the University of Michigan Medical School (GBH and VBY).
References
- 1. Hasegawa M, Kamada N, Jiao Y, Liu MZ, Nunez G, Inohara N. Protective role of commensals against Clostridium difficile infection via an IL‐1β‐mediated positive‐feedback loop. J Immunol 2012; 189:3085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hasegawa M, Yamazaki T, Kamada N, Tawaratsumida K, Kim YG, Nunez G et al Nucleotide‐binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 2011; 186:4872–80. [DOI] [PubMed] [Google Scholar]
- 3. Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88‐mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun 2012; 80:2989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McDermott AJ, Higdon KE, Muraglia R, Erb‐Downward JR, Falkowski NR, McDonald RA et al The role of Gr‐1+ cells and tumour necrosis factor‐α signalling during Clostridium difficile colitis in mice. Immunology 2015; 144:704–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sadighi Akha AA, Theriot CM, Erb‐Downward JR, McDermott AJ, Falkowski NR, Tyra HM et al Acute infection of mice with Clostridium difficile leads to eIF2α phosphorylation and pro‐survival signalling as part of the mucosal inflammatory response. Immunology 2013; 140:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sadighi Akha AA, McDermott AJ, Theriot CM, Carlson PE Jr, Frank CR, McDonald RA et al Interleukin‐22 and CD160 play additive roles in the host mucosal response to Clostridium difficile infection in mice. Immunology 2015; 144:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Price AB, Davies DR. Pseudomembranous colitis. J Clin Pathol 1977; 30:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kelly CP, Pothoulakis C, LaMont JT. Clostridium difficile colitis. N Engl J Med 1994; 330:257–62. [DOI] [PubMed] [Google Scholar]
- 9. Walker AS, Eyre DW, Wyllie DH, Dingle KE, Griffiths D, Shine B et al Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 2013; 56:1589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox JH, Kljavin NM, Ota N, Leonard J, Roose‐Girma M, Diehl L et al Opposing consequences of IL‐23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol 2012; 5:99–109. [DOI] [PubMed] [Google Scholar]
- 11. Godinez I, Raffatellu M, Chu H, Paixao TA, Haneda T, Santos RL et al Interleukin‐23 orchestrates mucosal responses to Salmonella enterica serotype Typhimurium in the intestine. Infect Immun 2009; 77:387–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ito R, Kita M, Shin‐Ya M, Kishida T, Urano A, Takada R et al Involvement of IL‐17A in the pathogenesis of DSS‐induced colitis in mice. Biochem Biophys Res Commun 2008; 377:12–6. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z, Zheng M, Bindas J, Schwarzenberger P, Kolls JK. Critical role of IL‐17 receptor signaling in acute TNBS‐induced colitis. Inflamm Bowel Dis 2006; 12:382–8. [DOI] [PubMed] [Google Scholar]
- 14. Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz‐Planillo R, Do N et al Interleukin‐22 regulates the complement system to promote resistance against pathobionts after pathogen‐induced intestinal damage. Immunity 2014; 41:620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest 2010; 120:2423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP‐2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d‐dependent manner. Blood 2005; 105:2543–8. [DOI] [PubMed] [Google Scholar]
- 17. Hunter CA. New IL‐12‐family members: IL‐23 and IL‐27, cytokines with divergent functions. Nat Rev Immunol 2005; 5:521–31. [DOI] [PubMed] [Google Scholar]
- 18. Dubin PJ, Kolls JK. IL‐23 mediates inflammatory responses to mucoid Pseudomonas aeruginosa lung infection in mice. Am J Physiol Lung Cell Mol Physiol 2007; 292:L519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dubin PJ, Martz A, Eisenstatt JR, Fox MD, Logar A, Kolls JK. Interleukin‐23‐mediated inflammation in Pseudomonas aeruginosa pulmonary infection. Infect Immun 2012; 80:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gasse P, Riteau N, Vacher R, Michel ML, Fautrel A, di Padova F et al IL‐1 and IL‐23 mediate early IL‐17A production in pulmonary inflammation leading to late fibrosis. PLoS ONE 2011; 6:e23185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Munoz M, Heimesaat MM, Danker K, Struck D, Lohmann U, Plickert R et al Interleukin (IL)‐23 mediates Toxoplasma gondii‐induced immunopathology in the gut via matrixmetalloproteinase‐2 and IL‐22 but independent of IL‐17. J Exp Med 2009; 206:3047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cowardin CA, Kuehne SA, Buonomo EL, Marie CS, Minton NP, Petri WA Jr. Inflammasome activation contributes to interleukin‐23 production in response to Clostridium difficile . MBio 2015; 6:e02386–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buonomo EL, Madan R, Pramoonjago P, Li L, Okusa MD, Petri WA Jr. Role of interleukin 23 signaling in Clostridium difficile colitis. J Infect Dis 2013; 208:917–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sonnenberg GF, Nair MG, Kirn TJ, Zaph C, Fouser LA, Artis D. Pathological versus protective functions of IL‐22 in airway inflammation are regulated by IL‐17A. J Exp Med 2010; 207:1293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keestra AM, Godinez I, Xavier MN, Winter MG, Winter SE, Tsolis RM et al Early MyD88‐dependent induction of interleukin‐17A expression during Salmonella colitis. Infect Immun 2011; 79:3131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P et al Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony‐stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med 2001; 194:519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y et al Interleukin‐22, a member of the IL‐10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 2005; 129:969–84. [DOI] [PubMed] [Google Scholar]
- 28. Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM et al IL‐22 is increased in active Crohn's disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 2006; 290:G827–38. [DOI] [PubMed] [Google Scholar]
- 29. Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q et al Interleukin‐22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 2008; 14:282–9. [DOI] [PubMed] [Google Scholar]
- 30. Perez J, Springthorpe VS, Sattar SA. Clospore: a liquid medium for producing high titers of semi‐purified spores of Clostridium difficile . J AOAC Int 2011; 94:618–26. [PubMed] [Google Scholar]
- 31. McDermott AJ, Frank CR, Falkowski NR, McDonald RA, Young VB, Huffnagle GB. Role of GM‐CSF in the inflammatory cytokine network that regulates neutrophil influx into the colonic mucosa during Clostridium difficile infection in mice. Gut Microbes 2014; 5:476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. Cefoperazone‐treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2011; 2:326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Reeves AE, Theriot CM, Bergin IL, Huffnagle GB, Schloss PD, Young VB. The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2011; 2:145–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sadighi Akha AA, Harper JM, Salmon AB, Schroeder BA, Tyra HM, Rutkowski DT et al Heightened induction of proapoptotic signals in response to endoplasmic reticulum stress in primary fibroblasts from a mouse model of longevity. J Biol Chem 2011; 286:30344–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A et al Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schmittgen TD, Livak KJ. Analyzing real‐time PCR data by the comparative CT method. Nat Protoc 2008; 3:1101–8. [DOI] [PubMed] [Google Scholar]
- 37. Hue S, Ahern P, Buonocore S, Kullberg MC, Cua DJ, McKenzie BS et al Interleukin‐23 drives innate and T cell‐mediated intestinal inflammation. J Exp Med 2006; 203:2473–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu Q, Martin RJ, Rino JG, Breed R, Torres RM, Chu HW. IL‐23‐dependent IL‐17 production is essential in neutrophil recruitment and activity in mouse lung defense against respiratory Mycoplasma pneumoniae infection. Microbes Infect 2007; 9:78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA et al IL‐22 mediates mucosal host defense against Gram‐negative bacterial pneumonia. Nat Med 2008; 14:275–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu J, Feng Y, Yang K, Li Q, Ye L, Han L et al Early production of IL‐17 protects against acute pulmonary Pseudomonas aeruginosa infection in mice. FEMS Immunol Med Microbiol 2011; 61:179–88. [DOI] [PubMed] [Google Scholar]