

Abstract
Dynein light chain LC8 is highly conserved among eukaryotes and has both dynein-dependent and dynein-independent functions. Interestingly, LC8 was identified as a subunit of the class I transcription factor A (CITFA), which is essential for transcription by RNA polymerase I (Pol I) in the parasite Trypanosoma brucei. Given that LC8 has never been identified with a basal transcription factor and that T. brucei relies on RNA Pol I for expressing the variant surface glycoprotein (VSG), the key protein in antigenic variation, we investigated the CITFA-specific role of LC8. Depletion of LC8 from mammalian-infective bloodstream trypanosomes affected cell cycle progression, reduced the abundances of rRNA and VSG mRNA, and resulted in rapid cell death. Sedimentation analysis, coimmunoprecipitation of recombinant proteins, and bioinformatic analysis revealed an LC8 binding site near the N terminus of the subunit CITFA2. Mutation of this site prevented the formation of a CITFA2-LC8 heterotetramer and, in vivo, was lethal, affecting assembly of a functional CITFA complex. Gel shift assays and UV cross-linking experiments identified CITFA2 as a promoter-binding CITFA subunit. Accordingly, silencing of LC8 or CITFA2 resulted in a loss of CITFA from RNA Pol I promoters. Hence, we discovered an LC8 interaction that, unprecedentedly, has a basal function in transcription.
INTRODUCTION
Dynein light chain LC8 was originally discovered as a component of the outer arm axonemal dynein in Chlamydomonas reinhardtii (1) but was later found to be present also in cytoplasmic dyneins 1 and 2 (2–4). LC8 is conserved throughout eukaryotic genomes (5). As a part of the dynein motor, LC8 is important for fundamental cellular processes, such as tubulin minus-end-directed intracellular transport, chromatid separation during mitosis, and nuclear migration (6), as well as flagellum-specific functions, namely, motility, intraflagellar transport (7), and ciliogenesis (8, 9). While not essential in Saccharomyces cerevisiae (10), mutation or knockdown of LC8 is embryonic lethal in animals (8, 9, 11, 12). Given that LC8 is more conserved between species than other components of the dynein motor and that LC8 is present in organisms which lack a dynein motor, it was likely that LC8 had nondynein functions (3, 5). LC8 has since been shown to interact with several different proteins and to affect various cellular processes, including protein localization and stability, transcription regulation, and apoptosis (13–15).
At physiological pH, LC8 exists almost exclusively as a dimer (16, 17), interacting with partner proteins via two identical sites generated at the dimer interface which bind to diverse short, linear motifs (16, 18, 19). LC8 promotes the dimerization of its binding partners through aligning dimerization domains present in the partner protein (13, 20–22). While it was previously hypothesized that LC8 functions as a linker, allowing attachment of the dynein motor to its cargo, the emerging view is that interaction with LC8 induces dimerization, imparting new structure and function, a view which is supported by detailed investigations of LC8 interactions (23–25). In addition to this improved understanding of LC8, interest in this molecule has grown as LC8 has been shown to have unique and important roles in an increasing number of human pathogens. These include viruses, such as HIV (26), Ebola (27), and rabies (28), and the protistan parasite Toxoplasma gondii (29).
Trypanosoma brucei, a member of the early-diverging phylogenetic order Kinetoplastida, is a vector-borne parasite that causes lethal disease in both humans and livestock (30). In T. brucei, LC8 was identified in mass spectrometry analysis of the flagellum (31) and, surprisingly, as a subunit of the class I transcription factor A (CITFA) (32, 33). As CITFA is a core promoter-binding factor required for initiation of RNA polymerase I (Pol I)-mediated transcription, this represents the first time that LC8 has been found to associate with basal transcription machinery. T. brucei is unique in that RNA Pol I not only transcribes rRNA gene (RRNA) units as in all other organisms but also transcribes gene arrays that encode its major cell surface proteins, namely, the variant surface glycoprotein, or VSG, in the mammalian-infective bloodstream form (BF) and procyclin in the insect-stage procyclic form (PF) (34). This production of functional mRNA by RNA Pol I is possible in T. brucei due to a unique mRNA processing mechanism, called spliced leader (SL) trans splicing, which caps mRNA posttranscriptionally by an RNA Pol II-independent process (35–37). By densely covering the cell in ∼10 million copies of the same VSG protein, T. brucei is able to shield invariant proteins from antibody recognition (38). The source of this massive protein expression is a single VSG gene located in one of ∼15 bloodstream expression sites (BESs) that are monoallelically expressed (39). Antigenic variation of VSG, which occurs by switching to the expression of another VSG gene, drawn from a large repertoire in the trypanosome genome, allows an infection to be maintained (40, 41). The importance of VSG to T. brucei is highlighted by the fact that RNA interference (RNAi) targeting VSG mRNA rapidly halted BF culture growth in the absence of immunological pressure and led to the clearance of trypanosomes from infected mice (42).
While it was previously shown that the CITFA complex, consisting of subunits CITFA1 to CITFA7 and LC8, is essential to RNA Pol I-mediated transcription and binds the BES promoter in purified form (32), the specific role of individual complex members, including knowledge of the LC8 binding partner, has remained unclear. Furthermore, given that all CITFA subunits except LC8 are conserved only among kinetoplastids and are without recognizable sequence motifs, that LC8 has never been implicated in the basal process of transcription initiation, and that LC8 has not been studied in a kinetoplastid organism, we set out to understand the specific role of LC8 in RNA Pol I-mediated transcription.
Kinetoplastids encode two distinct LC8 proteins, only one of which was found to be associated with CITFA. We found that this LC8, previously termed DYNLL1 (32, 33), is essential for cell viability in culture and that RNAi-mediated silencing of the gene led to defects in both cell cycle and transcription by RNA Pol I. To understand LC8's specific role in the latter, we identified an LC8 binding site near the N terminus of the essential CITFA2 subunit. Mutation of this site was lethal to trypanosomes, preventing the incorporation of this subunit into the CITFA complex. Moreover, we show that CITFA2 directly interacts with BES promoter DNA and is required for the CITFA complex to bind to the BES promoter in vivo, functions that crucially depend on the CITFA2-LC8 interaction. These data revealed an essential role for LC8 in T. brucei and the first evidence that LC8 is required for the formation of a transcription preinitiation complex (PIC) in any organism.
MATERIALS AND METHODS
DNAs and cell lines.
pT7-LC8-stl, for conditional silencing of LC8 genes (GeneDB and TriTrypDB [www.genedb.org and www.tritrypdb.org, respectively] accession numbers Tb927.11.18680 and Tb11.0845), was generated by inserting portions of the LC8 coding region and its adjacent 3′ untranslated region (UTR), nucleotides +97 to +602 relative to the translation initiation codon, into the pT7-stl vector (32) in a sense-stuffer-antisense arrangement, according to a previously published protocol (43). Transfection of SacII-linearized pT7-LC8-stl into single-marker BF (smBF) cells (44) generated smLC8 cells. pPURO-PTP-CITFA3 was generated by inserting 644 bp of the CITFA3 coding region (position 4 to position 647) into the NotI and ApaI sites of vector pN-PURO-PTP (45), which contains the sequence of the composite PTP tag, consisting of a tandem protein A domain (ProtA), a tobacco etch virus cleavage site, and a protein C (ProtC) epitope. pPURO-PTP-CITFA3, along with the previously described pCITFA7-PTP-NEO, pCITFA4-PTP-NEO (33), and pPURO-PTP-CITFA2 (32), was used to fuse the sequence of the composite PTP tag to endogenous alleles. Similarly, the gene-silencing vectors pT7-PTP-stl (46) and pT7-CITFA2-stl (32), along with accompanying cell lines, were described previously. smC2-PTP cells allow the conditional silencing of CITFA2 through targeting of the PTP tag coding sequence and were generated in two steps. Starting with a previously published smPTP cell line (46), which conditionally expresses double-stranded RNA (dsRNA) targeting the PTP tag sequence, we first used site-directed integration of SphI-linearized pPURO-PTP-CITFA2 into one CITFA2 allele to fuse the PTP tag sequence to the 5′ end of the CITFA2 coding region. In a second step, the remaining CITFA2 allele was replaced by a PCR product in which 100 bp of CITFA2 gene flanks surrounded the hygromycin phosphotransferase coding sequence. In order to conditionally express exogenous transgenes of CITFA2, pT7-trans was developed. Instead of a stem-loop construct, we inserted into pT7-stl a PCR product which contained (5′ to 3′) 490 bp of the intergenic region between HSP70 genes 2 and 3, NdeI and NotI restriction sites, a hemagglutinin (HA) tag sequence ending with a stop codon, and 741 bp of the β/α-tubulin intergenic region. pT7-CITFA2-HA was generated from pT7-trans through insertion of the full coding sequence of CITFA2 using NdeI and NotI restriction sites. pT7-NDel-HA was generated similarly, except that bases +4 through +31, corresponding to N-terminal amino acids 2 through 10 of CITFA2 (PEVGTQVYW), were deleted (NDel) by PCR. pT7-3Amut-HA was generated using a CITFA2 insert which had bases +16 through +24 (ACTCAAGTT, coding for amino acids TQV) replaced with GCCGCGGCA, which coded for a three-alanine substitution (3Amut). Transfection of these three plasmids, after linearization by EcoRV, into smC2-PTP cells generated cell lines smC2-PTP-CITFA2-HA, smC2-PTP-NDel-HA, and smC2-PTP-3Amut-HA. Transfection of these same plasmids into wild-type single-marker cells resulted in cell lines smC2-CITFA2-HA, smC2-NDel-HA, and smC2-3Amut-HA.
DNAs and recombinant protein.
To generate recombinant proteins for pulldown assays and sucrose gradients, seven different recombinant protein expression plasmids were created. pCITFA2-PTH, which allowed the expression of full-length wild-type recombinant CITFA2 (rCITFA2), was produced by inserting the full coding sequence of CITFA2 into the expression vector pET100/D-TOPO (Invitrogen) using NdeI and NotI restriction sites, as previously described (32). This resulted in rCITFA2, which had fused to its C terminus a ProtC epitope, followed by a thrombin cleavage site and a 6×His tag. Four additional plasmids were produced which differed only in the lengths of the integrated CITFA2 sequences: pCITFA2-N-PTH contains the sequence for only the N-terminal half of CITFA2 (bases +1 to +624), while pCITFA2-C-PTH codes for the C-terminal half (bases +621 to +1263). pCITFA2-N1/4-PTH, coding for the N-terminal quarter of CITFA2, contained bases +1 to +303, while pCITFA2-N2/4-PTH, coding for the second quarter, contained bases +289 to +624. p3Amut-PTH, however, uniquely coded for mutated full-length rCITFA2 that contained the same triple-alanine mutation as detailed above for pT7-C2-3Amut. These vectors were transformed into BL21 Escherichia coli, and protein expression was induced for 15 min to 1 h at 37°C by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). Shorter incubations were required for plasmids which included the C terminus of CITFA2 as its expression appears to be toxic to E. coli (data not shown). Recombinant LC8 was generated by placing the entire LC8 coding sequence downstream of the glutathione S-transferase (GST) sequence in the pGEX-4T-2 vector (GE Healthcare) using BamHI and NotI restriction sites. Recombinant GST-LC8 was expressed in BL21 E. coli and purified by glutathione affinity chromatography (GE Healthcare) according to the manufacturer's recommendations. Thrombin digest and elution were then performed such that, in all assays, LC8 was used as an untagged, recombinant protein (rLC8).
Generation of a purified anti-LC8 antibody.
Immune serum against LC8 was generated by immunization of Sprague-Dawley rats with rGST-LC8, according to a standard protocol (47). rGST-LC8-specific antibodies were purified from serum by blot-immobilized antigen, as previously detailed (46). In contrast to immune serum, the purified antibody did not detect a nonspecific band present in E. coli that comigrated with thrombin-digested LC8 (data not shown).
Protein analysis.
Immunoblot detections were performed using polyclonal antibodies directed against CITFA2 (32), CITFA6, CITFA7 (33), and TFIIB (47). The PTP tag was detected with a monoclonal anti-ProtC antibody (Roche), while HA-tagged proteins were detected with a rat monoclonal anti-HA antibody (Roche). Extract preparation and tandem affinity purifications of PTP-tagged CITFA2, CITFA4, and CITFA7 were conducted according to a standard protocol (45). Crude bacterial lysates of rCITFA2-PTH-expressing BL21 E. coli, used in protein pulldown assays with rLC8, were prepared as follows: after protein expression was induced with 1 mM IPTG for 30 min at 37°C, bacterial cultures were pelleted, and 0.2 g of the cell pellet was resuspended in 4 ml of HisTALON xTractor buffer (Clontech). Benzonase (250 units; Sigma), 400 ng of lysozyme, and 250 μl of a protease inhibitor solution, prepared by resuspending 1 tablet of protease inhibitor (Roche) in 1 ml of H2O, were then added. Following a 10-min incubation with shaking at 4°C, the mixture was centrifuged at 3,200 × g for 30 min at 4°C. The supernatant was taken and used as the crude bacterial lysate. For the pulldown assays, rCITFA2-PTH was purified from 100 μl of the crude lysate using 20 μl of equilibrated TALON metal affinity resin (Clontech), according to the manufacturer's specifications. A 40-μl binding reaction mixture containing the CITFA2-PTH-conjugated TALON resin, 100 mM KCl, 20 mM potassium glutamate, 20 mM HEPES-KOH (pH 7.7), 3 mM MgCl2, 100 ng/μl bovine serum albumin (BSA), 150 mM sucrose, 2.5 μl of the protease inhibitor solution, and 100 ng of rLC8 was incubated at 27°C for 1 h, with shaking. The resin was washed seven times with a buffer containing 400 mM KCl, 20 mM potassium glutamate, 20 mM HEPES-KOH (pH 7.7), 3 mM MgCl2, 10 ng/μl BSA, 0.5 mM dithiothreitol (DTT), and 0.1% Tween 20. Proteins were eluted using HisTALON elution buffer (Clontech) and investigated by immunoblotting. To analyze rCITFA2-rLC8 interactions by sedimentation, 500 ng of purified rLC8 was mixed with 200 μl of rCITFA2-PTH-containing crude bacterial lysates and incubated for 30 min at 27°C. The binding reaction was then loaded onto a 4-ml, 10 to 40% linear sucrose gradient, ultracentrifuged, and fractionated from top to bottom, as previously described (32). Coimmunoprecipitations (co-IPs) of CITFA2 were performed using trypanosome extract, as previously described (48). UV cross-linking and electrophoretic mobility shift assays (EMSAs) of a radiolabeled BES promoter probe and purified CITFA, both visualized by autoradiography, were conducted as previously detailed (32). Secondary structure analysis was carried out using PredictProtein (www.predictprotein.org/) (49).
RNA analysis.
To analyze the effect of LC8 silencing on transcription by RNA Pol I, total RNA was prepared by the hot-phenol method, as described previously (46). For the analysis of rRNA, total RNA was separated in Reliant precast 1.25% SeaKem Gold agarose RNA gels (Lonza), and rRNA was detected by ethidium bromide staining. For semiquantitative reverse transcription-PCR (RT-PCR) analysis, total RNA was reverse transcribed with Superscript reverse transcriptase II (Invitrogen) using an oligo(dT) primer. Semiquantitative PCR was performed using cycle numbers that were empirically determined to be within the linear amplification range for each primer pair; 5′-GATAAGCTTACGCGTTTCAACATTGAGAAGGATATTGC-3′ and 5′-GATTCTAGACTCGAGTCTTTGACTCATCCGTGCTGG-3′ were used to amplify the LC8 coding sequence and 3′ UTR. Primers amplifying α-tubulin and VSG2 sequences were published previously (33).
ChIP.
CITFA2-HA and CITFA3 promoter occupancy were analyzed by chromatin immunoprecipitation (ChIP) assays, as described previously (46), using monoclonal rat anti-HA antibody (Roche) and purified polyclonal anti-CITFA3 antibody, respectively. Negative-control precipitations were carried out using affinity beads not bound to antibody. Chromatin was sonicated until fragments averaged 200 to 400 bp in length. The precipitated DNA was analyzed by quantitative PCR (qPCR) using consensus primers for the slightly varying copies of the RRNA and BES promoters and a primer pair for the β/α-tubulin intergenic region, which were specified previously (33, 46). The percent immunoprecipitated DNA (percent IP) was calculated relative to the input material and corrected by subtracting the percent IP of the negative-control ChIP. Each ChIP experiment was independently carried out three to four times, and statistical analyses were performed using percent IP averages. Comparisons between corrected percent precipitations of noninduced and induced cells were performed using Student's t test. Prior to the application of the t test, an F test was performed to ensure that the assumption of equal variance between groups was not violated. If the F test indicated that the difference in variation was significant, then the t test was performed with the more conservative assumption of unequal variance. An unpaired, two-tailed test was used in all cases.
Microscopy.
To visualize changes in cell morphology and DNA content upon LC8 silencing, BFs were incubated with 4,6-diamidino-2-phenylindole (DAPI) at a final concentration of 2 ng/μl for 45 min and imaged using a Zeiss AxioVert 200 microscope and Zeiss Axiovision, version 4.6.3.0, software, as described previously (46). For quantification of the percentage of cells which were multiflagellated, multikinetoplastid, or multinucleated, cells were scored as abnormal if they met any one of the following criteria: ≥3 kinetoplasts, ≥3 nuclei, ≥3 flagella, or more nuclei than kinetoplasts. A total of 150 cells were scored from both the noninduced and induced populations.
Flow cytometry.
In order to assess changes in the size and DNA content of a large number of cells upon LC8 silencing, flow cytometry experiments were performed. Noninduced and 1-day-induced smLC8 cultures were stained with propidium iodide and counted using an LSR II flow cytometer (BD Biosciences), according to a previously described protocol (50). Thirty thousand cells were counted in each experiment, in triplicate, for each induction state, and ungated data were visualized using the FlowJo software package (Treestar, Inc.).
RESULTS
LC8 is essential for T. brucei viability, cytokinesis, and RNA Pol I-mediated transcription.
A survey of kinetoplastid genome databases revealed conservation of two distinct LC8 genes. One encodes an LC8 protein that is closely related to both human DYNLL1 and DYNLL2 (∼82% and 88% identity and similarity, respectively; the accession number for the T. brucei gene is Tb927.11.18680 at www.TritrypDB.org [51] or www.GeneDB.org [52]) and was previously termed DYNLL1 (32). A second LC8 is also present, yet this gene (GeneDB or TriTrypDB accession number Tb927.11.320) is more divergent from LC8 genes in other eukaryotes (∼61% and 85% identity and similarity, respectively, to human DYNLL) (see also Fig. S1 in the supplemental material). Since a phylogenetic analysis did not reveal that these two LC8 proteins resemble the DYNLL1 and DYNLL2 dichotomy found in chordates (see Fig. S1C), we propose to rename Tb927.11.18680 LC8 and name Tb927.11.320 LC8DV (where DV is for divergent). Trypsin-derived peptides of these two proteins are different except for a 4-amino-acid-long peptide (see Fig. S1A). Since previous mass spectrometric analyses of purified CITFA consistently identified LC8 but never LC8DV-derived peptides (32, 33), it is highly unlikely that LC8DV is a subunit of the transcription factor complex. We therefore concentrated our analysis on LC8.
To investigate the importance of LC8 to BFs, we reduced its expression by RNAi, using a conditional gene-silencing system which expressed double-stranded RNA (dsRNA) targeting both the LC8 coding region and 3′ UTR upon addition of doxycycline (44). In three different clonal cell lines, derived from smBF cells and termed smLC8 lines, culture growth arrested within 1 day, and the majority of cells were lost by 2 days of induction (Fig. 1A). By 3 days, no living cells could be identified by microscopic examination. RNA monitoring revealed that doxycycline induction resulted in a clear decline of LC8 mRNA abundance after 1 day, while the levels of LC8DV and α-tubulin mRNA were unaffected, confirming that the knockdown was specific to LC8 (Fig. 1B). To evaluate whether this rapid-death phenotype would allow us to detect effects on transcription by RNA Pol I, we measured levels of VSG mRNA derived from the active VSG2 gene and of rRNA. While the VSG2 mRNA level dropped considerably after 1 day of induction, the rRNA level decreased only modestly in the same experiment, possibly due to greater stability of rRNA (Fig. 1B). For LC8 protein analysis, we generated a rat immune serum against recombinant LC8 with an N-terminal GST tag that was expressed and purified from E. coli (see Fig. S2 in the supplemental material). Immunoblot monitoring of LC8-silenced cells showed a specific reduction in LC8 protein, while CITFA6 and TFIIB, an RNA Pol II-specific transcription factor, were either increased or unchanged during this short period (Fig. 1C), confirming the specificity of the knockdown. These data demonstrated that LC8 is essential for trypanosome viability, and they indicated that LC8 is also important, though perhaps indirectly, for transcription by RNA Pol I.
FIG 1.
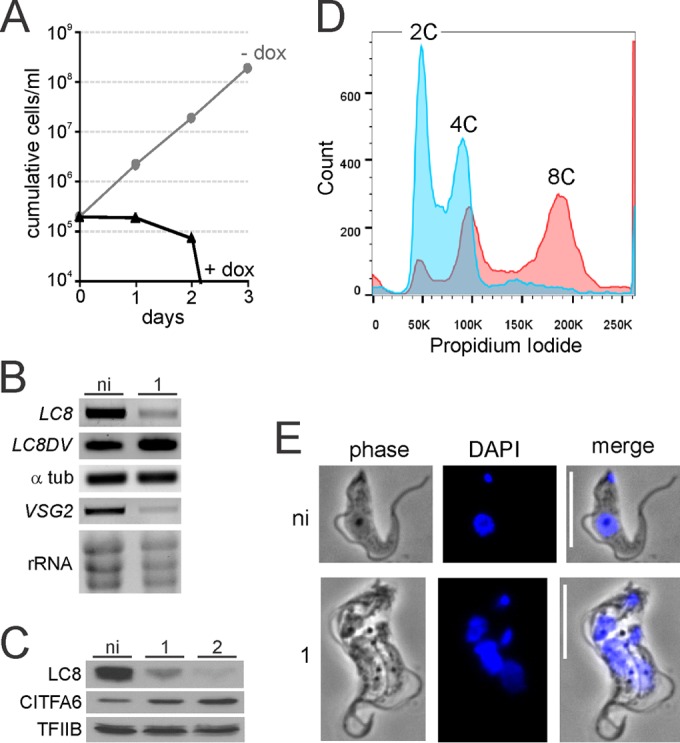
LC8 silencing has pleiotropic effects on BF trypanosomes. (A) Growth curve analysis of a representative clonal smLC8 BF cell line in the absence (−dox) and presence (+dox) of the LC8 knockdown-inducing compound doxycycline. (B) Total RNA prepared from noninduced (ni) and 1-day-induced cells was reverse transcribed with oligo(dT) and analyzed by semiquantitative PCR using LC8-, α-tubulin-, and VSG2-specific oligonucleotides. rRNA was detected by ethidium bromide staining of total RNA. (C) Whole-cell lysates of noninduced and 1- or 2-day-induced cells were analyzed by immunoblotting using LC8- or CITFA6-specific antibodies or, as a loading control, TFIIB-specific antibodies. (D) Flow cytometry analysis, without gating, of noninduced (blue) and 1-day-induced cultures (red). Propidium iodide staining intensity (x axis), which measures DNA content per cell, and cell counts (y axis) from one representative experiment are shown. K, thousands. (E) Indirect fluorescence microscopy of noninduced and 1-day-induced cells. Representative single cells from each culture were imaged using both phase-contrast and DAPI fluorescence. Small and large areas of DAPI intensity represent kinetoplasts and nuclei, respectively. Scale bars, 10 μm.
Since smBF cells and their derivatives have a doubling time of approximately 7 h in our hands (Fig. 1A, −dox), propidium iodide staining, which allows the quantification of the DNA content of individual cells, revealed a rapidly progressing second phenotype. After only 1 day of induction, the per-cell DNA content of induced cells had approximately tripled versus levels in noninduced cells, as revealed by flow cytometry of propidium iodide-stained cultures (Fig. 1D). Specifically, while noninduced cells demonstrated a curve with strong peaks representing cells with normal 2C and 4C DNA contents, induced cultures had smaller 2C and 4C peaks, with the majority of cells displaying polyploidy (8C). This result was confirmed by DAPI fluorescence microscopy, which demonstrated that the majority of cells in culture (64%, n = 150) had increased in size and were multiflagellated, multikinetoplastid, and multinucleated (Fig. 1E), a phenotype that was observed in only 2 out of 150 noninduced cells. The increase in size was confirmed by flow cytometry of noninduced and LC8-silenced trypanosomes (see Fig. S3 in the supplemental material). This phenotype is not likely due to the loss of LC8 from the CITFA complex as previously published knockdowns of other CITFA subunits failed to result in a similar phenotype (32, 33, 46, 53). Similarly, this phenotype does not resemble the precytokinesis defect in trypanosomes in which VSG is silenced since those cells did not become polyploid or increase in size (42). This phenotype would be consistent, however, with a role for LC8 in cell cycle progression, which has been reported previously in other organisms (14, 54).
LC8 binds to the N terminus of CITFA2.
In order to investigate the CITFA-specific role of LC8, we needed to determine LC8's binding partner within the CITFA complex since disrupting this interaction would likely only interfere with LC8's RNA Pol I-related function. To first verify that LC8 is a bona fide subunit of the CITFA complex, we PTP tagged the subunit CITFA3 at its N terminus in PF trypanosomes, tandem affinity purified the protein from crude extract, and visualized the final eluate by SDS-PAGE and protein staining. The banding pattern was consistent with previous tandem affinity purifications of CITFA, in which CITFA2, CITFA6, or CITFA7 was tagged (32, 33), and showed a single, clearly detectable band in the 10-kDa size range containing, as analyzed by liquid chromatography-tandem mass spectrometry, LC8 (Fig. 2A). After LC8's stable association with the CITFA complex was thus demonstrated, we sought to determine its binding partner within the complex. While LC8 binding sites are conserved from yeast to humans (5), a survey did not unambiguously reveal such a site in CITFA subunit sequences (data not shown). Previous sedimentation analyses of CITFA in 10 to 40% linear sucrose gradients, which were fractionated from top to bottom, consistently revealed a sedimentation peak of the CITFA complex in fractions 12 to 14 (32, 33). However, we recently discovered that CITFA2, in contrast to CITFA7 and other subunits, exhibited a narrower peak and was abundant only in fractions 13 and 14, possibly indicating the existence of CITFA complexes with partially different compositions (33). To compare the sedimentation profile of LC8 with the profiles of CITFA2 and CITFA7, we sedimented the purified complex through a linear sucrose gradient and detected these three proteins in individual fractions by immunoblotting with specific antibodies (Fig. 2B). While CITFA7's sedimentation peak spanned fractions 12 to 14, the peaks of CITFA2 and LC8 were restricted to fractions 13 and 14, suggesting that CITFA2 may be the binding partner of LC8.
FIG 2.

LC8 binds to the N terminus of CITFA2, promoting its dimerization. (A) The final eluate of a PTP-CITFA3 (P-CITFA3) tandem affinity purification (TAP eluate) was separated on an SDS-polyacrylamide (10 to 20%) gradient gel and stained with Coomassie blue. (B) Sedimentation of tandem affinity-purified CITFA by ultracentrifugation in a 10 to 40% linear sucrose gradient. Fractions, taken from top to bottom, were separated by SDS-PAGE and immunoblotted for specific detection of CITFA2, CITFA7, and LC8. CITFA7-P is so noted due to the presence of ProtC, which remains following tobacco etch virus protease cleavage. Arrows highlight the relative absence of CITFA2 and LC8 in fraction 12. Taq DNA polymerase (95 kDa), IgG (150 kDa), and apoferritin (444 kDa) were analyzed for molecular mass comparison (arrowheads). (C) Pulldown of recombinant, full-length (amino acid residues 1 to 422), wild-type CITFA2 with a C-terminal PTH tag, consisting of ProtC, a thrombin cleavage site (TCS), and a terminal 6×His tag, in the presence of recombinant LC8. Crude extract (input, Inp), supernatant (S), and precipitate (P) were analyzed in relative amounts of 1:1:8 by immunoblotting, using ProtC- and LC8-specific antibodies. A negative-control pulldown assay was conducted in the absence of CITFA2 (bottom panel). (D) Corresponding experiments with CITFA2-PTH fragments which are specified by residue numbers. (E) Alignment of the N-terminal CITFA2 sequences from Trypanosoma brucei brucei strain 427 (Tb427) (accession numbers are listed in Table S1 in the supplemental material), Trypanosoma congolense (Tcon), Trypanosoma vivax (Tv), Trypanosoma cruzi (Tc), Trypanosoma grayi (Tgr), Trypanosoma rangeli (Tran), Leishmania tarentolae (Lta), Leishmania mexicana (Lmx), Leishmania major (Lm), Leishmania infantum (Li), and Leishmania donovani (Ldon). Positions with more than 50% identity or similarity are highlighted in black or gray, respectively. The proposed LC8 binding site is marked below the alignment (asterisks), while the two mutants used for further investigation, NDel and 3Amut, are indicated above. (F) Pulldown assay of full-length rCITFA2-PTH carrying the 3Amut mutation (r3Amut-PTH). (G) Sucrose gradient sedimentation of rCITFA2-PTH alone, LC8 alone, or the two in combination, following coincubation (bottom two panels). Arrows indicate a cosedimentation peak in fraction 10 that was not present when either protein was analyzed on its own. Asterisks identify a truncated form of rCITFA2-PTH.
To directly test this, we generated in E. coli recombinant CITFA2 with a C-terminal composite PTH tag containing a ProtC epitope, a thrombin cleavage site, and six histidine residues (6×His). Incubating purified CITFA2-PTH, immobilized on beads, with purified LC8, after removal of the GST tag, coprecipitated LC8 in a CITFA2-PTH-dependent manner, strongly indicating a direct interaction between these two proteins (Fig. 2C). In order to confirm this result and to better specify the site of interaction, we repeated this pulldown experiment using recombinant protein portions of CITFA2. Precipitation of the N-terminal half of CITFA2, comprising amino acids 1 to 208, effectively precipitated LC8 from solution, while precipitation of the C-terminal half did not (Fig. 2D). We further divided the N-terminal half of CITFA2 and again found that the most-N-terminal portion, residues 1 to 101, precipitated LC8, while the second quarter of CITFA2 failed to precipitate LC8. We then attempted to identify the LC8 binding site within this reduced region of CITFA2 according to a previous study using motifs validated in other organisms (15) and another using a directed-evolution approach to quantitatively determine the affinity preferences of the LC8 binding site (18). In both of these publications, the LC8 binding site almost always contained a central glutamine (Q), with threonine (T) or valine (V) in the −1 and +1 positions. Three amino acids, threonine-glutamine-valine, at the N terminus of CITFA2 (amino acids 6 to 8) were identified as the likeliest site of interaction and are almost completely conserved among kinetoplastid CITFA2 sequences (Fig. 2E). In the course of this study two mutations of CITFA2 were pursued: an N-terminal deletion of amino acids 2 to 10 (NDel) and a replacement of amino acids 6 to 8 with alanines (3Amut). Precipitation of full-length recombinant 3Amut-PTH failed to precipitate LC8 from solution, confirming that LC8 binds CITFA2 via this N-terminal sequence (Fig. 2F).
LC8 binding promotes CITFA2 dimerization.
In other systems it was shown that LC8, acting as a dimer, binds to disordered regions of proteins, stabilizing their structures and allowing areas present in the binding partner, such as coiled-coil domains, to promote dimerization, thereby resulting in the formation of a heterotetramer (15, 20, 23). While secondary-structure prediction software identified the N terminus of CITFA2 as unstructured, no domains known to promote protein dimerization were recognized (data not shown). In order to investigate the possibility that LC8 binding induces dimerization of CITFA2, we performed sucrose gradient sedimentation of purified recombinant LC8 and crude bacterial lysate containing recombinant CITFA2-PTH. CITFA2-PTH, by itself, was found to peak in gradient fraction 7, which would be consistent with it existing as a monomer of 50 kDa (Fig. 2G), while the 20-kDa LC8 dimer was found at the top of the gradient. When we added excess amounts of LC8 to the bacterial lysate and allowed CITFA2-PTH and LC8 to interact before gradient sedimentation, however, both proteins exhibited a peak in fractions 9 and 10, consistent in size with a complex containing a 120-kDa CITFA2/LC8 heterotetramer. Serendipitously, when CITFA2-PTH was expressed in E. coli, immunoblotting against the C-terminal ProtC tag detected its full-length form and a truncated form which is missing approximately 5 kDa (Fig. 2G, asterisks). This putative N-terminal truncation was not shifted upon addition of LC8, indicating that LC8 cannot form a heterotetramer with the truncated CITFA2. These results strongly indicate that LC8 promotes the dimerization of CITFA2 by interacting with its N-terminal domain.
The CITFA2-LC8 interaction is essential for cell viability and RNA Pol I-mediated transcription.
In order to assess the importance of the CITFA2-LC8 interaction, we established BF cell lines in which doxycycline triggers both silencing of endogenous CITFA2 and expression of an RNAi-resistant CITFA2 transgene. In T. brucei, effective gene silencing via the RNAi pathway requires strong expression of an ∼500-bp-long dsRNA (43). We recently showed that targeting the heterologous PTP tag coding sequence effectively interfered with mRNAs carrying the PTP sequence, while having no deleterious off-target effect in BFs (46). The smPTP cell line, a derivative of the established smBF cell line for gene knockdowns (44), constitutively expresses the tetracycline (TET) repressor and T7 RNA Pol, and, upon induction, PTP dsRNA from a TET-controlled T7 promoter (46). To apply this system to CITFA2, we replaced one CITFA2 allele in smPTP cells by hygromycin phosphotransferase and inserted the PURO-PTP-CITFA2 plasmid into the second CITFA2 allele to obtain cell line smC2-PTP (Fig. 3A, left panel). As expected, induction of PTP dsRNA led to a loss of cell viability over 3 days of induction (Fig. 3A, middle panel), which matched previously performed CITFA2-silencing experiments that targeted the CITFA2 coding sequence (32). Immunoblotting of whole-cell lysates confirmed that CITFA2 was exclusively expressed as a PTP fusion and that depletion of PTP-CITFA2 was nearly complete after 1 day of induction (Fig. 3A, right panel). We next generated three CITFA2 rescue constructs which differed only in respect to the LC8 binding site (Fig. 3B). The constructs contained the complete wild-type, 3Amut, or NDel coding region with an HA tag sequence at the 3′ end (CITFA2-HA) and were under the control of a TET-regulated T7 promoter (note that due to SL trans splicing, trypanosomes can utilize T7 Pol for the effective production of functional mRNA [55]). The constructs were transfected into smC2-PTP cells and targeted to the ribosomal spacer region, a silent genomic locus commonly used for integration of exogenous plasmid constructs. As expected, the expression of wild-type CITFA2-HA almost completely rescued the growth defect which resulted from PTP-CITFA2 silencing (Fig. 3B, left panel). In contrast, neither the 3Amut nor NDel construct was able to rescue the knockdown of PTP-CITFA2 (Fig. 3B, middle and right panels). RNA analysis confirmed the reduction in PTP-CITFA2 mRNA in all three cell lines and the simultaneous expression of the HA-tagged transgenes (Fig. 3C). Furthermore, and consistent with the failed rescue of the PTP-CITFA2 knockdown by both mutant CITFA2-HA genes, RNA Pol I-derived rRNA and VSG2 mRNA transcripts were strongly reduced in induced cells, while such defects were absent in the wild-type rescue line (Fig. 3C). Immunoblot monitoring confirmed the knockdown of PTP-CITFA2 after 24 h of induction and the expression of the three different rescue transgenes within 8 h (Fig. 3D). While the wild-type transgene was stably expressed, mutant transgene-derived CITFA2 was only transiently detectable, and the mutant levels dropped substantially after 8 h of induction (Fig. 3D, arrows). This cannot be due to a decline in transgene mRNA as it was found to be well expressed at 48 h postinduction (Fig. 3C). Given that the decline in mutant CITFA2 levels coincided with the RNAi-mediated decline of PTP-tagged CITFA2, we hypothesize that wild-type CITFA2 might be stabilizing mutant CITFA2 through weak direct interaction. This would be consistent with the view that LC8 drives dimerization of proteins which already have a propensity toward dimerization (20). In summary, the lethality of mutating the LC8 binding site in CITFA2 in conjunction with the specific decline of rRNA and VSG2 transcripts in these rescue experiments strongly indicated that the interaction between LC8 and CITFA2 is essential for RNA Pol I-mediated transcription and BF viability in culture, notwithstanding the instability of the mutant transgene-derived CITFA2.
FIG 3.

Mutation of the LC8 binding site is lethal. (A) Schematic of the CITFA2 locus (left; not to scale) in BF cell line smC2-PTP in which one allele has been replaced with a hygromycin resistance cassette (HYG), while the remaining allele has been fused to the PTP tag sequence by integration of pPURO-PTP-CITFA2, which harbors a puromycin resistance cassette (PURO). Gray rectangles indicate gene flanks with essential RNA processing signals. Doxycycline-induced expression of PTP dsRNA specifically targets PTP-CITFA2 mRNA. An smC2-PTP culture growth curve in the presence (+dox) and absence (−dox) of doxycycline is shown (middle). Immunoblot monitoring of the CITFA2 knockdown using both anti-CITFA2 and anti-PTP antibodies, with TFIIB serving as a loading control, was performed (right). Note that the absence of an ∼55-kDa band in anti-CITFA2 antibody probing confirms exclusive expression of PTP-tagged CITFA2 in smC2-PTP cells. (B) At top is a schematic depiction (not to scale) of the construct that harbored the CITFA2-HA transgene and was targeted to the silent RRNA intergenic region to conditionally express RNAi-resistant CITFA2-HA mRNA and to rescue the PTP-CITFA2 knockdown. Culture growth curves of representative smC2-PTP cell lines whose PTP-targeted CITFA2 knockdown was rescued with wild-type (WT), 3Amut, or NDel CITFA2-HA expression are shown below. BLE, phleomycin resistance cassette. (C) RNA analysis of the rescue cell lines after either no induction (ni) or induction with 2 days of doxycycline treatment. (D) Immunoblotting of PTP-CITFA2 and CITFA2-HA proteins during a time course of doxycycline-induced PTP-CITFA2 knockdown (KD) and CITFA2-HA expression, with TFIIB serving as a loading control. Arrows indicate coreductions of 3Amut and NDel CITFA2-HA proteins with PTP-CITFA2 in the corresponding cell lines.
Mutant CITFA2 does not bind promoter DNA or interact with other CITFA subunits.
We sought to obviate the problem of mutant instability by generating a cell line in which mutant CITFA2-HA and wild-type CITFA2 were expressed simultaneously. We transfected our same three CITFA2-HA transgene constructs into BF cells that contained two wild-type CITFA2 alleles, generating cell lines C2-WT-HA, C2-NDel-HA, and C2-3Amut-HA. Immunoblot monitoring revealed that both wild-type and NDel CITFA2-HA were able to achieve long-term, high-level expression in the presence of wild-type CITFA2, while 3Amut CITFA2-HA, though durably expressed, never reached an equal protein level (Fig. 4A). This expression profile was consistent in two to four cell lines obtained with each construct. The low expression of the 3Amut protein could be due to a disruption in the secondary structure of CITFA2 causing instability, as the three alanine residues would strongly promote the formation of an α-helix (56) in a region predicted to form a β-strand.
FIG 4.

Mutation of the LC8 binding site prevents recruitment of CITFA2 to promoters and its assembly into the CITFA complex. (A) Comparison of constitutive wild-type (WT), NDel, or 3Amut CITFA-HA expression in individual BF cell lines by immunoblotting, with TFIIB serving as a loading control. (B) Anti-HA ChIP assays in cell lines constitutively expressing wild-type or NDel CITFA2-HA. Precipitated DNA was analyzed using primer pairs which amplified the consensus BES promoter (BES prom), the consensus RRNA promoter (RRNA prom), and the β/α-tubulin intergenic region. Error bars represent one standard deviation, with asterisks indicating a Student's t test P value of <0.05. (C) Anti-HA coimmunoprecipitation with the same cell lines. Blots monitoring wild-type (WT) or NDel CITFA2-HA immunoprecipitation were probed to detect the coimmunoprecipitation of LC8, CITFA6, CITFA7, and, as a loading control, TFIIB. Note that IgG light chain (IgG l.c.) was detected at the top of the CITFA6 immunoblot. (D) Sucrose gradient sedimentation of whole-cell extracts, prepared from cell lines that constitutively express wild-type (WT), NDel, or 3Amut CITFA2-HA cell lines, was analyzed by immunoblotting fractions 4 to 20 using either anti-HA or anti-CITFA6 immune serum.
Given that CITFA is a promoter-binding transcription factor, we sought to determine if the well-expressed NDel mutant was recruited to promoter DNA. ChIP using anti-HA antibody, specific for the transgene-derived CITFA2-HA, revealed that while wild-type CITFA2-HA was present at both the RRNA and BES promoters, NDel CITFA2-HA was not (Fig. 4B). This lack of promoter binding could be the result of either a loss of DNA binding by a complete CITFA complex or a lack of assembly of the mutant CITFA2 into the CITFA complex. To differentiate between these two possibilities, we immunoprecipitated both the wild-type and NDel CITFA2-HA proteins and analyzed for co-IP of other CITFA subunits (Fig. 4C). Precipitation of the wild-type protein resulted in the co-IP of LC8, CITFA6, and CITFA7, verifying that the introduced CITFA2-HA had assembled with other CITFA subunits. NDel CITFA2-HA, however, failed to co-IP any of these proteins, showing that the lack of promoter binding by the mutant was due to a lack of stable association with other CITFA subunits. To confirm this result and further investigate the assembly status of both wild-type and mutant CITFA2, we performed sucrose gradient sedimentation of BF extract. While wild-type CITFA2-HA had its major sedimentation peak in fractions 12 to 15, which coincided with the peak of CITFA6, both the 3Amut and NDel proteins peaked in fractions 6 and 7 and lacked a peak in fractions 12 to 15 (Fig. 4D). This result confirms that CITFA2 must bind LC8 for CITFA complex assembly. Given that sedimentation in fractions 6 and 7 would be consistent with a 50-kDa protein, it also appears likely that CITFA2 exists as a monomer in the absence of LC8 binding in our extracts. Note that wild-type CITFA2-HA has a minor peak in fractions 8 and 9, likely representing a CITFA2 dimer and/or a CITFA2-LC8 heterotetramer.
CITFA2 directly contacts BES promoter DNA and is required for promoter binding of CITFA in vivo.
CITFA2 was shown to be of crucial importance to the initiation of transcription by RNA Pol I from the RRNA, BES, and procyclin promoters, both in vivo and in vitro (32). However, its specific function in the complex has not been determined. Early UV cross-linking of partially purified CITFA and radiolabeled BES promoter DNA resulted in a major labeled protein band of ∼50 kDa (32), which is the approximate size of CITFA subunits 1 to 3. Accordingly, depletion of CITFA1 in BFs caused a loss of CITFA3 occupancy of the RRNA and BES promoters, indicating that CITFA1 is important for the transcription factor's ability to bind to RNA Pol I promoters (46). The BES promoter extends only to position −67 relative to the transcription initiation site (TIS) and harbors two distinct sequence elements (57, 58), both of which are required for efficient binding of CITFA to the BES promoter (32). The two elements are separated by 25 bp, indicating that more than one CITFA subunit mediates DNA binding. Thus, to determine a potential DNA-binding role of CITFA2, we first used the sucrose gradient fractions of the purified CITFA complex (Fig. 2A) for a gel shift assay with an 82-bp-long radiolabeled BES promoter probe. Consistent with the near absence of CITFA2 and LC8 from fraction 12 and their sedimentation peaks in fractions 13 and 14, fraction 12 shifted the promoter only faintly (Fig. 5A, arrow), while a strong shift signal was observed in fractions 13 and 14, indicating that CITFA2 and LC8 are important for the ability of CITFA to bind the BES promoter.
FIG 5.
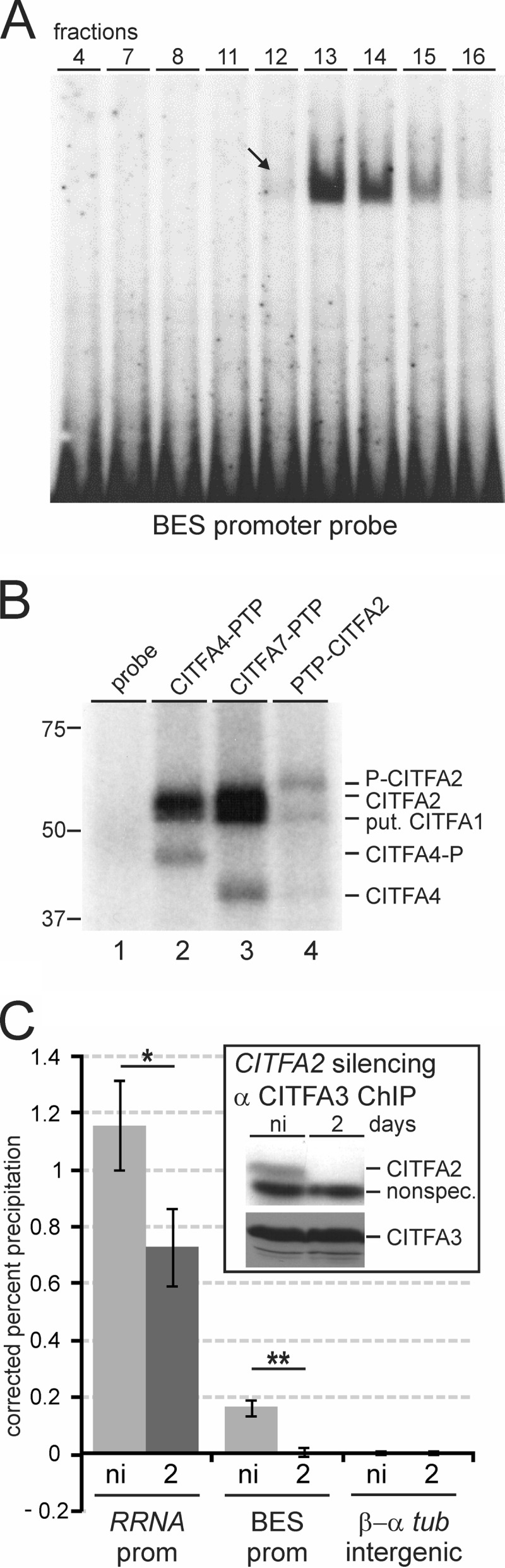
CITFA2 directly contacts the BES promoter and is required for CITFA to bind to RNA Pol I promoters in vivo. (A) Sucrose gradient fractions of purified CITFA, shown in Fig. 2A, were used in an EMSA with a radiolabeled BES promoter that was visualized by autoradiography. Fraction 12, which contains minimal LC8 and CITFA2 and an abundance of other CITFA subunits, barely binds to the probe (arrow), while fractions 13 and 14, which contain an abundance of all CITFA subunits, effectively bound the promoter probe. (B) UV cross-linking analysis using tandem affinity-purified CITFA with radiolabeled BES promoter. After DNA digest, proteins were separated by SDS-PAGE and visualized by autoradiography. On the right, tagged and untagged CITFA subunits are identified. As explained in the text, the band that did not shift is putatively CITFA1 (put. CITFA1). (C) Anti-CITFA3 ChIP assay in an smCITFA2 cell line which was either not induced or in which PTP-CITFA2 was silenced for 2 days. *, P < 0.05; **, P <0.01.
To better understand which CITFA subunits directly contact BES promoter DNA, we performed UV cross-linking using tandem affinity-purified CITFA from PFs that expressed PTP-CITFA7 (33). Cross-linking this high-purity eluate and a radiolabeled BES promoter revealed two specific protein bands in the 50- to 60-kDa range and a third band of ∼40 kDa (Fig. 5B, lane 3). Since the last band was likely CITFA4, given that no other CITFA subunit is 40 kDa, we repeated the experiment with a PF cell line that expressed CITFA4-PTP. After tandem affinity purification of a PTP-tagged protein, an ∼4 kDa portion of the tag (ProtC) remains on the purified protein. Accordingly, in the CITFA4-PTP purification, the 40-kDa band shifted up (Fig. 5B, lane 2, CITFA4-P), unequivocally identifying CITFA4 as a direct contactor of promoter DNA. Likewise, using purified CITFA containing a PTP-tagged CITFA2 resulted in a shift of the uppermost cross-linked band, identifying CITFA2 as having direct DNA contact (Fig. 5B, lane 4, P-CITFA2). Since we have been unable to verify functional PTP tagging of CITFA1, we repeated this experiment with PTP-tagged CITFA3. A failure of tagged CITFA3 to increase the size of any of the three bands (data not shown) makes it likely that CITFA1 is the third direct contactor of promoter DNA within the CITFA complex.
To confirm the importance of CITFA2 to CITFA promoter binding in vivo, we analyzed CITFA occupancy at the RRNA and BES promoters in noninduced BFs and in BFs in which CITFA2 was silenced for 2 days. Since CITFA2 depletion did not affect the abundance of CITFA3 (Fig. 5C, inset) and since absence of CITFA2 does not appear to disrupt the CITFA complex (Fig. 2A, fraction 12) (33), we performed ChIP using a purified, ChIP-grade polyclonal anti-CITFA3 antibody (46). Consistent with CITFA2 being a promoter-binding protein, CITFA3 occupancy of BES promoters was completely lost upon CITFA2 depletion (Fig. 5C). Although the CITFA3 occupancy of RRNA promoters was significantly reduced in the same experiments, CITFA3 association with RRNA promoters remained substantial. This might be due to structural differences between the RRNA and BES promoters. In contrast to the short BES promoter, the RRNA promoter extends to position −257 relative to the TIS, harboring four distinct promoter domains (59, 60). Thus, it is possible that additional factors present at the RRNA promoter interact with CITFA and stabilize it in the absence of CITFA2. In either case, these data clearly demonstrate that CITFA2 is important for CITFA binding to both the RRNA and BES promoters.
LC8 depletion affects CITFA occupancy of RRNA and BES promoters.
Finally, to verify that the recruitment failure of the CITFA2 NDel mutant to RNA Pol I promoters is due to a loss of the CITFA2-LC8 interaction, we analyzed whether LC8 silencing affected CITFA occupancy of RRNA and BES promoters. Using the same cell line as that used in the experiment shown in Fig. 1, we found a highly significant reduction in binding of CITFA3 to the BES promoter, despite the fact that LC8 silencing limited the analysis to 1 day of induction (Fig. 6). According to our observation that CITFA2 is less critical for CITFA3 occupancy of the RRNA promoter (Fig. 5C), LC8 depletion affected RRNA promoter precipitation only modestly (Fig. 6). These results are consistent with the CITFA2-LC8 interaction being crucial for CITFA function, and they verify LC8's important role in multifunctional RNA Pol I transcription in T. brucei.
FIG 6.
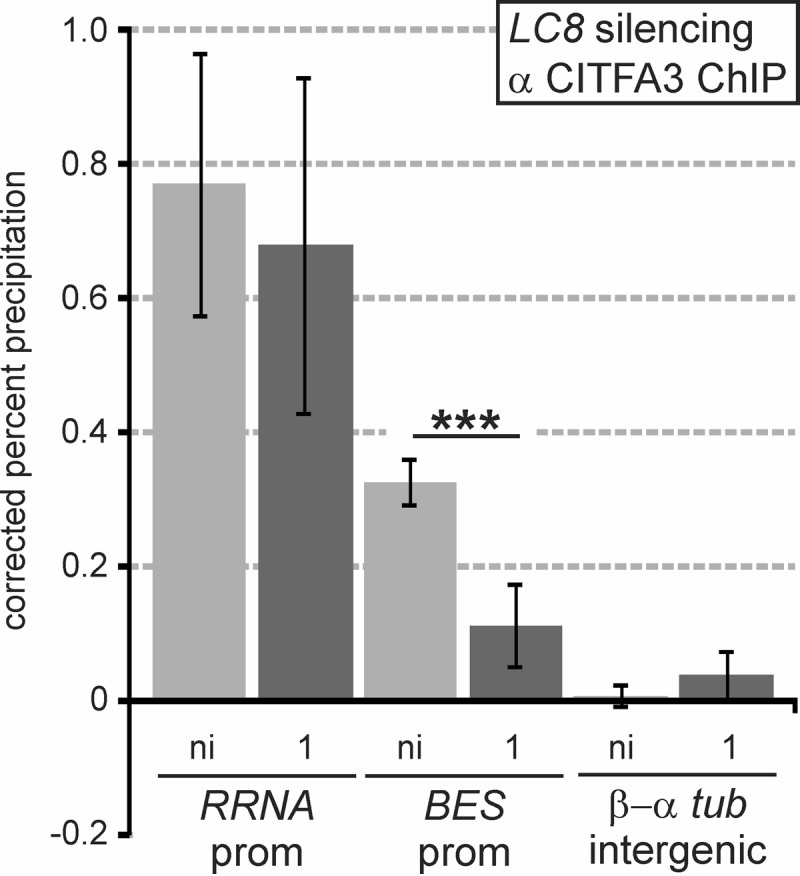
LC8 is required for recruitment of CITFA to the BES promoter. Anti-CITFA3 ChIP in smLC8 BFs without induction (ni) and after 1 day of LC8 silencing. Precipitated DNA was analyzed using the primer pairs previously noted. ***, P < 0.001.
DISCUSSION
Here, we have shown that LC8 has at least two essential functions in T. brucei, namely, in cell cycle progression and, as part of CITFA, in RNA Pol I transcription, which was the focus of this investigation. We found that LC8 directly interacts with the N terminus of CITFA2, requiring a conserved N-terminal TQV motif for binding. Sedimentation of recombinant CITFA2-LC8 complexes indicated that LC8 binding promotes the dimerization of CITFA2, resulting in a CITFA2-LC8 heterotetramer. Silencing of endogenous CITFA2 in conjunction with the expression of RNAi-resistant wild-type and mutant CITFA2 transgenes revealed that the CITFA2-LC8 interaction is essential for trypanosome viability in culture. Since CITFA2 with a mutated LC8 binding site could not be stably expressed in the absence of endogenous CITFA2, we expressed mutant CITFA2 in parallel to endogenous CITFA2 to analyze the specific function of the CITFA2-LC8 interaction in the RNA Pol I system. We found that CITFA2 is unable to bind promoter DNA or assemble with other CITFA subunits to form a complete CITFA complex in the absence of the CITFA2-LC8 interaction. Following up on a previous report which suggested a role for CITFA2 in promoter binding by CITFA (32), we found that CITFA2, CITFA4, and, likely, CITFA1 directly contact promoter DNA. Accordingly, CITFA2 silencing led to a defect in both RRNA and BES promoter binding of CITFA3, a result which was verified for the BES promoter by silencing LC8. These data suggest a model (Fig. 7) in which trypanosome LC8, by forming a dimer as in other organisms (16, 17), binds to the N termini of two CITFA2 molecules, promoting or stabilizing their dimerization. Since CITFA2 and LC8 remain stably associated with the CITFA complex after tandem affinity purification and sucrose gradient sedimentation, even at 400 mM KCl (Fig. 2A) (32, 33; also data not shown), it is likely that the full assembly of CITFA occurs independent of DNA. Finally, by contacting DNA directly through its CITFA2, CITFA4, and CITFA1 subunits, the transcription factor complex binds with high affinity to both elements of the BES promoter, which, in turn, leads to RNA Pol I recruitment and transcription initiation. Although we cannot exclude the possibility that CITFA2 dimerization leads to dimerization of the whole CITFA complex, the sedimentation profile of purified CITFA argues against this possibility. Consistent with an overall mass of CITFA of 323 kDa, which assumes a CITFA2-LC8 heterotetramer and monomers for all other subunits, full CITFA sedimented in between the 150- and 444-kDa size markers (Fig. 2A).
FIG 7.

Model of the CITFA2-LC8 interaction and function for BES promoter transcription. Formation of a CITFA2-LC8 heterotetramer is a prerequisite for its assembly into the CITFA complex. While a stable partial CITFA complex is formed without CITFA2-LC8, only a fully assembled CITFA complex that includes CITFA2 and LC8 is able to bind promoter DNA. The BES promoter, with its two sequence blocks essential for CITFA binding (yellow), is shown here along with the TIS. Please note that, for ease of visualization and understanding, LC8's site of interaction with CITFA2 was not shown at its dimer interface, where it actually occurs. Promoter-bound CITFA is able, either directly or indirectly, to recruit RNA Pol I and enable transcription initiation.
Our data and model are consistent with the view that LC8, rather than functioning as a linker between two different proteins, instead promotes homodimerization, thereby imparting new function (13, 20, 23, 61). Is LC8 essential for CITFA2 dimerization, or does it stabilize a CITFA2 dimer that has already formed? Sedimentation analysis of recombinant CITFA2 and LC8 proteins (Fig. 2F) and of extracts containing the NDel and 3Amut CITFA2-HA proteins (Fig. 4D) suggested that in the absence of LC8, CITFA2 cannot form a dimer. However, when endogenous, wild-type CITFA2 was depleted in trypanosomes, then the NDel and 3Amut CITFA2 proteins were concomitantly lost (Fig. 3D), whereas the same proteins could be constitutively expressed in the presence of wild-type CITFA2 (Fig. 4B). This stabilization of mutant CITFA2 by the wild-type protein suggests some degree of CITFA2 interaction in the absence of LC8. This finding is consistent with the demonstrated function of LC8 in structuring and stabilizing dimers of the human dynein intermediate chain (25, 62; reviewed in references 13 and 20), the Drosophila RNA-binding protein Swallow (24, 63), the human motor protein myosin Va (21, 22), and the yeast nucleoporin Nup159 (64, 65).
Given that some of LC8's nondynein binding partners, such as the human transcriptional repressor TRPS1 (66), estrogen receptor (67), and the NF-κB inhibitor IκBα (68), are involved in transcriptional regulation and since proper B cell maturation was recently demonstrated to directly depend on the DYNLL1 expression level (69), it is worth considering if LC8 might be regulating multifunctional RNA Pol I transcription in T. brucei. In accordance with this notion, our results indicated that depletion of CITFA2 (Fig. 5C) or LC8 (Fig. 6) affected CITFA3 occupancy of the BES promoter more than that of the RRNA promoter, indicating that LC8 may have a specific role in BES transcription. However, CITFA2 was shown in vitro and in vivo to be essential for RNA Pol I transcription initiating at the RRNA, procyclin gene, and BES promoters (32). In addition, the LC8 binding site within CITFA2 is at least partially conserved among all kinetoplastids (Fig. 2D), including those which do not display antigenic variation and are not known to utilize RNA Pol I for pre-mRNA synthesis, suggesting that if LC8 does have a regulatory role in RNA Pol I transcription, it is likely a general one. Nevertheless, the CITFA2-LC8 heterotetramer appears to have a crucial role in activating the CITFA complex. As previously determined, CITFA2 in BFs is expressed at about a 5-fold lower level than CITFA7, indicating that the majority of CITFA complexes in a trypanosome are inactive, requiring binding of the CITFA2-LC8 heterotetramer for activation (32, 33). Thus, it is possible that formation of a productive CITFA2-LC8 heterotetramer is quantitatively controlled in the cell. It has been shown in other systems that phosphorylation of either LC8 or its binding partner can alter their interaction, ranging from minor changes in kinetics to a complete elimination of binding (70–72). This regulatory phosphorylation occurs at S88 in human LC8, which is conserved as S89 in the genus Trypanosoma (see Fig. S1B in the supplemental material), allowing for such a regulation to exist. Furthermore, phosphorylation of CITFA2 at amino acid T6, one of the conserved residues in the LC8 binding motif, could also be used as a means to block LC8 binding, as is the case in Nek9, a kinase involved in mitotic progression (72).
Moreover, recent evidence suggests that CITFA is directly involved in the regulation of monoallelic VSG expression, which takes place outside the nucleolus in the so-called expression site body, or ESB (73, 74). ChIP assays consistently demonstrated that CITFA occupancy of the active BES promoter was several times higher than that of a silent BES promoter (53). Furthermore, the CITFA complex remained localized to the nucleolus and the ESB after depletion of CITFA1, which caused CITFA to dissociate from the RRNA and BES promoters (46). These results raised the possibility that sequestration of CITFA to the nucleolus and the ESB may be the trypanosomes' means to restrict productive RNA Pol I transcription to these compartments (75). Since LC8 has been implicated in subnuclear and subcellular localization of many of its binding partners (20), sequestration or spatially controlled formation of CITFA2-LC8 may be a mechanism for localizing CITFA function.
The RNA Pol I systems have long been known to be species specific (76). This is likely as a consequence of rapid evolutionary divergence, which is possible because this polymerase is recruited to only a single type of promoter. Nevertheless, conserved from yeast to humans, RRNA promoters have a bipartite structure with a TIS-proximal core promoter and an upstream control/promoter element that is ∼100 to 150 bp upstream of the TIS. The trypanosome BES promoter appears to comprise only the core promoter because it extends only to position −67 relative to the TIS, and both short promoter elements are required to bind CITFA. This makes it likely that CITFA is the functional homolog of the core promoter binding selectivity factor 1 (SL1) in humans (known as TIF1B in mouse) and the core factor (CF) in yeast. SL1 consists of the TATA-binding protein (TBP) and at least four TBP-associated factors termed TAF1A to TAF1D (77), whereas CF comprises the subunits RRN6, RRN7, and RRN11, which are weakly conserved orthologs of TAF1A to TAF1C (78). While it remains to be determined whether CITFA subunits represent highly divergent orthologs of TAFs, the lack of TBP and the essential role of the highly conserved LC8 for CITFA assembly and PIC formation clearly distinguish the trypanosome factor from its mammalian and yeast counterparts. Moreover, to our knowledge, LC8 has not been implicated in PIC formation of any of the three nuclear RNA Pols in any system so far.
In summary, this work is the first investigation of LC8 in a kinetoplastid organism and reveals a novel use of LC8 in the basal process of transcription initiation by RNA Pol I. It confirms the results from studies in other organisms regarding LC8's binding motif and its role in protein dimerization, demonstrating that this function of LC8 is of ancient evolutionary origin.
Supplementary Material
ACKNOWLEDGMENTS
We thank Evan Jellison, University of Connecticut Health Center (UCHC) Health Flow Cytometry Core, for his help in processing our samples, Ziyin Li, University of Texas, for providing us with the flow cytometry cell preparation protocol, James Grady, CICATS, for his insights on ChIP statistics, and Stephen King, UCHC, for his careful reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00705-15.
REFERENCES
- 1.Pfister KK, Fay RB, Witman GB. 1982. Purification and polypeptide composition of dynein ATPases from Chlamydomonas flagella. Cell Motil 2:525–547. doi: 10.1002/cm.970020604. [DOI] [PubMed] [Google Scholar]
- 2.Asante D, Stevenson NL, Stephens DJ. 2014. Subunit composition of the human cytoplasmic dynein-2 complex. J Cell Sci 127:4774–4787. doi: 10.1242/jcs.159038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.King SM, Patel-King RS. 1995. The Mr = 8,000 and 11,000 outer arm dynein light chains from Chlamydomonas flagella have cytoplasmic homologues. J Biol Chem 270:11445–11452. doi: 10.1074/jbc.270.19.11445. [DOI] [PubMed] [Google Scholar]
- 4.King SM, Dillman JF III, Benashski SE, Lye RJ, Patel-King RS, Pfister KK. 1996. The mouse t-complex-encoded protein Tctex-1 is a light chain of brain cytoplasmic dynein. J Biol Chem 271:32281–32287. doi: 10.1074/jbc.271.50.32281. [DOI] [PubMed] [Google Scholar]
- 5.Wickstead B, Gull K. 2007. Dyneins across eukaryotes: a comparative genomic analysis. Traffic 8:1708–1721. doi: 10.1111/j.1600-0854.2007.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridolfsson HN, Ly N, Meyerzon M, Starr DA. 2010. UNC-83 coordinates kinesin-1 and dynein activities at the nuclear envelope during nuclear migration. Dev Biol 338:237–250. doi: 10.1016/j.ydbio.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pazour GJ, Wilkerson CG, Witman GB. 1998. A dynein light chain is essential for the retrograde particle movement of intraflagellar transport (IFT). J Cell Biol 141:979–992. doi: 10.1083/jcb.141.4.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goggolidou P, Stevens JL, Agueci F, Keynton J, Wheway G, Grimes DT, Patel SH, Hilton H, Morthorst SK, DiPaolo A, Williams DJ, Sanderson J, Khoronenkova SV, Powles-Glover N, Ermakov A, Esapa CT, Romero R, Dianov GL, Briscoe J, Johnson CA, Pedersen LB, Norris DP. 2014. ATMIN is a transcriptional regulator of both lung morphogenesis and ciliogenesis. Development 141:3966–3977. doi: 10.1242/dev.107755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li W, Yi P, Ou G. 2015. Somatic CRISPR-Cas9-induced mutations reveal roles of embryonically essential dynein chains in Caenorhabditis elegans cilia. J Cell Biol 208:683–692. doi: 10.1083/jcb.201411041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stuchell-Brereton MD, Siglin A, Li J, Moore JK, Ahmed S, Williams JC, Cooper JA. 2011. Functional interaction between dynein light chain and intermediate chain is required for mitotic spindle positioning. Mol Biol Cell 22:2690–2701. doi: 10.1091/mbc.E11-01-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lightcap CM, Kari G, Arias-Romero LE, Chernoff J, Rodeck U, Williams JC. 2009. Interaction with LC8 is required for Pak1 nuclear import and is indispensable for zebrafish development. PLoS One 4:e6025. doi: 10.1371/journal.pone.0006025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dick T, Ray K, Salz HK, Chia W. 1996. Cytoplasmic dynein (ddlc1) mutations cause morphogenetic defects and apoptotic cell death in Drosophila melanogaster. Mol Cell Biol 16:1966–1977. doi: 10.1128/MCB.16.5.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbar E. 2008. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry 47:503–508. doi: 10.1021/bi701995m. [DOI] [PubMed] [Google Scholar]
- 14.Asthana J, Kuchibhatla A, Jana SC, Ray K, Panda D. 2012. Dynein light chain 1 (LC8) association enhances microtubule stability and promotes microtubule bundling. J Biol Chem 287:40793–40805. doi: 10.1074/jbc.M112.394353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rapali P, Szenes A, Radnai L, Bakos A, Pal G, Nyitray L. 2011. DYNLL/LC8: a light chain subunit of the dynein motor complex and beyond. FEBS J 278:2980–2996. doi: 10.1111/j.1742-4658.2011.08254.x. [DOI] [PubMed] [Google Scholar]
- 16.Benashski SE, Harrison A, Patel-King RS, King SM. 1997. Dimerization of the highly conserved light chain shared by dynein and myosin V. J Biol Chem 272:20929–20935. doi: 10.1074/jbc.272.33.20929. [DOI] [PubMed] [Google Scholar]
- 17.Barbar E, Kleinman B, Imhoff D, Li M, Hays TS, Hare M. 2001. Dimerization and folding of LC8, a highly conserved light chain of cytoplasmic dynein. Biochemistry 40:1596–1605. doi: 10.1021/bi002278+. [DOI] [PubMed] [Google Scholar]
- 18.Rapali P, Radnai L, Suveges D, Harmat V, Tolgyesi F, Wahlgren WY, Katona G, Nyitray L, Pal G. 2011. Directed evolution reveals the binding motif preference of the LC8/DYNLL hub protein and predicts large numbers of novel binders in the human proteome. PLoS One 6:e18818. doi: 10.1371/journal.pone.0018818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo KW, Naisbitt S, Fan JS, Sheng M, Zhang M. 2001. The 8-kDa dynein light chain binds to its targets via a conserved (K/R)XTQT motif. J Biol Chem 276:14059–14066. [DOI] [PubMed] [Google Scholar]
- 20.Barbar E, Nyarko A. 2014. NMR characterization of self-association domains promoted by interactions with LC8 hub protein. Comput Struct Biotechnol J 9:e201402003. doi: 10.5936/csbj.201402003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodi Z, Nemeth AL, Radnai L, Hetenyi C, Schlett K, Bodor A, Perczel A, Nyitray L. 2006. Alternatively spliced exon B of myosin Va is essential for binding the tail-associated light chain shared by dynein. Biochemistry 45:12582–12595. doi: 10.1021/bi060991e. [DOI] [PubMed] [Google Scholar]
- 22.Wagner W, Fodor E, Ginsburg A, Hammer JA III. 2006. The binding of DYNLL2 to myosin Va requires alternatively spliced exon B and stabilizes a portion of the myosin's coiled-coil domain. Biochemistry 45:11564–11577. doi: 10.1021/bi061142u. [DOI] [PubMed] [Google Scholar]
- 23.Barbar E, Nyarko A. 2015. Polybivalency and disordered proteins in ordering macromolecular assemblies. Semin Cell Dev Biol 37:20–25. doi: 10.1016/j.semcdb.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Hare M, Hays TS, Barbar E. 2004. Dynein light chain LC8 promotes assembly of the coiled-coil domain of swallow protein. Biochemistry 43:4611–4620. doi: 10.1021/bi036328x. [DOI] [PubMed] [Google Scholar]
- 25.Williams JC, Roulhac PL, Roy AG, Vallee RB, Fitzgerald MC, Hendrickson WA. 2007. Structural and thermodynamic characterization of a cytoplasmic dynein light chain-intermediate chain complex. Proc Natl Acad Sci U S A 104:10028–10033. doi: 10.1073/pnas.0703614104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jayappa KD, Ao Z, Wang X, Mouland AJ, Shekhar S, Yang X, Yao X. 2015. Human immunodeficiency virus type 1 employs the cellular dynein light chain 1 protein for reverse transcription through interaction with its integrase protein. J Virol 89:3497–3511. doi: 10.1128/JVI.03347-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luthra P, Jordan DS, Leung DW, Amarasinghe GK, Basler CF. 2015. Ebola virus VP35 interaction with dynein LC8 regulates viral RNA synthesis. J Virol 89:5148–5153. doi: 10.1128/JVI.03652-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan GS, Preuss MA, Williams JC, Schnell MJ. 2007. The dynein light chain 8 binding motif of rabies virus phosphoprotein promotes efficient viral transcription. Proc Natl Acad Sci U S A 104:7229–7234. doi: 10.1073/pnas.0701397104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qureshi BM, Hofmann NE, Arroyo-Olarte RD, Nickl B, Hoehne W, Jungblut PR, Lucius R, Scheerer P, Gupta N. 2013. Dynein light chain 8a of Toxoplasma gondii, a unique conoid-localized β-strand-swapped homodimer, is required for an efficient parasite growth. FASEB J 27:1034–1047. doi: 10.1096/fj.11-180992. [DOI] [PubMed] [Google Scholar]
- 30.Brun R, Blum J. 2012. Human African trypanosomiasis. Infect Dis Clin North Am 26:261–273. doi: 10.1016/j.idc.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 31.Broadhead R, Dawe HR, Farr H, Griffiths S, Hart SR, Portman N, Shaw MK, Ginger ML, Gaskell SJ, McKean PG, Gull K. 2006. Flagellar motility is required for the viability of the bloodstream trypanosome. Nature 440:224–227. doi: 10.1038/nature04541. [DOI] [PubMed] [Google Scholar]
- 32.Brandenburg J, Schimanski B, Nogoceke E, Nguyen TN, Padovan JC, Chait BT, Cross GA, Günzl A. 2007. Multifunctional class I transcription in Trypanosoma brucei depends on a novel protein complex. EMBO J 26:4856–4866. doi: 10.1038/sj.emboj.7601905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen TN, Nguyen BN, Lee JH, Panigrahi AK, Günzl A. 2012. Characterization of a novel class I transcription factor A (CITFA) subunit that is indispensable for transcription by the multifunctional RNA polymerase I of Trypanosoma brucei. Eukaryot Cell 11:1573–1581. doi: 10.1128/EC.00250-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Günzl A, Bruderer T, Laufer G, Schimanski B, Tu LC, Chung HM, Lee PT, Lee MG. 2003. RNA polymerase I transcribes procyclin genes and variant surface glycoprotein gene expression sites in Trypanosoma brucei. Eukaryot Cell 2:542–551. doi: 10.1128/EC.2.3.542-551.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wirtz E, Hartmann C, Clayton C. 1994. Gene expression mediated by bacteriophage T3 and T7 RNA polymerases in transgenic trypanosomes. Nucleic Acids Res 22:3887–3894. doi: 10.1093/nar/22.19.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudenko G, Chung HM, Pham VP, Van der Ploeg LH. 1991. RNA polymerase I can mediate expression of CAT and neo protein-coding genes in Trypanosoma brucei. EMBO J 10:3387–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zomerdijk JC, Kieft R, Borst P. 1991. Efficient production of functional mRNA mediated by RNA polymerase I in Trypanosoma brucei. Nature 353:772–775. doi: 10.1038/353772a0. [DOI] [PubMed] [Google Scholar]
- 38.Schwede A, Jones N, Engstler M, Carrington M. 2011. The VSG C-terminal domain is inaccessible to antibodies on live trypanosomes. Mol Biochem Parasitol 175:201–204. doi: 10.1016/j.molbiopara.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hertz-Fowler C, Figueiredo LM, Quail MA, Becker M, Jackson A, Bason N, Brooks K, Churcher C, Fahkro S, Goodhead I, Heath P, Kartvelishvili M, Mungall K, Harris D, Hauser H, Sanders M, Saunders D, Seeger K, Sharp S, Taylor JE, Walker D, White B, Young R, Cross GA, Rudenko G, Barry JD, Louis EJ, Berriman M. 2008. Telomeric expression sites are highly conserved in Trypanosoma brucei. PLoS One 3:e3527. doi: 10.1371/journal.pone.0003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cross GA, Kim HS, Wickstead B. 2014. Capturing the variant surface glycoprotein repertoire (the VSGnome) of Trypanosoma brucei Lister 427. Mol Biochem Parasitol 195:59–73. doi: 10.1016/j.molbiopara.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 41.Horn D. 2014. Antigenic variation in African trypanosomes. Mol Biochem Parasitol 195:123–129. doi: 10.1016/j.molbiopara.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sheader K, Vaughan S, Minchin J, Hughes K, Gull K, Rudenko G. 2005. Variant surface glycoprotein RNA interference triggers a precytokinesis cell cycle arrest in African trypanosomes. Proc Natl Acad Sci U S A 102:8716–8721. doi: 10.1073/pnas.0501886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi H, Djikeng A, Mark T, Wirtz E, Tschudi C, Ullu E. 2000. Genetic interference in Trypanosoma brucei by heritable and inducible double-stranded RNA. RNA 6:1069–1076. doi: 10.1017/S1355838200000297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wirtz E, Leal S, Ochatt C, Cross GA. 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99:89–101. doi: 10.1016/S0166-6851(99)00002-X. [DOI] [PubMed] [Google Scholar]
- 45.Schimanski B, Nguyen TN, Günzl A. 2005. Highly efficient tandem affinity purification of trypanosome protein complexes based on a novel epitope combination. Eukaryot Cell 4:1942–1950. doi: 10.1128/EC.4.11.1942-1950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park SH, Nguyen BN, Kirkham JK, Nguyen TN, Günzl A. 2014. A new strategy of RNA interference that targets heterologous sequences reveals CITFA1 as an essential component of class I transcription factor A in Trypanosoma brucei. Eukaryot Cell 13:785–795. doi: 10.1128/EC.00014-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schimanski B, Brandenburg J, Nguyen TN, Caimano MJ, Günzl A. 2006. A TFIIB-like protein is indispensable for spliced leader RNA gene transcription in Trypanosoma brucei. Nucleic Acids Res 34:1676–1684. doi: 10.1093/nar/gkl090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nguyen TN, Schimanski B, Günzl A. 2007. Active RNA polymerase I of Trypanosoma brucei harbors a novel subunit essential for transcription. Mol Cell Biol 27:6254–6263. doi: 10.1128/MCB.00382-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rost B, Yachdav G, Liu J. 2004. The PredictProtein server. Nucleic Acids Res 32:W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Z, Umeyama T, Wang CC. 2009. The aurora kinase in Trypanosoma brucei plays distinctive roles in metaphase-anaphase transition and cytokinetic initiation. PLoS Pathog 5:e1000575. doi: 10.1371/journal.ppat.1000575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, Gardner MJ, Gingle A, Grant G, Harb OS, Heiges M, Hertz-Fowler C, Houston R, Innamorato F, Iodice J, Kissinger JC, Kraemer E, Li W, Logan FJ, Miller JA, Mitra S, Myler PJ, Nayak V, Pennington C, Phan I, Pinney DF, Ramasamy G, Rogers MB, Roos DS, Ross C, Sivam D, Smith DF, Srinivasamoorthy G, Stoeckert CJ Jr, Subramanian S, Thibodeau R, Tivey A, Treatman C, Velarde G, Wang H. 2010. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res 38:D457–D462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Logan-Klumpler FJ, De SN, Boehme U, Rogers MB, Velarde G, McQuillan JA, Carver T, Aslett M, Olsen C, Subramanian S, Phan I, Farris C, Mitra S, Ramasamy G, Wang H, Tivey A, Jackson A, Houston R, Parkhill J, Holden M, Harb OS, Brunk BP, Myler PJ, Roos D, Carrington M, Smith DF, Hertz-Fowler C, Berriman M. 2012. GeneDB–an annotation database for pathogens. Nucleic Acids Res 40:D98–D108. doi: 10.1093/nar/gkr1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nguyen TN, Müller LS, Park SH, Siegel TN, Günzl A. 2014. Promoter occupancy of the basal class I transcription factor A differs strongly between active and silent VSG expression sites in Trypanosoma brucei. Nucleic Acids Res 42:3164–3176. doi: 10.1093/nar/gkt1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu B, Xiang X, Lee YR. 2003. The requirement of the LC8 dynein light chain for nuclear migration and septum positioning is temperature dependent in Aspergillus nidulans. Mol Microbiol 47:291–301. doi: 10.1046/j.1365-2958.2003.03285.x. [DOI] [PubMed] [Google Scholar]
- 55.Stewart M, Haile S, Jha BA, Cristodero M, Li CH, Clayton C. 2010. Processing of a phosphoglycerate kinase reporter mRNA in Trypanosoma brucei is not coupled to transcription by RNA polymerase II. Mol Biochem Parasitol 172:99–106. doi: 10.1016/j.molbiopara.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 56.Pace CN, Scholtz JM. 1998. A helix propensity scale based on experimental studies of peptides and proteins. Biophys J 75:422–427. doi: 10.1016/S0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanhamme L, Pays A, Tebabi P, Alexandre S, Pays E. 1995. Specific binding of proteins to the noncoding strand of a crucial element of the variant surface glycoprotein, procyclin, and ribosomal promoters of Trypanosoma brucei. Mol Cell Biol 15:5598–5606. doi: 10.1128/MCB.15.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pham VP, Qi CC, Gottesdiener KM. 1996. A detailed mutational analysis of the VSG gene expression site promoter. Mol Biochem Parasitol 75:241–254. doi: 10.1016/0166-6851(95)02513-8. [DOI] [PubMed] [Google Scholar]
- 59.Janz L, Clayton C. 1994. The PARP and rRNA promoters of Trypanosoma brucei are composed of dissimilar sequence elements that are functionally interchangeable. Mol Cell Biol 14:5804–5811. doi: 10.1128/MCB.14.9.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schimanski B, Laufer G, Gontcharova L, Günzl A. 2004. The Trypanosoma brucei spliced leader RNA and rRNA gene promoters have interchangeable TbSNAP50-binding elements. Nucleic Acids Res 32:700–709. doi: 10.1093/nar/gkh231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.King SM. 2008. Dynein-independent functions of DYNLL1/LC8: redox state sensing and transcriptional control. Sci Signal 1:pe51. doi: 10.1126/scisignal.147pe51. [DOI] [PubMed] [Google Scholar]
- 62.Nyarko A, Hare M, Hays TS, Barbar E. 2004. The intermediate chain of cytoplasmic dynein is partially disordered and gains structure upon binding to light-chain LC8. Biochemistry 43:15595–15603. doi: 10.1021/bi048451+. [DOI] [PubMed] [Google Scholar]
- 63.Kidane AI, Song Y, Nyarko A, Hall J, Hare M, Lohr F, Barbar E. 2013. Structural features of LC8-induced self-association of swallow. Biochemistry 52:6011–6020. doi: 10.1021/bi400642u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nyarko A, Song Y, Novacek J, Zidek L, Barbar E. 2013. Multiple recognition motifs in nucleoporin Nup159 provide a stable and rigid Nup159-Dyn2 assembly. J Biol Chem 288:2614–2622. doi: 10.1074/jbc.M112.432831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stelter P, Kunze R, Flemming D, Hopfner D, Diepholz M, Philippsen P, Bottcher B, Hurt E. 2007. Molecular basis for the functional interaction of dynein light chain with the nuclear-pore complex. Nat Cell Biol 9:788–796. doi: 10.1038/ncb1604. [DOI] [PubMed] [Google Scholar]
- 66.Kaiser FJ, Tavassoli K, Van den Bemd GJ, Chang GT, Horsthemke B, Moroy T, Ludecke HJ. 2003. Nuclear interaction of the dynein light chain LC8a with the TRPS1 transcription factor suppresses the transcriptional repression activity of TRPS1. Hum Mol Genet 12:1349–1358. doi: 10.1093/hmg/ddg145. [DOI] [PubMed] [Google Scholar]
- 67.Rayala SK, den Hollander P, Balasenthil S, Yang Z, Broaddus RR, Kumar R. 2005. Functional regulation of oestrogen receptor pathway by the dynein light chain 1. EMBO Rep 6:538–544. doi: 10.1038/sj.embor.7400417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jung Y, Kim H, Min SH, Rhee SG, Jeong W. 2008. Dynein light chain LC8 negatively regulates NF-κB through the redox-dependent interaction with IκBα. J Biol Chem 283:23863–23871. doi: 10.1074/jbc.M803072200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jurado S, Gleeson K, O'Donnell K, Izon DJ, Walkley CR, Strasser A, Tarlinton DM, Heierhorst J. 2012. The Zinc-finger protein ASCIZ regulates B cell development via DYNLL1 and Bim. J Exp Med 209:1629–1639. doi: 10.1084/jem.20120785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lei K, Davis RJ. 2003. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci U S A 100:2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song Y, Benison G, Nyarko A, Hays TS, Barbar E. 2007. Potential role for phosphorylation in differential regulation of the assembly of dynein light chains. J Biol Chem 282:17272–17279. doi: 10.1074/jbc.M610445200. [DOI] [PubMed] [Google Scholar]
- 72.Gallego P, Velazquez-Campoy A, Regue L, Roig J, Reverter D. 2013. Structural analysis of the regulation of the DYNLL/LC8 binding to Nek9 by phosphorylation. J Biol Chem 288:12283–12294. doi: 10.1074/jbc.M113.459149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chaves I, Rudenko G, Dirks MA, Cross M, Borst P. 1999. Control of variant surface glycoprotein gene-expression sites in Trypanosoma brucei. EMBO J 18:4846–4855. doi: 10.1093/emboj/18.17.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Navarro M, Gull K. 2001. A Pol I transcriptional body associated with VSG mono-allelic expression in Trypanosoma brucei. Nature 414:759–763. doi: 10.1038/414759a. [DOI] [PubMed] [Google Scholar]
- 75.Günzl A, Kirkham JK, Nguyen TN, Badjatia N, Park SH. 2015. Mono-allelic VSG expression by RNA polymerase I in Trypanosoma brucei: expression site control from both ends? Gene 556:68–73. doi: 10.1016/j.gene.2014.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grummt I. 2003. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev 17:1691–1702. doi: 10.1101/gad.1098503R. [DOI] [PubMed] [Google Scholar]
- 77.Goodfellow SJ, Zomerdijk JC. 2013. Basic mechanisms in RNA polymerase I transcription of the ribosomal RNA genes. Subcell Biochem 61:211–236. doi: 10.1007/978-94-007-4525-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Knutson BA, Hahn S. 2013. TFIIB-related factors in RNA polymerase I transcription. Biochim Biophys Acta 1829:265–273. doi: 10.1016/j.bbagrm.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.