

Abstract
The transcellular signaling of neurotrophins is postulated, but evidence is scarce. We now show that a small number of NT4- and BDNF-overexpressing neurons in the cortical explant of thalamocortical cocultures rapidly evoked a Trk receptor-dependent upregulation of neuropeptide Y (NPY) mRNA in interneurons. In contrast to BDNF, the action of NT4 was independent of calcium influx through NMDA receptors and L-type calcium channels. NPY neurons vastly outnumbered the neurotrophin-overexpressing neurons (mostly pyramidal cells), arguing for a spread of the neurotrophin signal via axonally connected neuronal populations. Furthermore, NT4 transfection of one explant of axonally connected corticocortical cocultures evoked significantly larger numbers of NPY neurons in both explants. Delivery of the signal was not by diffusion of neurotrophins via the medium. Moreover, cortical NPY neuron numbers increased after NT4 and BDNF transfection of a cocultured tectal explant innervated selectively by cortical layer V pyramidal neurons. The transcellular induction of NPY suggests a source-to-sink model for axonal transport and a local cortical redistribution of TrkB ligands to interneurons competent for NPY expression.
Keywords: activity, anterograde and retrograde transport, biolistics, neurotrophin release, rat visual system
In the neocortex, neurotrophins are master regulators of ontogenetic differentiation and adult function. Local production and local intercellular actions of neurotrophins are well accepted, and, e.g., BDNF is released from dendrites of cortical pyramidal cells and in this way becomes available to afferent pyramidal cells and inhibitory interneurons (1). However, neurotrophins have long-range and widespread effects as a result of neuronal connectivity. For instance, BDNF is axonally exported along the corticostriatal projection (2). Cortical NT4 has been shown to prevent lateral geniculate neurons from shrinkage after monocular deprivation by means of retrograde transport (3). Furthermore, NT4 is anterogradely transported in the retinotectal projection and prevents developmental cell death in the colliculus (4). NT4 delivered by the corticotectal projection accelerates the growth of tectal interneurons (5). Neurons contacted by axons supplying neurotrophins accumulate the peptides, as shown for retinotectal delivery of NT3 (6) and transcellular transfer of BDNF (7). Whether neurotrophins acquired by retrograde or anterograde transport become degraded upon internalization or become recycled to second-order neurons is unknown.
Neuropeptide Y (NPY) is transiently expressed by many interneurons in the early postnatal rat visual cortex (VC) and gradually down-regulated during the second month. This phenotype restriction is evoked by afferent innervation, and it shapes the regional and laminar-specific pattern of NPY mRNA expression of the mature VC (8, 9). The phenotype restriction proceeds despite postnatally increasing and high adult steady-state levels of BDNF (8) and strong expression of TrkB receptors on interneurons (10). This result has led to the hypothesis (9) that an early postnatal specification process renders many putative NPY neurons insensitive to adult steady-state levels of endogenous neurotrophins. However, because the neurons have expressed NPY once during development, they are later capable of up-regulating NPY in a use-dependent manner under conditions of higher than normal steady-state levels of neurotrophins. In the adult, NPY expression is highly sensitive to and tightly regulated by TrkB ligands (11–13) and becomes up-regulated by pathophysiological activity that increases BDNF expression.
We now used the NPY expression in cortical interneurons as a cellular readout for an increased supply of TrkB ligands via the axonal projections of pyramidal cells in organotypic coculture systems. Overproduction of BDNF and NT4 was evoked by biolistic transfection (14) followed by expression and release of the neurotrophins from a small number of transfectants. We provide evidence that neurotrophins are axonally imported from transfected tectal explants and that neurotrophin-mediated signals are transcellularly delivered to cortical interneurons competent for NPY expression, suggesting a functional recycling of neurotrophins.
Materials and Methods
Organotypic Cultures (OTC). Roller-tube cultures were prepared as described in ref. 8 from newborn Long-Evans hooded rats. In brief, VC, dorsolateral thalamus, or superior colliculus (SC) was chopped in 350-μm-thick (VC) or 500-μm-thick (thalamus, SC) slices that were arranged as corticocortical, thalamocortical, or corticotectal cocultures. Cultures were kept for up to 70 days. The medium was changed every third day. Cocultures connected with visible tissue bridges were transfected; they contain the axonal projections that become reestablished within the first 2 weeks (8, 15, 16). Retrograde DiI (Molecular Probes) tracing was used to prove that connections form in cocultures from the experimental batches and, in particular, that the corticotectal projection develops under activity deprivation.
Biolistic Transfection. The enhanced green fluorescent protein pEGFP-N1 plasmid (Clontech) was used as the reporter. The BDNF plasmid (pCMV5-BDNF) was kindly provided by Y. A. Barde (Max Planck Institute, Martinsried). The NT4 plasmid (pCMX-hNT4myc) was kindly provided by G. D. Yancopoulos (Regeneron). All plasmids carry the cytomegalovirus promoter. DNA coating of gold particles, preparation of cartridges, and transfection with the hand-held Helios Gene Gun (Bio-Rad) were performed as described in ref. 14. EGFP fluorescence was detectable up to 21 days after blasting, indicating that the neurotrophins also were present during the 10-days in vitro (DIV) time period maximally allowed for expression, and both factors were transcribed and translated (14). Transfected cocultures were visually inspected, and only those displaying at least 20 transfected neurons were processed for in situ hybridization.
Experimental Conditions. Cortices of thalamocortical cocultures were transfected with EGFP, BDNF/EGFP, or NT4/EGFP at 65 DIV and analyzed for NPY mRNA expression at 70 DIV. The K252a tyrosine kinase inhibitor (40 nM, Calbiochem/Merck) was applied concurrently to the transfection with BDNF and NT4. To block calcium influx 50 μM d,l-2-amino-phopshonovaleric acid (APV, an NMDA receptor blocker) and 100 μM nifedipine (L-type voltage-gated calcium channel blocker; both from Sigma-Aldrich) were applied concurrently to the transfection. APV reduces but does not completely abolish neuronal activity in OTC (17), and neither drug interferes with neurotrophin release (18). To probe for a diffusible action of the neurotrophins, “conditioned” medium harvested from NT4 transfected cocultures was fed to age-matched untreated cocultures. To test for axonal delivery, one explant of corticocortical cocultures or the SC in corticotectal cocultures was selectively transfected with NT4 or BDNF plasmids (expression from 65–70 or 20–25 DIV, respectively; see Fig. 7, which is published as supporting information on the PNAS web site) by using an aperture in front of the gene-gun muzzle. Neutralizing antibodies against NT4 and BDNF (PeproTech) were used to control for direct versus indirect effects of the transfected neurotrophins. Antibodies were applied to corticotectal cocultures once daily at 300 ng/ml medium starting 1 day after transfection, and their efficiency has previously been proven (19).
In Situ Hybridization and Detection of the Reporter Protein. Freefloating OTC were processed for in situ hybridization with digoxigenin-UTP-labeled riboprobes specific for the NPY signal sequence as described in refs. 8, 9, and 13. EGFP expression reached peak levels 24 h after transfection (FITC filter; excitation at 490 nm, emission at 520 nm). For double-labeling, EGFP was detected by immunofluorescence after NPY in situ hybridization with mouse αGFP-20 antibody (Sigma) as described in ref. 14, followed by Alexa Fluor 594-conjugated secondary antibody (Molecular Probes).
Analysis. The number of NPY-mRNA-expressing neurons was plotted by using the Eutectics Neuron Tracing system at ×200 magnification. After thionin counterstaining, neuronal nuclei were counted, and the percentage of NPY-expressing neurons was calculated (8, 13). For every condition, 6–10 OTC from different culture batches were analyzed. The mean percentages were used to construct the graphs with SEMs. A nonparametric Mann–Whitney U test was performed, and for multiple testing, the α-value was corrected according to Holm (20).
PCR. Semiquantitative PCR was used to assess the cortical expression status of NT4 and BDNF mRNA after tectal transfections. RT-PCR for the neurotrophin mRNAs was performed as described in ref. 21 in three cDNA libraries each prepared from three cortical explants of corticotectal cocultures after tectal transfection of EGFP, BNDF, or NT4. With each library three PCR reactions were run (in total, nine reactions for each amplicon and experimental condition). Expression levels were normalized to actin mRNA expression. Graphs represent the means with SEMs relative to BDNF and NT4 mRNA expression after EGFP transfection (control set to 1).
Results
BDNF and NT4 Overexpression Induces NPY in Cortices of Aged Thalamocortical Cocultures. The cortical explants of thalamocortical cocultures were selectively transfected at 60 DIV (after the phenotype restriction) (8). NPY mRNA expression was analyzed at 70 DIV. Cultures transfected with EGFP served as controls and at DIV 70 revealed 2.28 ± 0.66% NPY neurons (Fig. 1 A and E) (8).
Fig. 1.

NPY-mRNA-expressing neurons in cortical explants of 70-DIV thalamocortical cocultures. (A) NPY neuron numbers remained low after 60- to 70-DIV EGFP expression. Transfection of EGFP+BDNF (B) and EGFP+NT4 (C) (expression from 60 to 70 DIV) strongly increased NPY neuron numbers. The Insets show that EGFP-fluorescent BDNF and NT4 transfectants (Right Inset)do not express NPY mRNA (Left Inset). (D) NPY neuron numbers had increased already 16 h after NT4 transfection. (E) Mean percentage with SEM of NPY-mRNA-expressing neurons in the experimental conditions of A–D. Letters on the x axis match the labeling of the pictures in this and the following figures. White matter is oriented downwards. (Scale bar: 300 μm.) **, P < 0.01; ***, P < 0.001.
Cocultures transfected with BDNF plasmids displayed 5.13 ± 0.49% NPY neurons (Fig. 1 B and E; significantly different from the EGFP-transfected cocultures, P = 0.0013), and NPY neurons occurred in all layers. The major neuronal fraction of cortical transfectants are pyramidal cells (14). In vivo, pyramidal cells produce BDNF and supply the factor to interneurons to regulate the quality of inhibition. Fig. 1B Inset shows that transfectants and NPY neurons formed separate populations. Cocultures transfected with NT4 plasmids displayed 5.67 ± 0.66% NPY neurons (Fig. 1 C and E; significantly different from the EGFP-transfected cocultures, P < 0.0001, but not from BDNF-transfected cocultures, P = 0.138). The staining intensities and distribution of the NPY neurons were similar to the BDNF-transfected cultures. Again, there was no coexpression of EGFP/NT4 and NPY mRNA (Fig. 1C Inset). The natural producers of NT4 are as yet unknown, but overexpressed NT4 can be used by pyramidal cells in an autocrine fashion to accelerate dendritic growth (14), suggesting that it can be processed and released by pyramidal cells.
The NT4-induced up-regulation of the NPY expression was observed already 16 h after transfection (4.59 ± 0.68% NPY neurons; Fig. 1 D and E; significantly different from the EGFP control, P < 0.001, but also different from “long-term” NT4 overexpressing cultures, P = 0.007). Putative NPY neurons thus reacted fast to NT4 overexpression, and the NPY response persisted as long as the NT4 transfectants remained active producers.
BDNF and NT4 Act via Tyrosine Kinases, and BDNF Requires Ca2+ Influx. The effects of BDNF and NT4 overexpression were mediated by Trk receptors because both factors failed in cultures treated with K252a. BDNF overexpression with K252a revealed 1.9 ± 0.29% NPY neurons (Fig. 2 A and G; not different from EGFP control, P = 0.129, but significantly different from BDNF overexpression without K252a, P = 0.002). NT4 overexpression with K252a revealed 1.78 ± 0.78 NPY neurons (Fig. 2 B and G; not different from EGFP control, P = 0.143, but highly significantly different from the NT4 overexpression without K252a, P = 0.001).
Fig. 2.

Dependence on Trk receptors and calcium influx. (A) Low density of NPY neurons in BDNF-transfected cultures treated with K252a. (B) Low density of NPY neurons in NT4-transfected cultures treated with K252a. (C) Blocking L-type calcium channels with nifedipine and NMDA receptors with APV efficiently prevented NPY up-regulation after BDNF transfection but not after NT4 transfection (D). (E) Representative plot of the distribution of EGFP/NT-4-expressing neurons (triangles) and nonneuronal cells (mostly glia; circles) and NPY neurons (small dots) in a thalamocortical coculture at 70 DIV treated with nifedipine and APV concurrent to transfection. (F) NT4 overexpression failed to induce NPY in cultures activity-deprived with 10 mM Mg2+ (chr. Mg). (G) Mean percentage (with SEM) of NPY neurons in the experimental conditions of A–F. White matter is oriented downward (A–E). (Scale bars: A–D, 300μm; E, 500 μm.) ***, P < 0.001.
Next, the role of calcium influx was tested. Nifedipine and APV were applied concurrent with transfection of BDNF. Only 2.92 ± 0.97% of the neurons expressed NPY (Fig. 2 C and G; not different from EGFP control, P = 0.426, but significantly different from the BDNF overexpression alone, P = 0.004). In contrast, NT4 overexpression in the presence of APV and nifedipine evoked 6.48 ± 0.29% NPY neurons (Fig. 2 D, E, and G; significantly different from EGFP control, P = 0.0001, but not NT4 alone, P = 0.101). This finding suggested that the release and the action of overexpressed NT4 is independent of calcium influx through L-type channels and NMDA receptors. Fig. 2E shows the high density of NPY neurons together with NT4-overexpressing neurons and nonneuronal transfectants. NPY neurons outnumbered the transfectants by an order of magnitude. This finding argued against autocrine actions and for axonal delivery of the overexpressed neurotrophins.
However, the NT4 action was not entirely independent of neuronal activity because NT4 overexpression failed to induce NPY expression in corticocortical cocultures exposed to 10 mM MgSO4, which prevents action potential activity in the network (22) (Fig. 2 F and G). The same occurred after NT4 transfection into tectal explants in corticotectal cocultures (see below; 1.92 ± 0.22% NPY neurons, n = 7 cultures).
NT4 Acts Transcellularly on NPY Neurons. The finding that transfectants and NPY neurons were separate populations led to the question how neurons competent for NPY expression in mature cortex cultures are affected by the extra amount of neurotrophins. First, we ruled out a supply via the medium. Conditioned medium was collected from 60- to 70-DIV NT4-transfected thalamocortical cocultures that were subsequently found to display the NPY up-regulation. However, the medium failed to increase the percentage of NPY neurons in nontransfected age-matched cocultures (2.02 ± 0.82% NPY neurons; not different from EGFP control but significantly different from NT4 transfected OTC, P = 0.0025; Fig. 3 A and D). This finding suggested an axonal delivery route.
Fig. 3.

Circumstantial evidence for axonal delivery of NT4 in corticocortical cocultures. (A) Conditioned medium from NT4-transfected cocultures fails to promote NPY in untransfected age-matched cocultures. This result argues against a diffusible action of the neurotrophin. (B) Transfection of NT4 (expression from 60 to 70 DIV) strongly up-regulated the NPY expression in the transfected explant of corticocortical OTC and also in the nontransfected explant (C). (D) Mean percentage with SEM of NPY neurons in the experimental conditions of A–C in comparison with untransfected visual corticocortical OTC (VC-VC control). White matter is oriented downward. (Scale bar: 300 μm.) **, P < 0.01.
The plots (Fig. 2E) revealed no obvious clustering of NPY neurons around nearby transfectants. This finding argued against a local uptake of overexpressed neurotrophins by interneurons. We therefore analyzed 60- to 70-DIV corticocortical cocultures that also undergo a phenotype restriction down to 3.73 ± 0.36% NPY neurons (8) (Fig. 3D). At 60 DIV, we transfected one of the two reciprocally connected cortices with NT4 plasmids and analyzed both cortices at 70 DIV. As expected, the transfected cortex displayed 7.13 ± 0.74% NPY neurons (Fig. 3 B and D), which was significantly different from nontransfected corticocortical cocultures that served as control (P = 0.002). Even when blasting transfectants selectively in only one half of an explant, the NPY neurons were frequent throughout the entire explant (data not shown), again arguing against local actions. Strikingly, the nontransfected cocultured cortices also displayed 5.97 ± 0.56% NPY neurons (Fig. 3C; fourth bar in Fig. 3D). This result was significantly different from control (P = 0.003) but not different from the transfected cortex (P = 0.284). Obviously, the NT4 from the transfected cortex influenced the nontransfected cortex. Because only pyramidal cells make both the reciprocal long-range and the local intrinsic connections (8, 15), the delivery appears to occur via retrograde and/or anterograde axonal projections followed by a transfer of NT4 to local interneurons.
Effects of NT4 and BDNF Imported from Tectal Explants. Because corticocortical cocultures are reciprocally innervated, it was impossible to distinguish between an effect due to afferents or efferents. We therefore used corticotectal cocultures. Here, SC becomes unidirectionally innervated by layer V pyramidal neurons (16). Unfortunately, without a backprojection no phenotype restriction proceeds in the cortex (8). However, we knew that activity deprivation for the first 2 weeks will permanently reduce the percentage of cortical NPY neurons (13). Indeed, 15-DIV activity-deprived cortex monocultures reveal 2.13 ± 0.17% NPY neurons (Fig. 4A). When such cultures are returned to normal medium, they instantly recover neuronal activity (22), but the NPY mRNA expression remained at 1.85 ± 0.18% (Fig. 4A). The same was true for corticotectal cultures grown activitydeprived from 0 to 15 DIV, followed by a recovery of activity from 15 to 25 DIV and transfection of the tectal explant at 20 DIV with EGFP. At 25 DIV, the cortical explants displayed 2.12 ± 0.33% NPY neurons (Fig. 4 A and B). Despite the absence of neuronal activity, we can conclude the axonal projections formed in an organotypic manner because DiI tracing at 20 DIV labeled the layer V pyramidal neurons (Fig. 4E).
Fig. 4.

Circumstantial evidence for axonal delivery of NT4 in corticotectal cocultures. (A) NPY expression remained low in activity-deprived VC cultures (VC deprived). Even resumption of activity failed to increase the NPY expression (20 DIV under 10 mM Mg2+ and subsequently 10 DIV recovery in 2 mM Mg2+; VC recovery). (B) The cortical explant of corticotectal cocultures displays low numbers of NPY neurons after 15-DIV activity deprivation and transfection with EGFP from 20 to 25 DIV. (C) After NT4 transfection of the tectal explant, NPY neuron numbers increased in the cortical explant. (D) After BDNF transfection of the tectal explant, NPY neuron numbers increased in the cortical explant, and BDNF was more effective than NT4 (see A). (E) DiI injection into the tectal explant of a corticotectal coculture activity-deprived for 20 DIV. Despite the absence of activity, layer V pyramidal cells with apical dendrites reaching layer I were retrogradely labeled, suggesting that their morphological differentiation was not impaired (19). White lines, borders of the cortical explant; black line, border of the tectal explant. Roman numerals mark cortical layers. White matter is oriented downward. (F and G) The cortical expression of NT4 and BDNF mRNA was analyzed by RT-PCR. (F) The NT4 mRNA expression was not altered after tectal transfection with BDNF or NT4 plasmids. Transfection with EGFP plasmids in 25-DIV spontaneously active cultures and in recovery cultures (15 DIV deprived, then 10 DIV active; level was set to 1 for normalization) were used as controls, and the initial activity deprivation did not alter cortical NT4 expression. (G) The same was observed for cortical BDNF mRNA expression in the control conditions, and cortical BDNF mRNA was not changed after tectal NT4 transfection. However, cortical BDNF mRNA expression increased after tectal BDNF transfection. EGFP, transfection with EGFP plasmid alone; BDNF, transfection with EGFP+BDNF plasmid; NT4, transfection with EGFP+NT4 plasmid. (Scale bars: 300 μm.) **, P < 0.01; ***, P < 0.001.
Therefore, corticotectal cocultures were maintained from 0–15 DIV under activity deprivation to knock down NPY neuron numbers. Neuronal activity was allowed to resume until 20 DIV to avoid potential deficits in axonal transport. At 20 DIV, the tectal explants were selectively transfected (Fig. 7) with NT4 plasmids, followed by 5 days of expression. At 25 DIV, the cortices displayed 3.79 ± 0.29% NPY neurons in all layers (Fig. 4 A and C; significantly different from EGFP control corticotectal cocultures, P = 0.008). The NPY induction was even stronger after BDNF transfection of tectal explants: now 6.3 ± 0.45% cortical neurons were expressed NPY (Fig. 4 A and D; significantly different from control, P < 0.001).
Neurotrophins are known to induce neurotrophin expression (21). Therefore, in corticotectal cocultures the cortical expression level of BDNF and NT4 mRNA was measured by RT-PCR (Fig. 4 F and G). Expression of cortical NT4 mRNA was not altered after tectal transfection and import of BDNF or NT4 (Fig. 4F). The expression of cortical BDNF mRNA was not altered after tectal transfection of NT4. However, cortical BDNF mRNA expression increased after tectal transfection of BDNF (Fig. 4G; P = 0.052).
Transcellular signaling requires the release of the factors. To interfere with signaling, neutralizing antibodies against the transfected neurotrophins were applied. As expected, they prevented the NPY induction (3.0 ± 0.19% NPY neurons for NT4 transfection/NT4 neutralization, Fig. 5 A and E; 2.9 ± 0.15% NPY neurons for BDNF transfection/BDNF neutralization, Fig. 5 B and E). In contrast, NT4 induced the NPY expression in the presence of neutralizing anti-BDNF antibodies (Fig. 5 C and E; 5.57 ± 0.64% NPY neurons; significantly different from EGFP control corticotectal cocultures, P < 0.001). Surprisingly, however, BDNF failed to induce NPY expression in the presence of neutralizing anti-NT4 antibodies (Fig. 5 D and E; 3.29 ± 0.37% NPY neurons).
Fig. 5.

Effects of neutralization of BDNF and NT4. (A) Neutralizing NT4 after tectal NT4 transfection prevented the induction of NPY. (B) Neutralizing BDNF after tectal BDNF transfection prevented the induction of NPY. (C) Neutralization of BDNF after tectal transfection of NT4 did not prevent the induction of NPY. (D) It seems that BDNF needs endogenous NT4 signaling as a cofactor. In contrast, neutralization of BDNF in corticotectal OTC does not prevent the up-regulation of NPY by tectal NT4 transfection. (E) Mean percentage (with SEM) of NPY neurons in the experimental conditions of A–D. White matter is oriented downward. (Scale bar: 300 μm.) ***, P < 0.001, tested against VC-SC-deprived (Fig. 4B).
Discussion
Cortically transfected NT4 quickly induces NPY expression. NT4 must have been released because the action depended on Trk receptors that activate only upon binding extracellular ligands. A contribution of enhanced electrical activity is unlikely because NT4 infusions in vivo do not alter spontaneous activity (23). In activity-deprived OTC, exogenous NT4 at 20 ng/ml medium activates NPY expression (13), and overexpressed NT4 enhances pyramidal cell dendritic growth in an autocrine fashion (19). These findings argue for an activity-independent action and a constitutive release from dendrites (1, 24). The present results imply axonal delivery of NT4 to interneurons, and unexpectedly, NT4 now failed to induce NPY expression in activity-deprived OTC. This finding suggests that, in contrast to dendritic release, the axonal transport or release of NT4 might be activity-dependent.
Cortical transfection of BDNF also increased NPY neuron numbers. BDNF is predominantly released via the regulated secretory pathway (1, 18). This requires neuronal activity, which is present in our cultures (25). BDNF and activity mutually depend on each other. In the absence of activity, exogenous BDNF fails to up-regulate NPY (13, 26) and fails to promote dendritic growth (19, 27). As expected, BDNF overexpression failed to activate NPY expression in the presence of APV and nifedipine, although electrical activity is not completely abolished. The failure of BDNF to show an effect was presumably due to an impaired release. Although the antagonists do not interfere with the release or recycling of vesicles, they reduce electrical activity that likely reduces the calcium influx required for BDNF release (1, 18). Moreover, an impaired calcium influx decreases endogenous BDNF production, and the virally driven BDNF in few producers might just be too low to promote NPY.
The results from the corticocortical and corticotectal cocultures argue for axonal delivery. In the latter case, NT4 and BDNF have to be secreted by tectal transfectants and retrogradely transported by layer V pyramidal neurons (Fig. 6). We suggest that retrograde import also played a major role in the corticocortical condition, because the number of neurons projecting to the transfected explant was certainly higher than the number of transfected neurons with “callosal” projections. TrkB receptors on pyramidal cell axon terminals contacting transfectants could retrieve NT4 followed by retrograde transport of the signaling endosome (1, 2, 28, 29). Our results suggest a direct effect of NT4 imported into the cortical circuitry. First, the cortical NT4 and BDNF mRNA expression remained unchanged after tectal NT4 transfection, and NT4 concentrations were possibly too low to promote neurotrophin expression (21). Second, the NT4 action persisted when BDNF was neutralized. Imported neurotrophins are then distributed to local interneurons. For recycling, the factors dissociate from the receptor and become repacked into vesicles (29). Either interneurons retrieve NT4 via their axons contacting the importing pyramidal cell, or the pyramidal cell serves as transcellular transport device and delivers NT4 anterogradely (7) via cortical axon collaterals making synapses with somata or dendrites of interneurons. In addition to the retrograde transport postulated in our study, NT4 is anterogradely transported in the retinotectal (4) and corticotectal projection (19). A neurotrophic factor may thus be transported in both directions along the same projection, and the transport capacities are apparently not saturated by the endogenous factors. We suggest a “source-to-sink model” in which the net transport rate is from sources to sinks and is driven by the unequal availability of the neurotrophins in the CNS centers (Fig. 6). It remains to be determined whether the same neurotrophin molecules are transcytosed from tectal neurons to cortical pyramidal neurons and then to cortical interneurons or whether the internalized neurotrophins induce the release of endogenously produced cortical factors.
Fig. 6.

The source-to-sink model. NT4 overexpressers in the tectal explant ( ) become a source of NT4. Nonoverexpressing pyramidal neurons in layer V of the cortical explant (
) become a source of NT4. Nonoverexpressing pyramidal neurons in layer V of the cortical explant (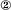 ) represent a first sink of NT4 and retrogradely import the factor (a). Now, layer V pyramidal neurons () accumulate NT4 and represent a source of NT4 for cortical interneurons (
) represent a first sink of NT4 and retrogradely import the factor (a). Now, layer V pyramidal neurons () accumulate NT4 and represent a source of NT4 for cortical interneurons (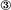 ). They, in turn, represent the second sink of NT4 and either retrieve somatic or dendritic released imported NT4 (b) or receive imported NT4 by anterograde delivery via pyramidal cell axon collaterals (c). The model also explains the results obtained in corticocortical cocultures; in addition, transfected projection neurons may directly deliver NT4 anterogradely to neurons in nontransfected explants.
). They, in turn, represent the second sink of NT4 and either retrieve somatic or dendritic released imported NT4 (b) or receive imported NT4 by anterograde delivery via pyramidal cell axon collaterals (c). The model also explains the results obtained in corticocortical cocultures; in addition, transfected projection neurons may directly deliver NT4 anterogradely to neurons in nontransfected explants.
The latter may hold for BDNF. Cortical BDNF mRNA increased after BDNF transfection of the tectal explants. This finding suggests that higher levels of endogenous BDNF contribute to the NPY induction. The BDNF action was lost after NT4 neutralization, suggesting that a release of endogenous NT4 is involved in mediating the effects of imported BDNF or that NT4 is a signaling cofactor for BDNF. BDNF promotes its own expression (21) and activity, and activity, in turn, promotes BDNF expression (23, 30, 31). BDNF and activity are master regulators for NPY expression (11, 32, 33). In fact, NPY neuron numbers were higher after tectal BDNF transfection than after NT4 transfection. NPY is a potent antiepileptic peptide that reduces glutamate release while concurrently strengthening inhibition (34–36). NPY knockout mice die from seizures (37), and the high mortality is likely not due to the lack of NPY in a minor subset of interneurons but rather to the failure of use-dependent NPY up-regulation. With regard to pathophysiological conditions, our results suggest that an aberrantly high NPY expression may not only characterize epileptic foci. NPY expression may also be increased in axonally connected areas that had imported, for example, BDNF from epileptic foci and had prophylactically responded with an up-regulation of NPY expression.
Supplementary Material
Acknowledgments
We thank Regeneron Pharmaceuticals for the pCMX-hNT4myc expression plasmid and Dr. Yves Alain Barde for pCMV5-BDNF expression plasmid. This work was supported by Deutsche Forschungsgemeinschaft Neurovision (P.W.) and Graduiertenkolleg 736.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: DIV, days in vitro; NPY, neuropeptide Y; OTC, organotypic cultures; SC, superior colliculus; VC, visual cortex.
References
- 1.Lessmann, V., Gottmann, K. & Malcangio, M. (2003) Prog. Neurobiol. 69, 341-374. [DOI] [PubMed] [Google Scholar]
- 2.Altar, C. A. & DiStefano, P. S. (1998) Trends Neurosci. 21, 433-437. [DOI] [PubMed] [Google Scholar]
- 3.Riddle, D. R., Lo, D. C. & Katz, L. C. (1995) Nature 378, 189-191. [DOI] [PubMed] [Google Scholar]
- 4.Spalding, K. L., Tan, M. M., Hendry, I. A. & Harvey, A. R. (2002) Mol. Cell. Neurosci. 19, 485-500. [DOI] [PubMed] [Google Scholar]
- 5.Wahle, P., Di Cristo, G., Schwerdtfeger, G., Engelhardt, M., Berardi, N. & Maffei, L. (2003) Development (Cambridge, U.K.) 130, 611-622. [DOI] [PubMed] [Google Scholar]
- 6.Wang, X., Butowt, R., Vasko, M. R. & von Bartheld, C. S. (2002) J. Neurosci. 22, 931-945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kohara, K., Kitamura, A., Morishima, M. & Tsumoto, T. (2001) Science 291, 2419-2423. [DOI] [PubMed] [Google Scholar]
- 8.Obst, K. & Wahle, P. (1997) Eur. J. Neurosci. 9, 2571-2580. [DOI] [PubMed] [Google Scholar]
- 9.Wahle, P., Gorba, T., Wirth, M. J. & Obst-Pernberg, K. (2000) Development (Cambridge, U.K.) 127, 1943-1951. [DOI] [PubMed] [Google Scholar]
- 10.Gorba, T. & Wahle, W. (1999) Eur. J. Neurosci. 11, 1179-1190. [DOI] [PubMed] [Google Scholar]
- 11.Carnahan, J. & Nawa, H. (1995) Mol. Neurobiol. 10, 135-149. [DOI] [PubMed] [Google Scholar]
- 12.Marty, S. & Onteniente, B. (1999) Eur. J. Neurosci. 11, 1647-1656. [DOI] [PubMed] [Google Scholar]
- 13.Wirth, M. J., Obst, K. & Wahle, P. (1998) Eur. J. Neurosci. 10, 1457-1464. [DOI] [PubMed] [Google Scholar]
- 14.Wirth, M. J. & Wahle, P. (2003) J. Neurosci. Methods 125, 45-54. [DOI] [PubMed] [Google Scholar]
- 15.Bolz, J., Novak, N., Gotz, M. & Bonhoeffer, T. (1990) Nature 346, 359-362. [DOI] [PubMed] [Google Scholar]
- 16.Cardoso de Oliveira, S. & Hoffmann, K. P. (1995) Eur. J. Neurosci. 7, 599-612. [DOI] [PubMed] [Google Scholar]
- 17.Corner, M. A., van Pelt, J., Wolters, P. S., Baker, R. E. & Nuytinck, R. H. (2002) Neurosci. Biobehav. Rev. 26, 127-185. [DOI] [PubMed] [Google Scholar]
- 18.Balkowiec, A. & Katz, D. M. (2002) J. Neurosci. 22, 10399-10407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wirth, M. J., Brün, A., Grabert, J., Patz, S. & Wahle, P. (2003) Development (Cambridge, U.K.) 130, 5827-5838. [DOI] [PubMed] [Google Scholar]
- 20.Holm, S. (1979) Scand. J. Stat. 6, 65-70. [Google Scholar]
- 21.Patz, S. & Wahle, P. (2004) Eur. J. Neurosci. 20, 1-8. [DOI] [PubMed] [Google Scholar]
- 22.Gorba, T., Klostermann, O. & Wahle, W. (1999) Cereb. Cortex 9, 864-877. [DOI] [PubMed] [Google Scholar]
- 23.Lodovichi, C., Berardi, N., Pizzorusso, T. & Maffei, L. (2000) J. Neurosci. 20, 2155-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hibbert, A. P., Morris, S. J., Seidah, N. G. & Murphy, R. A. (2003) J. Biol. Chem. 278, 48129-48136. [DOI] [PubMed] [Google Scholar]
- 25.Klostermann, O. & Wahle, P. (1999) Neuroscience 92, 1243-1259. [DOI] [PubMed] [Google Scholar]
- 26.Marty, S., Berninger, B., Carrol, P. & Thoenen, H. (1996) Neuron 16, 565-570. [DOI] [PubMed] [Google Scholar]
- 27.McAllister, A. K., Katz, L. C. & Lo, D. C. (1996) Neuron 17, 1057-1064. [DOI] [PubMed] [Google Scholar]
- 28.Heerssen, H. M., Pazyra, M. F. & Segal, R. A. (2004) Nat. Neurosci. 7, 596-604. [DOI] [PubMed] [Google Scholar]
- 29.von Bartheld, C. S. (2004) J. Neurobiol. 58, 295-314. [DOI] [PubMed] [Google Scholar]
- 30.Binder, D. K., Croll, S. D., Gall, C. M. & Scharfman, H. E. (2001) Trends Neurosci. 24, 47-53. [DOI] [PubMed] [Google Scholar]
- 31.Kafitz, K. W., Rose, C. R., Thoenen, H. & Konnerth, A. (1999) Nature 401, 918-921. [DOI] [PubMed] [Google Scholar]
- 32.Gall, C., Lauterborn, J., Isackson, P. & White, J. (1990) Prog. Brain Res. 83, 271-390. [DOI] [PubMed] [Google Scholar]
- 33.Schwarzer, C., Sperk, G., Samanin, R., Rizzi, M., Gariboldi, M. & Vezzani, A. (1996) Brain Res. Rev. 22, 27-50. [PubMed] [Google Scholar]
- 34.Colmers, W. F. & Bleakman, D. (1994) Trends Neurosci. 17, 373-379. [DOI] [PubMed] [Google Scholar]
- 35.Woldbye, D. P., Larsen, P. J., Mikkelsen, J. D., Klemp, K., Madsen, T. M. & Bolwig, T.-G. (1997) Nat. Med. 3, 761-764. [DOI] [PubMed] [Google Scholar]
- 36.Bacci, A., Huguenard, J. R. & Prince, D. A. (2002) Proc. Natl. Acad. Sci. USA 99, 17125-17130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baraban, S. C., Hollopeter, G., Erickson, J. C., Schwartzkroin, P. A. & Palmiter, R. D. (1997) J. Neurosci. 17, 8927-8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.