

Abstract
After mild ischemic insults, many neurons undergo delayed neuronal death. Aberrant activation of the cell cycle machinery is thought to contribute to apoptosis in various conditions including ischemia. We demonstrate that loss of endogenous cyclin-dependent kinase (Cdk) inhibitor p16INK4a is an early and reliable indicator of delayed neuronal death in striatal neurons after mild cerebral ischemia in vivo. Loss of p27Kip1, another Cdk inhibitor, precedes cell death in neocortical neurons subjected to oxygen–glucose deprivation in vitro. The loss of Cdk inhibitors is followed by upregulation of cyclin D1, activation of Cdk2, and subsequent cytoskeletal disintegration. Most neurons undergo cell death before entering S-phase, albeit a small number (∼1%) do progress to the S-phase before their death. Treatment with Cdk inhibitors significantly reduces cell deathin vitro. These results show that alteration of cell cycle regulatory mechanisms is a prelude to delayed neuronal death in focal cerebral ischemia and that pharmacological interventions aimed at neuroprotection may be usefully directed at cell cycle regulatory mechanisms.
Keywords: cell cycle, cerebral ischemia, cyclin-dependent kinases, delayed neuronal cell death, p16 INK4a, p27Kip1
Neurons enter and remain in a terminally differentiated or “resting” state after final cell division. If the control of the resting state breaks down, neurons may reenter the cell cycle, a process that is presumably lethal (Lee et al., 1992; Herrup and Busser, 1995; Gill and Windebank, 1998). Thus, aberrant activation of the cell cycle machinery is thought to cause apoptosis in postmitotic neurons after various insults, including cerebral ischemia and Alzheimer's disease (Busser et al., 1998; Osuga et al., 2000; Sakurai et al., 2000). Increases in cyclin D1 and cyclin-dependent kinase (Cdk) 4 expression occur after focal ischemia in mice and rats (Li et al., 1997), in global ischemia in rats (Timsit et al., 1999), and in a rabbit spinal cord ischemia model (Sakurai et al., 2000). In these models the induction of cyclin D1 occurs in neurons either before nuclear condensation and the appearance of chromosomal DNA fragmentation (Timsit et al., 1999; Osuga et al., 2000) or after the appearance of an apoptotic phenotype (Guégan et al., 1997). Neuronally differentiated PC12 cells, sympathetic neurons, and primary neuronal cells in culture upregulate cell cycle proteins before their apoptotic death when DNA damage occurs or when trophic support is withdrawn (Farinelli and Greene, 1996; Park et al., 1996, 1997a,b,1998; Padmanabhan et al., 1999).
On the other hand, expression of cell cycle markers after ischemia might be a sign of potentially beneficial mechanisms. Recovery from ischemic damage may recapitulate ontogeny, and the expression of developmental proteins in the penumbra zone to some degree may indicate active reconditioning that could promote cell survival (Li et al., 1997, 1998; Cramer and Chopp, 2000).
The aim of the present study was to define the temporal and spatial relationship between expression of cell cycle proteins including cyclin D1, Cdk2, Cdk4, the endogenous Cdk inhibitors p16INK4a and p27Kip1, and delayed neuronal death after mild focal cerebral ischemia in mice (Endres et al., 1998b). We also sought to determine the cell cycle event during which neuronal death is triggered to gain an understanding of the link between cell cycle regulation and delayed neuronal death. We found that loss of endogenous Cdk inhibitors is a likely trigger for reentry of postmitotic neurons into the cell cycle. Neurons that survive the ischemia do not show any loss of Cdk inhibitors.
MATERIALS AND METHODS
Animal experiments. All experimental procedures that were performed on laboratory animals conformed to institutional guidelines for the care and use of laboratory animals. 129/SvEvTacBr wild-type mice (18–22 gm; Taconic Farms, Germantown, NY) were administered 1 mg · hr−1 · kg−1bromodeoxyuridine (BrdU; Sigma, St. Louis, MO) via subcutaneously implanted osmotic mini-pumps (flow rate 1 μl/hr; Alzet, Cupertino, CA). This dose is nontoxic (Gould et al., 1997; Liu et al., 1998;Kempermann et al., 1998). The administration of BrdU via osmotic mini-pumps was at least as effective as a more established method of daily intraperitoneal injections (50 mg/kg body weight) as determined by comparing the number of BrdU-positive cells using the two protocols (data not shown).
Ischemia model. Mice were anesthetized for induction with 1.5% halothane and maintained in 1.0% halothane in 70% N2O and 30% O2 using a vaporizer. Cerebral ischemia was induced with a 8.0 nylon monofilament coated with a silicone resin/hardener mixture (Xantopren M Mucosa and Activator NF Optosil Xantopren; Haereus Kulzer) as described previously (Endres et al., 1998a,b, 1999, 2000). The filament was introduced into the left internal carotid artery up to the anterior cerebral artery. Thereby the middle cerebral artery and anterior choroidal arteries were occluded. Filaments were withdrawn after 30 min of ischemia to allow reperfusion. Regional cerebral blood flow measured using laser-Doppler-flowmetry (Perimed, Jarfälla, Sweden) fell to <20% during ischemia and returned to ∼100% within 5 min after reperfusion in either group (p > 0.05). Core temperature during the experiment was maintained at 36.5°C ± 0.5°C with a feedback temperature control unit.
Primary neuronal cell culture. Primary neuronal cultures of cerebral cortex were obtained from E17 Wistar rats (Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin, Berlin, Germany). Cultures were prepared according to Brewer (1995) with some modifications as described previously (Bruer et al., 1997; Lautenschlager et al., 2000). Whole cerebral cortices were dissected, incubated for 15 min in trypsin/EDTA (0.05/0.02% w/v in PBS) at 36.5°C, rinsed twice with PBS and once with dissociation medium (modified Eagle's medium with 10% fetal calf serum, 10 mm HEPES, 44 mmglucose, 100 U penicillin + streptomycin/ml, 2 mml-glutamine, 100 IE insulin/l), dissociated by Pasteur pipette in dissociation medium, pelleted by centrifugation (at 210 × g for 2 min at 21°C), redissociated in starter medium (neurobasal medium with supplement B27, 100 U penicillin + streptomycin/ml, 0.5 mml-glutamine, 25 μmglutamate), and plated in 24-well plates at a density of 200,000 cells/cm2. Wells were pretreated by incubation with poly-l-lysine (0.5% w/v in PBS) at room temperature for 1 hr, then rinsed with PBS, followed by incubation with coating medium (dissociation medium with 0.03‰ w/v collagen G) for 1 hr at 37°C, then rinsed twice with PBS, before cells were seeded in starter medium. Cultures were kept at 36.5°C and 5% CO2 and were fed with cultivating medium (starter medium without glutamate) by replacing half of the medium twice a week beginning from the fourth day in vitro (DIV). By choosing a serum-free culture condition, we were able to maintain cultures with a very low percentage of glia (Lautenschlager et al., 2000). Neurobasal medium and supplement B27 were obtained from Life Technologies (Eggenstein, Germany); modified Eagle's medium, PBS, HEPES buffer trypsin/EDTA, penicillin/streptomycin,l-glutamine, collagen G, and poly-l-lysine were from Biochrom (Berlin, Germany), and multi-well plates were from Falcon (Franklin Lakes, NJ).
Oxygen–glucose deprivation. Serum-free primary neuronal cultures were treated after 10–14 DIV. The condition of cells at various time points after treatment was determined morphologically by phase-contrast microscopy. Before oxygen–glucose deprivation (OGD), the medium was removed from the cultures and preserved. Cultures were rinsed twice with PBS, then subjected to OGD for 90 min in a balanced salt solution at PO2 < 2 mmHg, followed by replacement of the preserved medium as described previously (Bruer et al., 1997; Harms et al., 2000). For control conditions, cells were placed in a balanced salt solution with 20 mmd-glucose for 120 min in normoxic atmosphere with 5% CO2. Neuronal injury was quantitatively assessed by the measurement of lactate dehydrogenase (LDH) at various time points in the medium (Koh and Choi, 1987). The enzyme standard for kinetic LDH test was obtained from Sigma Chemie GmbH (Deisenhofen, Germany). Representative photographs were taken with phase-contrast microscopy 24 hr after OGD.
Olomoucine treatment protocol. The Cdk inhibitor olomoucine (Alexis, Grünberg, Germany), dissolved in DMSO (50 mm), was used at final concentrations of 1, 10, and 100 μm in the medium. Olomoucine was applied to cortical cell cultures 1 hr before and during OGD. The vehicle-treated cultures received 0.2% DMSO.
Tissue preparation and immunocytochemistry. In the in vivo experiments, the brains were perfusion fixed in 4% paraformaldehyde in 0.1 m PBS, pH 7.4, and post-fixed in the same fixative overnight at 4°C. Coronal 40 μm sections were cut on a Vibratome (Technical Products, St. Louis, MO). The sections were incubated in a blocking solution containing 10% normal goat serum and 0.3% Triton X-100 for 30 min followed by the primary antibodies [rabbit polyclonal anti-p16INK4a, anti-p27Kip1, anti-cyclin D1 (Santa Cruz, Heidelberg, Germany); 1:250] overnight at 4°C. After three washes with PBS, the sections were incubated in biotinylated secondary antibody (goat anti-rabbit, 1:250; Vector Laboratories, Burlingame, CA) for 90 min at room temperature and developed with Texas Red-labeled streptavidin (1:200; Molecular Probes, Leiden, Holland). Sections were washed in PBS and processed for double labeling with neuronal markers as follows. The sections were incubated with antibodies against neuronal markers, mouse monoclonal anti-MAP-2 (1:2000; Roche, Grenzach-Wyhlen, Germany) or mouse monoclonal anti-NeuN (1:100; Chemicon, Hofheim, Germany) overnight at room temperature. This was followed by incubation in Alexa 488-conjugated goat anti-mouse IgG (1:250; Molecular Probes) for 90 min at room temperature.
For terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) histochemistry, unfixed brains were snap-frozen in isopentane at −40°C. Coronal 10 μm sections were cut on a cryostat (Microm, Heidelberg, Germany), thaw-mounted on glass slides, and stored at −20°C. TUNEL was performed using a fluorescence ApopTag Kit (Biogen, Heidelberg, Germany) according to the manufacturer's instructions. For double labeling with TUNEL and cell-type specific markers, sections were first incubated in blocking solution containing 10% normal serum and 0.1% Triton in PBS and then incubated overnight at 4°C with anti-NeuN (mouse monoclonal, 1:100; Chemicon), anti-GFAP (astroglial marker, rabbit polyclonal, 1:500; Dako, Hamburg, Germany), or anti-MAC-1 (microglial marker, rat monoclonal, 1:1000; Serotec, Oxford, UK) antibodies, followed by incubation with corresponding biotinylated secondary antibodies, and finally with Texas Red-labeled streptavidin (Molecular Probes; 1:250). The sections were then thoroughly rinsed in PBS and processed for TUNEL staining.
For BrdU and TUNEL double labeling, the sections were first processed for TUNEL staining. Then, the DNA was hydrolyzed by 2N HCl into single strands, and the sections were incubated with rat monoclonal anti-BrdU antibody (1:500; Harlan, Borchen, Germany) overnight at 4°C and reacted with biotinylated rabbit anti-rat IgG followed by Texas Red-labeled streptavidin. For immunocytochemical analysis of cell cultures, cells were seeded onto glass coverslips, fixed with freshly prepared 4% paraformaldehyde in PBS for 15 min, permeabilized with 0.3% Triton X-100 in PBS, and exposed to blocking solution (PBS containing 10% goat serum and 1% bovine serum albumin) for 30 min at room temperature. Cultures then were incubated with the rabbit polyclonal antibodies to p16INK4a, p27Kip1, or cyclin D1 (1:100) for 1 hr at room temperature and developed with Texas Red-labeled goat anti-rabbit IgG (1:500) for 30 min at room temperature. After rinsing with PBS, the glass coverslips were incubated with anti-NeuN (1:100) for 1 hr at room temperature, followed by incubation in Alexa 488-conjugated goat anti-mouse (1:500). For control studies, sections were treated the same way except that TdT (for TUNEL studies) or primary antibodies (for immunoreactivity studies) were omitted, resulting in no visible staining.
Confocal laser scan microscopy. A Nikon Optiphot/Bio-Rad MRC 600 (Hempstead, UK) confocal laser scanning microscope equipped with an argon/krypton laser was used. The images were acquired using CoMOS software program (Version 7.0a, Bio-Rad) and imported into Adobe Photoshop 5.0 (Adobe Systems, Mountain View, CA).
Cell counting protocols. To estimate the number of p16INK4a-negative MAP-2-positive neurons, four sections 80 μm apart at the level of anterior commissure were chosen from each animal. Seven randomly chosen nonoverlapping high-power fields (400×) from the striatum were examined from each section (n = 3 for each reperfusion time points at 9, 18, 48, and 72 hr as well as for sham-operated controls). All MAP-2-positive neurons in each high-power field were counted, and the number of MAP-2 positive that were p16INK4 negative was recorded. TUNEL-positive cells were counted in a single section at the same level in seven randomly chosen, nonoverlapping high-power fields (n = 3 for each time point). The cells were classified as TUNEL positive only when they showed strong nuclear signal with condensed nuclei with clumped chromatin without cytoplasmic staining (see Fig. 1B, arrowheads). Colocalization of TUNEL with BrdU was examined 72 hr after 30 min middle cerebral artery occlusion (MCAo)/reperfusion (n = 4) in three randomly chosen sections at the level of anterior commissure, whereby 12 high-power fields within each ischemic striatum were examined. The proportion of TUNEL and BrdU double-labeled cells was calculated.
Fig. 1.
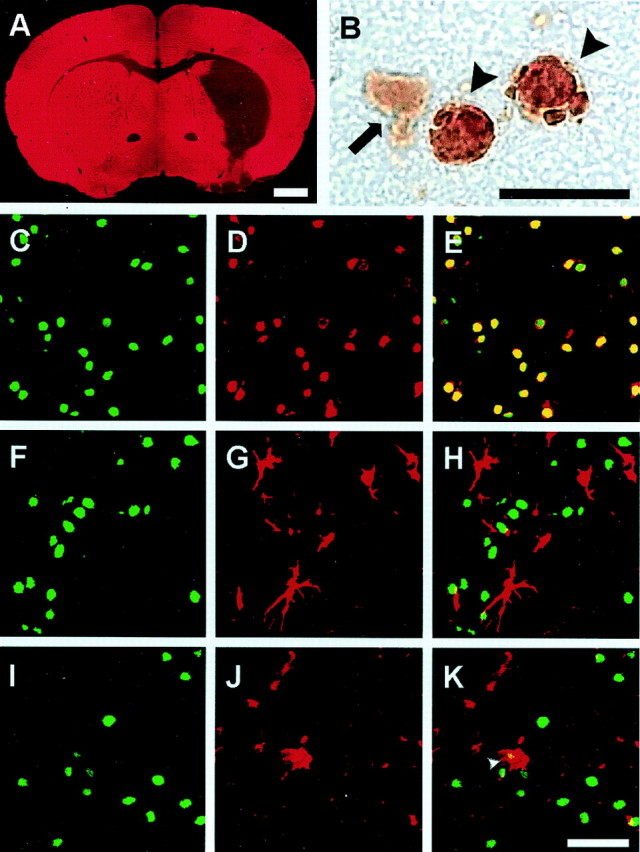
Selective neuronal death in the mouse striatum 72 hr after an episode of 30 min MCAo and reperfusion. The low-power view of MAP-2 immunostaining indicates striatal lesion and cortical sparing (A). At higher magnification (B), TUNEL-positive cells (arrowheads) show in DAB staining condensed nuclei with clumped chromatin. Cells showing weak diffuse DAB-positive cytoplasmic staining are not considered TUNEL positive (arrow). Sections were double stained for TUNEL (C,F, I) and cell type-specific markers NeuN (D), GFAP (G), or MAC-1 (J) and examined in a confocal microscope. Cell-specific labeling was visualized using antibodies conjugated with fluorescein (C, F,I: green) or Texas Red (D,G, J: red). No double labeling was detected for TUNEL and the astrocytic marker, GFAP (H). Moreover, there was no double labeling for TUNEL and the microglial marker, MAC-1 (K), with the exception of some cells that most likely represent engulfed nuclei of dead neurons (K,arrowhead). By contrast, most of the TUNEL-positive cells were also immunoreactive for the neuronal marker NeuN (E), indicating neuronal origin of the TUNEL-positive cells. Scale bars: A, 1 mm;B, 10 μm; C–K, 30 μm.
Electron microscopy. The mice were transcardially perfused with 0.1% glutaraldehyde/2% paraformaldehyde. After overnight post-fixation, 60-μm-thick Vibratome sections were processed for BrdU immunohistochemistry using ABC kit (Vector) and DAB (Sigma) as a chromagen. The sections were post-fixed in 2.5% glutaraldehyde in PBS and 1% osmium tetroxide, rinsed in 0.1 m sodium acetate, and stained with 2% uranyl acetate for 1 hr. The sections were dehydrated in ethanol and embedded in resin. Ultrathin sections were examined in a Zeiss EM10 electron microscope.
Immunoblots. Proteins were denatured by boiling in 30 μl sample buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1% w/v DTT, 2% w/v SDS, and 0.01% w/v bromophenol blue) for 3 min. Samples (30 μl) were subjected to 10% SDS-PAGE, and the dried gels were subjected to autoradiography. Cells were lysed in 130 μl RIPA buffer (10 mmNa2HPO4, pH 7.0, 300 mm NaCl, 0.1% w/v SDS, 1% v/v NP40, 1% w/v Na-deoxycholate, 2 mm EDTA, 1 mm DTT, and protease/phosphatase inhibitors as described) for 30 min on ice. Proteins were denatured by boiling in 30 μl sample buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1% w/v DTT, 2% w/v SDS, and 0.01% w/v bromophenol blue) for 3 min. Samples (15 μl) were electrophoretically separated, transferred to polyvinylidene difluoride membranes and blocked, and primary antibodies (0.2–1.0 μg/ml) were incubated overnight at 4°C on a rotary platform with gentle agitation. They were subsequently probed with secondary HRP-conjugated anti-mouse or anti-rabbit IgG antibodies (diluted 1:5000; Amersham Pharmacia Biotech, Braunschweig, Germany). Equal loading was confirmed by resolving 20 μg total protein by SDS-PAGE and probing with anti-actin antibody (1:2000). Detection was performed using the enhanced chemiluminescence assay (Amersham). To provide semiquantitative analysis of band intensity, band densitometry was determined from scanned images of nonsaturated immunoblot films, using Scion Image, version Beta 4.0.2 software (Scion Corporation, Frederick, MD). To compare at least three different experiments, for each protein and brain region, pixel intensities of the bands obtained in each experiment were added and set as 100%. The individual band was calculated as percentage of total signals.
Histone kinase assays. Anti-Cdk2 immunocomplexes were washed twice in lysis buffer and once with ice-cold kinase buffer (50 mm Tris-HCl, 10 mmMgCl2, 1.0 mm DTT) and resuspended in 50 μl kinase buffer supplemented with 10 μg lysine-rich histone HIIIS (Sigma), 10 μCi [γ-32P]ATP (111 MBq/mmol; NEN, Boston, MA). Control reactions were run in parallel with the omission of antibody. After incubation for 60 min at 37OC with continuous agitation, the reaction was terminated by addition of 25 μl SDS sample buffer. Boiled samples (30 μl) were separated by 15% SDS-PAGE and Coomassie stained for evaluation of equal protein loading, and the amount of incorporated radioactive label was quantified using a phosphorimager and TINA software (Raytest).
Statistical evaluation. Data are shown as mean ± SEM. To avoid possible variations of the cell cultures depending on the quality of dissection and seeding procedures, data were pooled from at least three representative experiments. For statistical analyses, one-way ANOVA was followed by Tukey's post hoc test.p < 0.05 was considered statistically significant.
RESULTS
Mild ischemia leads to delayed neuronal death
After 30 min MCA occlusion and reperfusion in 129/SV mice, cell death was prominent in the striatum and spared the cortex (Fig.1A). TUNEL staining first appeared in the striatum at 18 hr, increased by 36 hr, and peaked by 72 hr (Fig. 1B). When double labeling for TUNEL and cell-type specific markers was performed at the 72 hr time point, almost 100% of the TUNEL-positive cells were also NeuN positive (Fig.1C–E). However, not any of the TUNEL-positive cells were GFAP positive (Fig.1F–H). Also, there was no MAC-1/TUNEL double labeling detected, with the exception of some cells that most likely represent engulfed nuclei of dead neurons (Fig.1I–K). Interestingly, TUNEL and MAP-2 double labeling was not detected at 72 hr. MAP-2, a neuron-specific cytoskeletal marker, was profoundly downregulated in the ischemic region at the 72 hr time point (Fig.1A); it is known to be extremely sensitive to ischemia (Li et al., 1998; Endres et al., 1999). MAP-2-positive cells were TUNEL negative and morphologically intact. These cells most likely represent neurons that survive the ischemic insult. We have reported previously that only ∼15% of the neurons, identified as such by morphological criteria, survived at 72 hr, and this percentage did not change significantly at 21 d (Endres et al., 1998b, 2000; Fink et al., 1998).
Loss of p16INK4a and p27Kip1 after ischemia/reperfusion
We analyzed cellular localization of p16INK4a and p27Kip1, endogenous Cdk inhibitors, in the striata of normal mice in vivo. We detected strong immunoreactivity for the Cdk4 inhibitor p16INK4a, a member of the INK4 family, in striatal neurons. In fact, p16INK4a/NeuN double-labeling experiments confirmed that all striatal neuronal nuclei expressed p16INK4a (Fig.2A–C). No staining was obtained with antibodies against p27Kip1, a member of the CIP/KIP family that inhibits a wide range of Cdks (data not shown). By contrast, cortical neurons in culture demonstrated nuclear staining for both p16INK4a and p27Kip1. p27Kip1/NeuN double labeling confirmed strong nuclear expression of p27Kip1 in cultured neurons (Fig. 2J–L), whereas p16INK4a immunoreactivity was weaker (data not shown). Expression of p16INK4a and p27Kip1 protein was confirmed by immunoblot analysis.
Fig. 2.
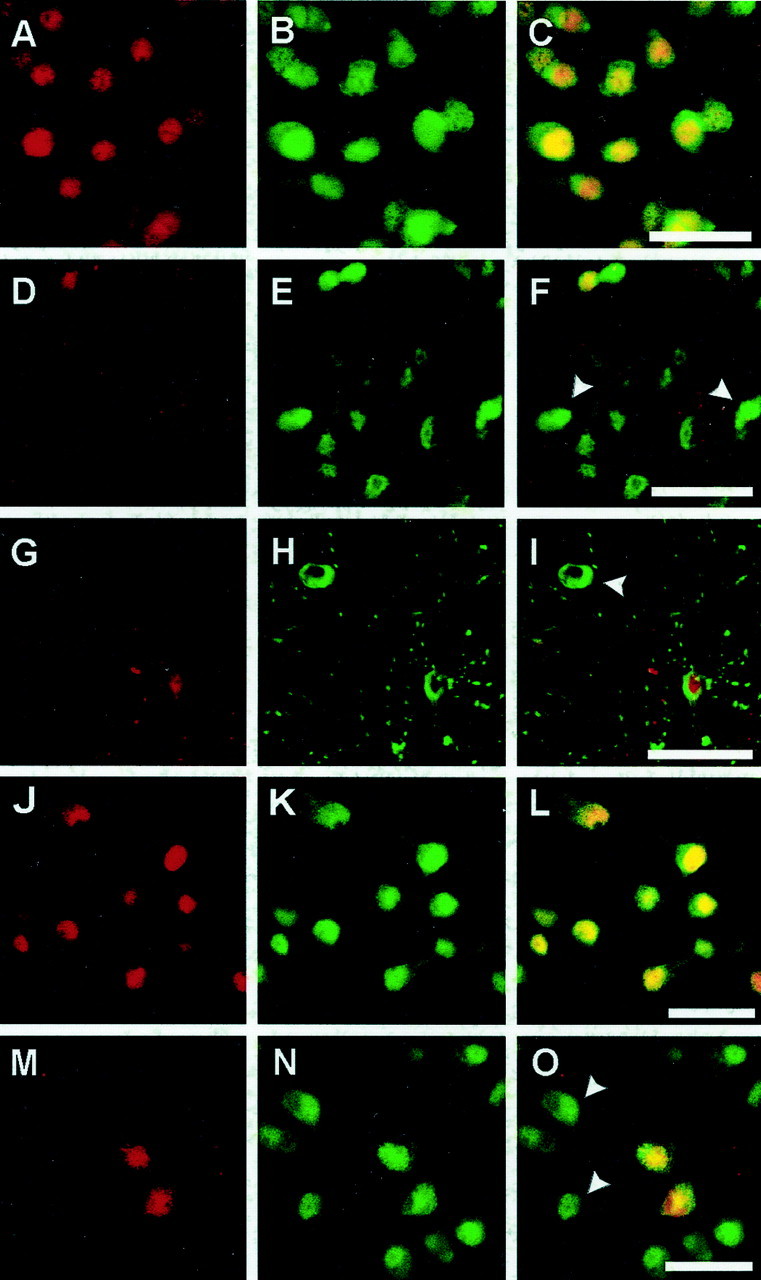
Expression of p16INK4a and p27Kip1, endogenous inhibitors of cyclin-dependent kinases, after 30 min MCAo and reperfusion in the mouse striatum (A–I, p16INK4a) and after OGD in rat primary cortical neurons (J–O, p27Kip1). Immunoreactivity for p16INK4a/p27Kip1was visualized with Texas Red (A, D,G, J, M:red), and neuronal marker NeuN was visualized with Alexa 488 (B, E, H,K, N: green), with colocalization resulting in a yellow color (C, F, I,L, O). Strong nuclear expression of p16INK4a was seen in all neurons in the normal (non-ischemic) striatum as shown by double labeling with p16INK4a and NeuN (A–C). p16INK4aimmunoreactivity was lost in ischemic striatal neurons at 9 hr after MCAo/reperfusion as shown by the appearance of NeuN-positive p16-negative cells (D–F,arrowheads). Double labeling of p16INK4a with the ischemia-sensitive neuronal marker MAP-2 demonstrated that the loss of p16INK4aexpression occurred in cytoarchitectonically intact neurons at 9 hr (G–I; arrowhead inI). Strong nuclear p27Kip1immunoreactivity was detected in all neurons in primary neuronal culture (J–L). Two hours after OGD the majority of neurons downregulated p27 Kip1, as indicated byarrowheads (M–O). Scale bars, 30 μm.
After cerebral ischemia there was a profound and early downregulation of endogenous Cdk inhibitors. As early as 9 hr after 30 min MCAo/reperfusion, p16INK4a was downregulated in the ischemic striatum (Fig.2D–F). This early downregulation did not simply reflect cell death or cytoskeletal disintegration because p16INK4a-negative neurons were strongly MAP-2 positive and intact by morphological criteria at 9 and 18 hr (Fig. 2G–I). We analyzed the temporal and spatial relationship between p16INK4a downregulation, MAP-2 staining, and markers of cell death. At 9 hr, when no TUNEL-positive cells were detected in the ischemic striatum, 48.0 ± 8.3% of all MAP-2-positive cells were p16INK4anegative. Thus, some MAP-2-expressing, apparently intact neurons downregulated p16INK4a well before TUNEL labeling became apparent. Concomitant with the increase of TUNEL-positive cells, the amount of p16-negative and morphologically intact neurons decreased (23.3 ± 1.8% at 18 hr vs 16.7 ± 8.3% at 48 hr) (Fig. 3). At 72 hr all remaining morphologically intact MAP-2-positive neurons were p16INK4a positive. Moreover, TUNEL and p16INK4a double-labeled cells were not detected at any time point. Thus, p16INK4adownregulation preceded MAP-2 downregulation and neuronal death.
Fig. 3.

Time course of p16INK4adownregulation and delayed neuronal death after 30 min MCAo.A, The number of p16INK4a-negative and MAP-2 positive cells is presented as a percentage of all MAP-2-positive cells at 9, 18, 48, and 72 hr after MCAo as well as in sham-operated, control mice. In controls and at 72 hr after MCAo, all MAP-2-positive cells were also p16INK4apositive. B, The number of TUNEL-positive cells per square millimeters was determined at the same time points as inA. No TUNEL positivity was observed in sham-operated animals and at 9 hr after MCAo, whereas the maximum number of TUNEL-positive cells was observed at 72 hr after MCAo;n = 3–6. Data are mean values ± SEM. *p < 0.05; ***p < 0.001.
Similar to the early downregulation of p16INK4ain vivo, p16INK4a and p27Kip1 were downregulated in cultured cortical neurons as early as 2 hr after OGD, as shown by immunohistochemistry (Fig. 2M–O) and immunoblots (p16INK4a: 44.9 ± 9.9% decrease at 4 hr, p < 0.001; p27Kip1: 34.6 ± 9.0 and 57.2 ± 6.5% decrease at 2 and 4 hr, p < 0.05 andp < 0.001, respectively) (Fig.4).
Fig. 4.
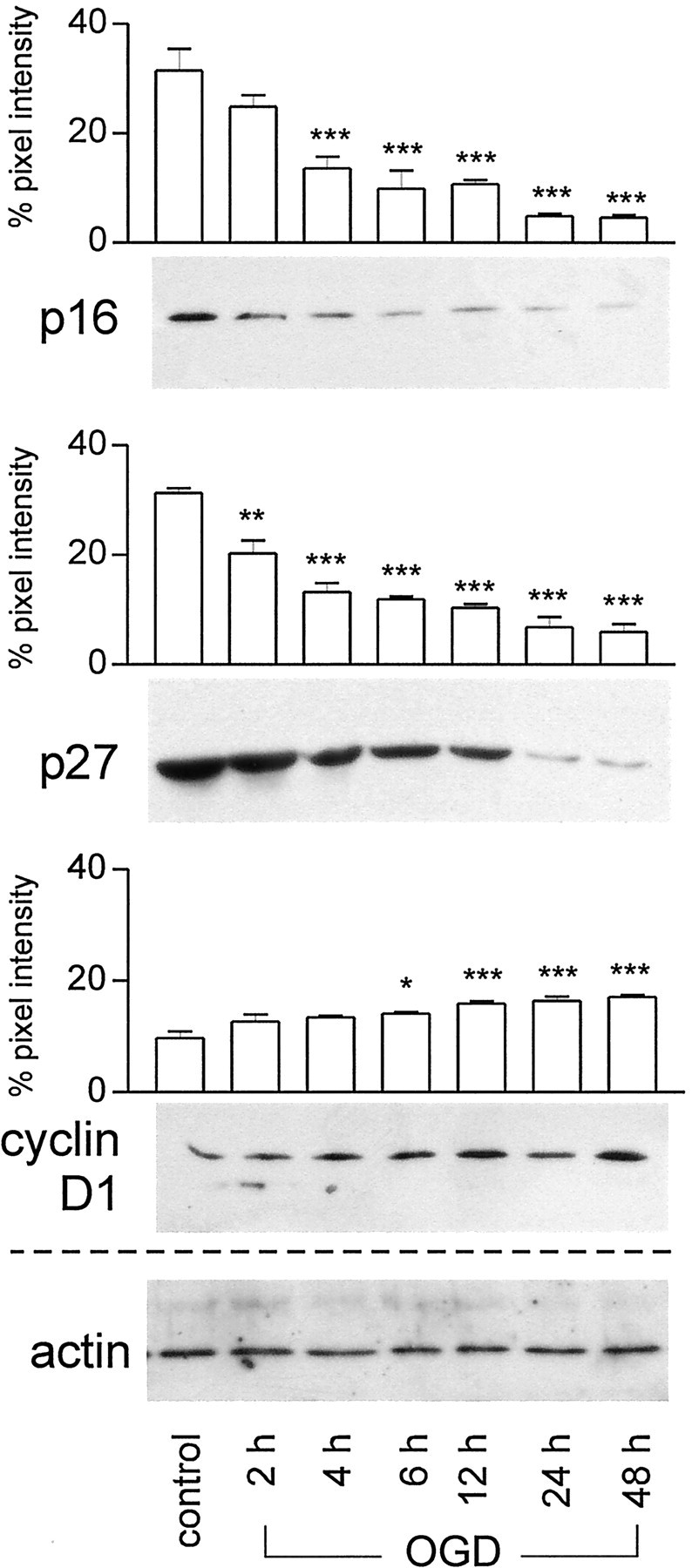
Immunoblots showing time-dependent changes of cell cycle-related proteins in primary cortical neurons after 90 min oxygen–glucose deprivation. Cell lysates (20 μg) were subjected to SDS-PAGE, and membranes were probed with antibodies against p16INK4a, p27Kip1, and cyclin D1 (0.2–1.0 μg/ml). Actin served as internal control. The experiment was repeated three times; a representative experiment is shown. For semiquantitative analysis, the intensity of each band was quantitated from scanned images of nonsaturated immunoblot films using Scion Image (Scion Corporation, Frederick, MD). The pixel intensity of the bands obtained in each experiment was summed and set as 100%, and the individual band was calculated as percentage of total signals. The graphs show a significant downregulation of p27Kip1starting at 2 hr and of p16INK4a at 4 hr after OGD compared with controls. Cyclin D1 levels were significantly upregulated at 6 hr and further increased at 48 hr. Mean value ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
When our in vivo and in vitro data are taken together, endogenous Cdk inhibitors such as p16INK4a or p27Kip1 appear to be reliable markers of neuronal survival and their loss a reliable indicator of neuronal death after cerebral ischemia.
Cell cycle protein expression after ischemia/hypoxia
Next, we investigated the expression of the G1 phase cyclin, cyclin D1, in striatal neuronsin vivo after focal ischemia and in neocortical neuronsin vitro after OGD. We detected no cyclin D1 immunoreactivity in striatal neurons from sham-operated controls (data not shown), whereas cortical neurons in control cultures expressed cyclin D1 in the cytosol (Fig.5A–C). Cyclin D1 is thought to be inactive in the cytosol and requires nuclear translocation to activate Cdks (Yang and Kornbluth, 1999). Indeed, after cerebral ischemia in vivo and OGD in vitro, we detected nuclear expression and an increase in immunoreactivity of cyclin D1 (Fig. 5D–I). Z-series confocal images confirmed nuclear expression of cyclin D1 (Fig.5J). Immunoblot analyses confirmed the increase in cyclin D1 in vitro detected by immunohistochemistry (150.8 ± 21.0% at 6 hr, p < 0.05) (Fig. 4). As expected, some glial cells exhibited strong cyclin D1 immunoreactivityin vivo.
Fig. 5.
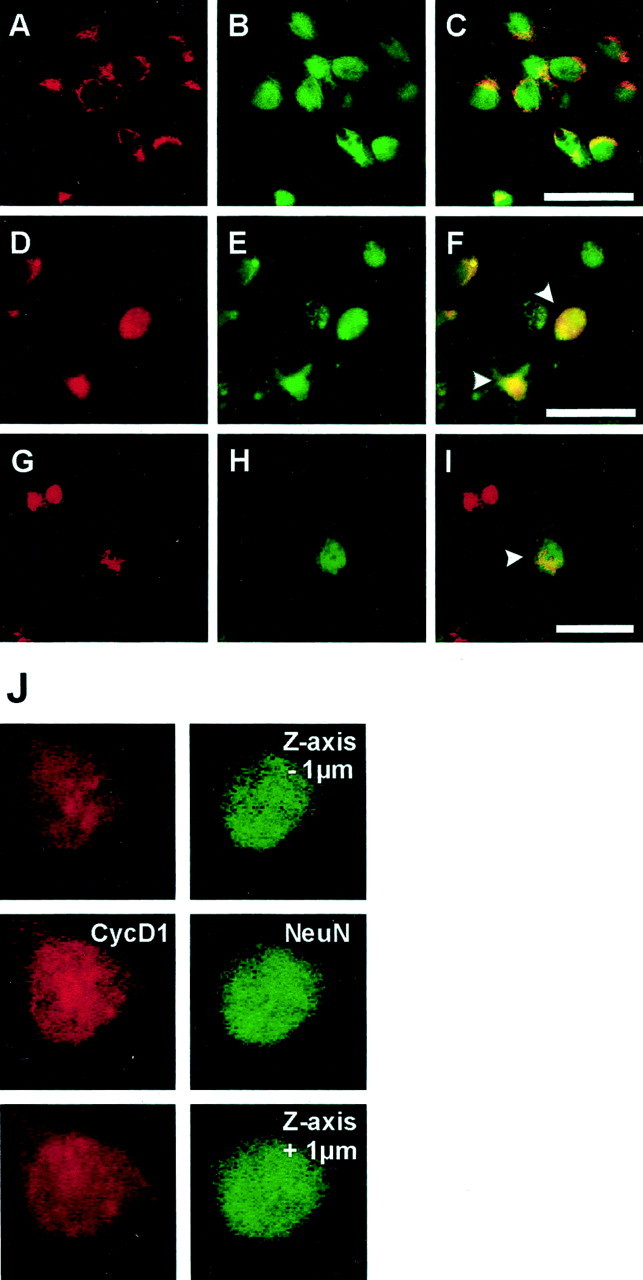
Cyclin D1 expression in primary cortical neurons in culture after OGD (A–F) and in striatal neurons of mice after MCAo/reperfusion (G–I). Immunoreactivity for cyclin D1 was visualized with Texas Red (A,D, G; red), and neuronal marker NeuN was visualized with Alexa 488 (B,E, H; green). Double labeling for cyclin D1 and NeuN demonstrates that cyclin D1 is expressed exclusively in the cytoplasm of cultured neurons under control conditions (A–C). Two hours after OGD cyclin D1 is strongly upregulated, and its immunoreactivity translocates to the nucleus (D–F,arrowhead). Confocal Z-series images (step = 1 μm) confirm the nuclear expression of cyclin D1 in OGD-treated neurons (J). Cyclin D1 is not expressed in normal striatal neurons (data not shown). Forty-eight hours after MCAo/reperfusion, nuclear cyclin D1 immunoreactivity is detected in neurons in the ischemic striatum (G–I,arrowhead). Scale bars:A–I, 30 μm.
Cyclin D1 activates Cdk4, which in turn phosphorylates the retinoblastoma protein in mid G1 phase, followed by the activation of Cdk2 around the G1–S transition. We performed histone kinase assays to determine Cdk2 activity in cultured cortical neurons after OGD to determine whether the molecular machinery necessary for G1–S transition was being mobilized (Fig. 6). Cdk2 activity increased at 12 hr (658.3 ± 4.3% increase,p < 0.05) and peaked at 24 hr (1171.0 ± 137.5% increase, p < 0.01). When cells were pretreated with olomoucine (1–100 μm), a synthetic Cdk inhibitor, Cdk activation at 12 and 24 hr was completely abolished (Fig. 6). Hence, we demonstrate that Cdks are activated in neurons after ischemia/hypoxia, and olomoucine administration effectively inhibits Cdk activation in this model. Immunoblot experiments showed that protein levels of Cdk4 and Cdk2 did not change significantly over time (data not shown).
Fig. 6.

Cdk2 activity increases in cortical neurons in culture after 90 min OGD. Cell lysates were immunoprecipitated with anti-Cdk2 antibody, and the resultant complexes were allowed to incubate with [γ-32P]ATP and histone HIIIS as substrate. In each lane, 30 μg protein was separated. There was a clear increase in kinase activity at 12 and 24 hr after OGD as compared with control. This kinase activation at 12 and 24 hr was completely abolished when the cells were pretreated with 10 μmolomoucine (olo) 1 hr before and during OGD. To estimate Cdk2 activity, the amount of incorporated radioactive label was quantified using a phosphorimager and TINA software. The values of three independent experiments are graphically represented as mean value ± SEM. *p < 0.01; **p < 0.001 versus control.
Inhibition of Cdk activity protects from oxygen–glucose deprivation
Olomoucine is a purine derivative that inhibits Cdks 1, 2, and 5 and ERK1/MAP-kinase and blocks G1–S-phase transition (Vesely et al., 1994;Abraham et al., 1995). Cultured cortical neurons were pretreated with olomoucine (1, 10, or 100 μm) before OGD. Cell damage after OGD was determined by quantifying LDH release into the culture medium. Olomoucine pretreatment significantly protected neurons from OGD and reduced LDH release at 24 hr (Fig.7A). The strongest protection was achieved with 10 μm concentration (∼75% reduction in LDH release; 41.5 ± 7.6 vs 9.2 ± 2.6 U/ml medium; n = 24 in three independent experiments;p = 0.004). The decrease in LDH release (Fig.7A) correlated with higher numbers of viable neurons at 72 hr as assessed by phase-contrast microscopy (Fig.7B–D). Thus, inhibition of Cdk activity protects cultured cortical neurons against OGD.
Fig. 7.

The Cdk inhibitor olomoucine protects primary neuronal cultures subjected to OGD. A, Quantitative assessment of neuronal injury by measurement of LDH release 24 hr after OGD presented as the difference between control cultures and cultures subjected to OGD. Olomoucine treatment produced significant protection at 10 and 100 μm, whereas higher concentrations were toxic to the cultures (data not shown). The results are presented as the mean value ± SEM from three independent experiments performed in triplicate. *p < 0.05 and **p < 0.01 versus vehicle-pretreated sister cultures exposed to OGD; one-way ANOVA followed by Tukey's post hoc test. B, Phase-contrast micrograph of primary cortical neurons in culture. C, Twenty-four hours after 90 min OGD and pretreatment with vehicle (0.02% DMSO).D, Cultures exposed to the same insult as inC but pretreated with olomoucine (10 μm). Scale bar, 70 μm.
Progression to S-phase is a rare event
We used BrdU as a marker for DNA synthesis (Nowakowski et al., 1989) to determine whether downregulation of Cdk inhibitors and upregulation of cyclin D1 caused striatal neurons to enter S-phase after ischemic damage in vivo. After 30 min MCAo and reperfusion, BrdU-positive cells first appeared in the ischemic striatum at 36 hr. Their numbers increased at 48 and 72 hr. Most of the BrdU-labeled cells had the morphological appearance of microglia/macrophages and could be labeled with antibodies against MAC-1. A small number of NeuN-labeled cells were BrdU positive at 72 hr (<1%; data not shown). In fact, electron microscopy confirmed that some BrdU-positive cells were indeed neurons (Fig.8A). A small fraction of TUNEL-positive cells was BrdU positive (0.957 ± 0.172% of all TUNEL-positive cells; 72 hr; n = 4 animals), indicating that some of the TUNEL-positive cells had entered S-phase (Fig.8B–G). As demonstrated in Figure 1, virtually all TUNEL-positive cells are NeuN positive at 72 hr. Together, these data suggest that some neurons indeed entered S-phase before their death.
Fig. 8.
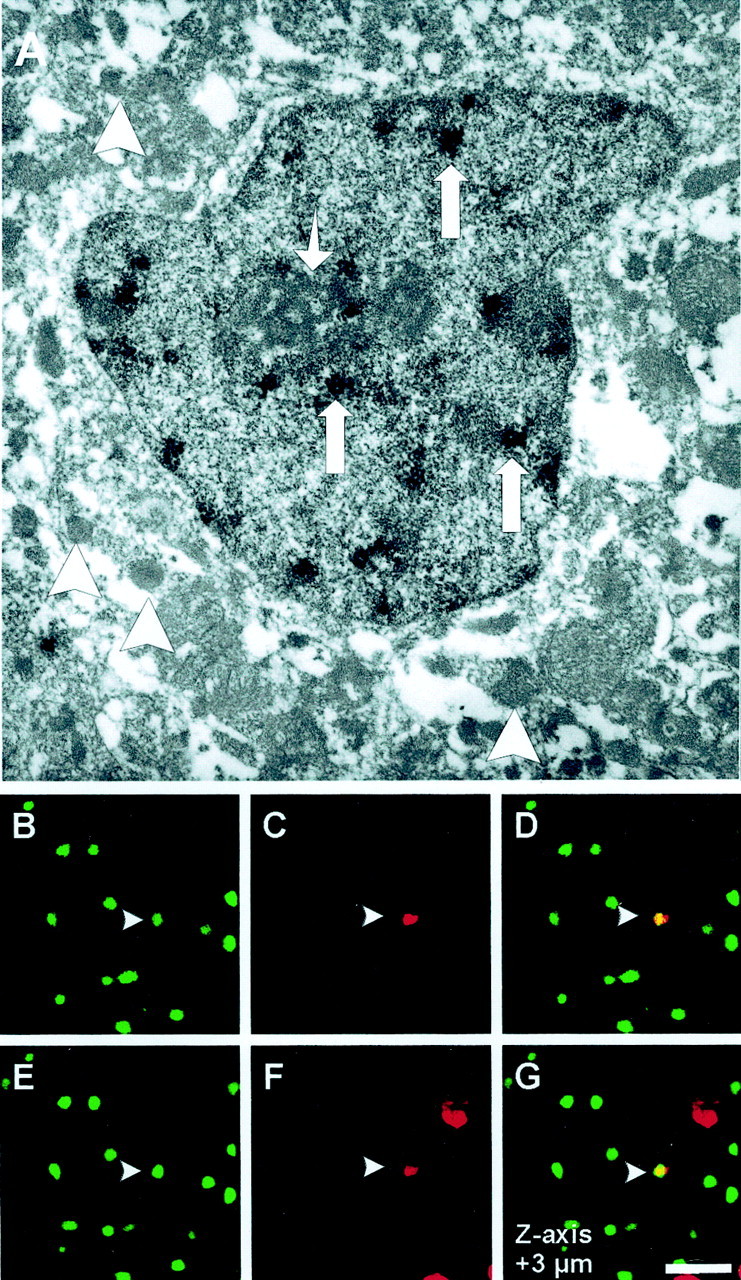
TUNEL/BrdU double labeling. BrdU was administered via subcutaneous osmotic mini-pumps in 129/SV mice (1 mg · hr−1 · kg−1 body weight). Mice were subjected to 30 min of MCAo and 72 hr reperfusion.A, For electron microscopy studies, Vibratome sections of ischemic striatum were immunostained for BrdU using DAB as a chromagen. Electron micrograph shows a cell with numerous electron-dense osmiophilic granules (large arrows) within the nucleus corresponding to the BrdU labeling. The cell has a central nucleus with a prominent nucleolus (small arrow) and multiple vesicles (arrowheads) in its cytoplasm and lacks glial filaments, indicating that it is of neuronal origin. Original magnification: 11,000×. TUNEL/BrdU double labeling was performed on fresh-frozen cryosections (10 μm) using ApopTag Kit and rat monoclonal anti-BrdU antibody. TUNEL was visualized with fluorescein (B, E: green), and Texas Red was used for BrdU immunoreactivity (C,F: red). Of all TUNEL-positive cells, 0.957 ± 0.172% stain positive for the S-phase marker BrdU (n = 4 animals). Z-series of confocal images through the nucleus (1 μm steps) confirms the costaining for both markers (D, G: yellow). Scale bar, 30 μm.
We were not able to detect any mitotic figure, however, either in vivo or in vitro, although the presence of mitotic figures has been reported after a hypoxic insult in vitro(Bossenmeyer-Puorié et al., 1999). It appears that although some neurons enter S-phase, they do not proceed beyond the G2–M checkpoint within 72 hr after the insult.
DISCUSSION
We provide evidence that cell cycle protein expression is altered in postmitotic neurons in response to focal cerebral ischemia. The earliest event is the downregulation of the Cdk inhibitors p16INK4a and p27Kip1 followed by upregulation and nuclear expression of cyclin D1 and activation of Cdks. We consider these events as “attempts” at cell cycle reentry. Because none of the neurons enters M-phase within the 72 hr period, we conclude that these attempts fail. These findings show that neurons attempt cell cycle reentry after cerebral ischemia, most likely because of loss of endogenous Cdk inhibitors, and that the loss of Cdk inhibitors is a preamble for neuronal death rather than for cell division or survival, or both.
Loss of endogenous cell cycle inhibitors may be an early trigger for cell cycle activation
P16INK4a and p27Kip1 induce cell cycle arrest by inhibiting Cdk activity (Johnson and Walker, 1999; Vidal and Koff, 2000), and they promote cell cycle exit during development (Zindy et al., 1997; Watanabe et al., 1998). We show that virtually all neurons in the striatum express p16INK4a and that all postmitotic neurons in cortical cultures express p27Kip1 in the nucleus. Nuclear expression of p16INK4ain vivo and p27Kip1in vitro, but not vice versa, may relate to differences in cell maturation. The in vivo studies were performed on 6-week-old mice, whereas the neurons in vitro were obtained from embryonic day 17 rats and cultured for up to 14 d. p27Kip1is the predominant Cdk inhibitor and the only Cdk inhibitor to decrease 5 hr after KCl withdrawal in primary cultures of cerebellar granule neurons at 5–6 d in vitro (Padmanabhan et al., 1999). By contrast, increased levels of p16INK4a are associated with cell senescence (Huschtscha and Reddel, 1999), and its expression may relate to a more mature state.
After an ischemic insult, p16INK4a and p27Kip1 were profoundly downregulated. We propose that the downregulation acts as an early trigger of attempted cell cycle reentry and subsequent neuronal death. The loss of p16INK4a is a specific event and not related to general protein degradation, because at early time points, p16INK4a-negative neurons are morphologically intact and express MAP-2. Most neurons downregulate p16INK4a between 9 and 18 hr after ischemia (Figs. 3, 4). Later on, p16INK4a-negative neurons undergo cytoskeletal disintegration and become TUNEL positive, whereas virtually every neuron that maintains high levels of p16INK4a remains viable at least for 72 hr. Hence, p16INK4a may be a survival factor, and its early downregulation may predict neuronal death. Similarly, Sindbis virus-induced expression of p16INK4a/p27Kip1 protected sympathetic and cortical neurons from death induced by DNA-damaging compounds (Park et al., 1998).
Our in vitro evidence showed unequivocally that the loss of p27Kip1 was paralleled by nuclear expression of cyclin D1, closely followed by Cdk2 activation, which is thought to be crucial for G1–S transition (Sherr, 1994). Some ischemic neurons progressed to S-phase in ourin vivo model (see below). These data are in agreement with several studies on brain tumors showing that the loss of endogenous Cdk inhibitors is sufficient to precipitate uncontrolled cell proliferation (Nishikawa et al., 1995; Ueki et al., 1996). Similarly, expression of Cdk inhibitors was investigated in Alzheimer's disease and in support of the hypothesis that an aborted attempt to activate the cell cycle in terminally differentiated neurons might be a critical event in the pathomechanism of Alzheimer's disease (Arendt et al., 1996, 1998;McShea et al., 1997, 1999; Nagy et al., 1997).
Cell cycle protein expression and delayed neuronal death
We demonstrate upregulation and, more importantly, nuclear expression of cyclin D1 in neurons after ischemia/hypoxia, extending previous reports (Freeman et al., 1994; Kranenburg et al., 1996; Li et al., 1997, 1998; McShea et al., 1997; Nagy et al., 1998; Timsit et al., 1999). Nuclear expression of cyclin D1 was followed by Cdk activation. Inhibition of Cdk activation protected neurons from death, a finding that is in accordance with studies that exposed cultured neurons to various insults, including DNA damage, trophic factor deprivation, and β-amyloid toxicity (Park et al., 1997a; Bossenmeyer-Puorié et al., 1999; Stefanis et al., 1999). In preliminary experiments, we also detected Cdk4 expression in striatal neurons in vivo. Although we did not study the effects of olomoucine in vivo, a recent study convincingly demonstrated that Cdk inhibitors protect neurons from death after focal cerebral ischemia (Osuga et al., 2000).
The mechanisms that cause cell death after Cdk activation have remained elusive. They may relate to release of cytochrome c and activation of caspase 9 and eventually caspase-3 (Stefanis et al., 1999). We have demonstrated previously that activation of caspase 3 contributes to cell death after mild ischemia (Endres et al., 1998b;Fink et al., 1998) and OGD (Harms et al., 2000), and the time course of caspase activation is compatible with cell cycle events reported in this study. Another possibility is that Cdk activation might mediate cell death by converting p35 to neurotoxic p25 (Patrick et al., 1999)
S-phase progression after cerebral ischemia
The expression of cell cycle-related proteins after ischemia may signify their function in the apoptotic machinery rather than in the cell cycle (Heintz, 1993; Padmanabhan et al., 1999; Park et al., 1998; Stefanis et al., 1999). To further analyze cell cycle progression, we used BrdU as S-phase marker (Nowakowski et al., 1989;Takahashi et al., 1993; Gage et al., 1995). After ischemia some cells were double labeled with TUNEL and BrdU in our in vivomodel. Because virtually all TUNEL cells were NeuN positive by 72 hr (Fig. 1), we postulate that the TUNEL and BrdU double-labeled cells are neurons. Indeed, we identified BrdU-positive cells as neurons by electron microscopy (Fig. 8A). Similarly, TUNEL/BrdU double labeling was also used in previous studies in the developing nervous system to demonstrate S-phase progression before apoptotic death (Thomaidou et al., 1997; ElShamy et al., 1998). Reentry of neurons into S-phase before cell death was a rare event: ∼1% of the TUNEL-positive cells were BrdU positive. Thus, the majority of neurons died before S-phase after Cdk activation. Accordingly, in preliminary experiments we did not detect any expression of cyclin A and cyclin B in ischemic neurons, which are indicative of S–G2 and G2–M transition, respectively. Another caveat may be that BrdU incorporation indicated DNA repair rather than DNA synthesis. However, a significant number of TUNEL-positive cells (i.e., >99% at 72 hr) are not labeled with BrdU (which would be the case if BrdU were the marker of DNA damage) (Thomaidou et al., 1997). Moreover, other markers of proliferation such as cyclin D1 are expressed. There is good evidence in the literature from both in vitro as well as in vivo experiments that the concentration of BrdU used in this study is not sensitive enough to detect DNA repair (Gobbel et al., 1998;Parent et al., 1999; Palmer et al., 2000). Thus, we show that a small number of neurons enter S-phase before undergoing delayed cell death.
The fact that we were not able to identify mitotic figures argues against the possibility that BrdU labeled-neurons were newly born during the 72 hr period (Gu et al., 2000). Furthermore, it is unlikely that 72 hr would be a sufficient interval for a progenitor cell to give rise to a mature neuron (Kuhn et al., 1996; Palmer et al., 2000). Moreover, we were not able to detect any obvious migration of BrdU-positive cells from the subventricular zone of neuronal progenitor cells to the ischemic striatum by 72 hr.
In conclusion, we show that endogenous Cdk inhibitors are constitutively expressed in quiescent neurons but that they are downregulated early after cerebral ischemia/hypoxia. We show that the loss of Cdk inhibitors may be the trigger for cell destruction. The downstream events leading to delayed neuronal death after Cdk activation remain to be determined.
Footnotes
This research was supported by grants from the Deutsche Forschungsgemeinschaft (En343/4-1 and En 343/6-1) to M.E., by the Hermann and Lilly Schilling Stiftung (U.D.), and by United States Public Health Service Grant NS 32657 (P.G.B.). We thank Michael A. Moskowitz and Verne S. Caviness Jr. for advice, for critical comments, and for providing access to laboratory equipment and facilities. We are also indebted to Gerd Kempermann for critically reading this manuscript, to Felix Engel for advice, and to Hannelore Glatte and Ute Kannbley for assistance with illustrations.
J.K. and C.H. contributed equally to this manuscript.
Correspondence should be addressed to Dr. Matthias Endres, Experimentelle Neurologie, Klinik und Poliklinik für Neurologie der Charité, Humboldt-Universität zu Berlin, Schumannstrasse 20/21, D-10098 Berlin, Germany. E-mail:matthias.endres@charite.de.
REFERENCES
- 1.Abraham RT, Acquarone M, Andersen A, Asensi A, Belle R, Berger F, Bergounioux C, Brunn G, Buquet-Fagot C, Fagot D, Glab N, Goudeau H, Goudeau M, Guerrier P, Houghton P, Hendriks H, Kloareg B, Lippai M, Marie D, Maro B, Meijer L, Mester J, Mulner-Lorillon O, Poulet SA, Schierenberg E, Schutte B, Vaulot D, Verlhac MH. Cellular effects of olomoucine, an inhibitor of cyclin-dependent kinases. Biol Chem. 1995;83:105–120. doi: 10.1016/0248-4900(96)81298-6. [DOI] [PubMed] [Google Scholar]
- 2.Arendt T, Rödel L, Gärtner U, Holzer M. Expression of the cyclin-dependent kinase inhibitor p16 in Alzheimer's disease. NeuroReport. 1996;7:3047–3049. doi: 10.1097/00001756-199611250-00050. [DOI] [PubMed] [Google Scholar]
- 3.Arendt T, Holzer M, Gärtner U. Neuronal expression of cyclin dependent kinase inhibitors of the INK4 family in Alzheimer's disease. J Neural Transm. 1998;105:949–960. doi: 10.1007/s007020050104. [DOI] [PubMed] [Google Scholar]
- 4.Bossenmeyer-Puorié C, Chihab R, Schroeder H, Daval JL. Transient hypoxia may lead to neuronal proliferation in the developing mammalian brain: from apoptosis to cell cycle completion. Neuroscience. 1999;91:221–231. doi: 10.1016/s0306-4522(98)00565-x. [DOI] [PubMed] [Google Scholar]
- 5.Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- 6.Bruer U, Weih MK, Isaew NK, Meisel A, Ruscher K, Bergk A, Trendelenburg G, Wiegand F, Victorov IV, Dirnagl U. Induction of tolerance in rat cortical neurons: hypoxic preconditioning. FEBS Lett. 1997;414:117–121. doi: 10.1016/s0014-5793(97)00954-x. [DOI] [PubMed] [Google Scholar]
- 7.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brains. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cramer SC, Chopp M. Recovery recapitulates ontogeny. Trends Neurosci. 2000;23:265–271. doi: 10.1016/s0166-2236(00)01562-9. [DOI] [PubMed] [Google Scholar]
- 9.El Shamy WM, Fridvall LK, Ernfors P. Growth arrest failure, G1 restriction point override, and S phase death of sensory precursor cells in the absence of neurotrophin-3. Neuron. 1998;21:1003–1015. doi: 10.1016/s0896-6273(00)80619-4. [DOI] [PubMed] [Google Scholar]
- 10.Endres M, Laufs U, Huang Z, Nakamura T, Huang PL, Moskowitz MA, Liao JK. Stroke protective effects of HMG-CoA reductase inhibitors mediated by upregulation of endothelial nitric oxide synthase. Proc Natl Acad Sci USA. 1998a;95:1880–1885. doi: 10.1073/pnas.95.15.8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal cerebral ischemia by inhibitors of the caspase family. J Cereb Blood Flow Metab. 1998b;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 12.Endres M, Fink K, Zhu J, Stagliano NE, Bondada V, Geddes JW, Azuma T, Mattson MP, Kwiatkowski DJ, Moskowitz MA. Neuroprotective effects of gelsolin and analogues during murine stroke. J Clin Invest. 1999;103:347–354. doi: 10.1172/JCI4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Endres M, Meisel A, Biniskiewicz D, Namura S, Prass K, Ruscher K, Lipski A, Jaenisch R, Moskowitz MA, Dirnagl U. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fink K, Zhu J, Namura S, Shimizu-Sasamata M, Endres M, Ma J, Yuan J, Moskowitz MA. Prolonged therapeutic window for ischemic brain damage due to delayed caspase-3 activation. J Cereb Blood Flow Metab. 1998;18:1071–1076. doi: 10.1097/00004647-199810000-00003. [DOI] [PubMed] [Google Scholar]
- 16.Freeman RS, Estun S, Johnson EM. Analysis of cell cycle-related gene expression in post-mitotic neurons: selective induction of cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 17.Gage FH, Coates PW, Palmer TD, Kuhn HG, Fisher LJ, Suhnen JO, Peterson DA, Suhr ST, Ray J. Survival and differentiation of adult progenitor cells transplanted to the adult brain. Proc Natl Acad Sci USA. 1995;92:11879–11883. doi: 10.1073/pnas.92.25.11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, Chan PH. Response of postmitotic neurons to X-radiation: implications for the role of DNA damage in neuronal apoptosis. J Neurosci. 1998;18:147–155. doi: 10.1523/JNEUROSCI.18-01-00147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–2850. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu W, Brannstrom T, Wester P. Cortical neurogenesis in adult rats after reversible photothrombotic stroke. J Cereb Blood Flow Metab. 2000;20:1166–1173. doi: 10.1097/00004647-200008000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Guégan C, Lévy V, David J-P, Ajchanbaum-Cymbalista F, Sola B. c-Jun and cyclin D1 proteins as mediators of neuronal death after focal ischaemic insults. NeuroReport. 1997;8:1003–1007. doi: 10.1097/00001756-199703030-00037. [DOI] [PubMed] [Google Scholar]
- 23.Harms C, Lautenschlager M, Bergk A, Freyer D, Weih MK, Dirnagl U, Weber JR, Hörtnagl H. Melatonin is protective in necrotic but not in caspase-dependent, free-radical-independent apoptotic neuronal cell death in primary cortical cultures. FASEB J. 2000;14:1814–1824. doi: 10.1096/fj.99-0899com. [DOI] [PubMed] [Google Scholar]
- 24.Heintz N. Cell death and the cell cycle: a relationship between transformation and neurodegeneration? Trends Biochem Sci. 1993;18:157–159. doi: 10.1016/0968-0004(93)90103-t. [DOI] [PubMed] [Google Scholar]
- 25.Herrup K, Busser JC. The induction of multiple cell cycle events precedes target-related neuronal death. Development. 1995;121:2385–2395. doi: 10.1242/dev.121.8.2385. [DOI] [PubMed] [Google Scholar]
- 26.Huschtscha LI, Reddel RR. p16INK4a and the control of cellular proliferative life span. Carcinogenesis. 1999;20:921–926. doi: 10.1093/carcin/20.6.921. [DOI] [PubMed] [Google Scholar]
- 27.Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annu Rev Pharmacol Toxicol. 1999;39:295–312. doi: 10.1146/annurev.pharmtox.39.1.295. [DOI] [PubMed] [Google Scholar]
- 28.Kempermann G, Kuhn HG, Gage FH. Experience-induced neurogenesis in the senescent dentate gyrus. J Neurosci. 1998;18:3206–3212. doi: 10.1523/JNEUROSCI.18-09-03206.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase eflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 30.Kranenburg O, van der Eb AJ, Zatema A. Cyclin D1 is an essential mediator of apoptotic neuronal cell death. EMBO J. 1996;15:46–54. [PMC free article] [PubMed] [Google Scholar]
- 31.Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;15:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lautenschlager M, Onufriev MV, Gulyaeva NV, Harms C, Freyer D, Sehmsdorf U-S, Ruscher K, Moiseeva MV, Arnswald A, Vicotorov I, Dirnagl U, Weber JR, Hörtnagl H. Role of nitric oxide in the ethylcholine aziridium (AF64A) model of delayed apoptotic neurodegeneration in vivo and in vitro. Neuroscience. 2000;97:383–393. doi: 10.1016/s0306-4522(99)00599-0. [DOI] [PubMed] [Google Scholar]
- 33.Lee EYHP, Chang C-Y, Hu N, Wang YCJ, Lai CC, Herrup K, Le WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;349:288–293. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Chopp M, Powers C, Jiang N. Immunoreactivity of cyclin D1/cdk4 in neurons and oligodendrocytes after focal cerebral ischemia in rat. J Cereb Blood Flow Metab. 1997;17:846–856. doi: 10.1097/00004647-199708000-00003. [DOI] [PubMed] [Google Scholar]
- 35.Li Y, Jiang N, Powers C, Chopp M. Neuronal damage and plasticity identified by microtubule-associated proteins 2, growth-associated protein 43, and cyclin D1 immunoreactivity after focal cerebral ischemia in rats. Stroke. 1998;29:1972–1981. doi: 10.1161/01.str.29.9.1972. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Solway K, Messing RO, Sharp FR. Increased neurogenesis in the dentate gyrus after transient global ischemia in gerbils. J Neurosci. 1998;18:7768–7778. doi: 10.1523/JNEUROSCI.18-19-07768.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McShea A, Harris PLR, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators p16 and Cdk4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 38.McShea A, Wahl AF, Smith MA. Re-entry into the cell cycle: a mechanism for neurodegeneration in Alzheimer disease. Medical Hypotheses. 1999;52:525–527. doi: 10.1054/mehy.1997.0680. [DOI] [PubMed] [Google Scholar]
- 39.Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol. 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- 40.Nagy Z, Esiri MM, Smith AD. The cell division cycle and the pathophysiology of Alzheimer's disease. Neuroscience. 1998;87:731–739. doi: 10.1016/s0306-4522(98)00293-0. [DOI] [PubMed] [Google Scholar]
- 41.Nishikawa R, Furnari FB, Lin H, Arap W, Berger MS, Cavence WK, Huang HJ. Loss of p16 INK4 expression is frequent in high grade gliomas. Cancer Res. 1995;55:1941–1945. [PubMed] [Google Scholar]
- 42.Nowakowski RS, Lewin SB, Miller MW. Bromodeoxyuridine immunohistochemical determination of the lengths of the cell cycle and the DNA-synthetic phase for an anatomically defined population. J Neurocytol. 1989;18:311–318. doi: 10.1007/BF01190834. [DOI] [PubMed] [Google Scholar]
- 43.Osuga H, Osuga S, Wang F, Hogan MJ, Slack RS, Hakim AM, Ikeda J-E, Park DS. Cyclin-dependent kinases as a therapeutic target for stroke. Proc Natl Acad Sci USA. 2000;97:10254–10259. doi: 10.1073/pnas.170144197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Padmanabhan J, Park DS, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palmer TD, Wilhoite AR, Gage FH. Vascular niche for adult hippocampal neurogenesis. J Comp Neurol. 2000;425:479–494. doi: 10.1002/1096-9861(20001002)425:4<479::aid-cne2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 46.Parent JM, Tada E, Fike JR, Lowenstein DH. Inhibition of dentate granule cell neurogenesis with brain irradiation does not prevent seizure-induced mossy fiber synaptic reorganization in the rat. J Neurosci. 1999;19:4508–4519. doi: 10.1523/JNEUROSCI.19-11-04508.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996;271:8161–8169. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 48.Park DS, Levine B, Ferrari G, Greene LA. Cyclin-dependent kinase inhibitors and dominant negative cyclin-dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997a;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin-dependent kinases suppress camptothecin-induced neuronal apoptosis. J Neurosci. 1997b;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin-dependent kinases participate in death of neurons evoked by DNA-damaging agents. J Cell Biol. 1998;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai L-H. Conversion of p35 to p25 deregulates cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 52.Sakurai M, Hayashi T, Abi K, Toyama Y, Tabayashi K. Cyclin D1 and cdk4 protein induction in motor neurons after transient spinal cord ischemia in rabbits. Stroke. 2000;31:200–207. doi: 10.1161/01.str.31.1.200. [DOI] [PubMed] [Google Scholar]
- 53.Sherr CJ. G1 progression: cyclin on cue. Cell. 1994;79:551–555. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 54.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi T, Nowakowski RS, Caviness VS., Jr Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone of the fetal mouse. J Neurosci. 1993;13:820–833. doi: 10.1523/JNEUROSCI.13-02-00820.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Thomaidou D, Mione MC, Cavanagh JFR, Parnavelas JG. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay É, Ben-Ari Y, Khrestchatisky M. Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur J Neurosci. 1999;11:263–278. doi: 10.1046/j.1460-9568.1999.00434.x. [DOI] [PubMed] [Google Scholar]
- 58.Ueki K, Ono Y, Henson JW, Efird JT, von Deimling A, Louis DN. CDKN2/p16 or RB alterations occur in the majority of glioblastomas and are inversely correlated. Cancer Res. 1996;56:150–153. [PubMed] [Google Scholar]
- 59.Vesely J, Havlicek L, Strand M, Blow JJ, Donella-Deana A, Pinna L, Letham DS, Kato J, Detivaud L, Leclerc S. Inhibition of cyclin-dependent kinases by purine analogues. Eur J Biochem. 1994;224:771–786. doi: 10.1111/j.1432-1033.1994.00771.x. [DOI] [PubMed] [Google Scholar]
- 60.Vidal A, Koff A. Cell-cycle inhibitors: three families united by a common cause. Gene. 2000;247:1–15. doi: 10.1016/s0378-1119(00)00092-5. [DOI] [PubMed] [Google Scholar]
- 61.Watanabe G, Pena P, Shambaugh GE, III, Haines GK, III, Pestell RG. Regulation of cyclin dependent kinase inhibitor proteins during neonatal cerebella development. Dev Brain Res. 1998;108:77–87. doi: 10.1016/s0165-3806(98)00032-7. [DOI] [PubMed] [Google Scholar]
- 62.Yang J, Kornbluth S. All aboard the cyclin train: subcellular trafficking of cyclins and their cdk partners. Trends Cell Biol. 1999;9:207–210. doi: 10.1016/s0962-8924(99)01577-9. [DOI] [PubMed] [Google Scholar]
- 63.Zindy F, Soares H, Herzog K-H, Morgan J, Sherr CJ, Roussel MF. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. 1997;8:1139–1150. [PubMed] [Google Scholar]