Summary
Clinical characteristics.
IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome is characterized by systemic autoimmunity, typically beginning in the first year of life, which includes the triad of enteropathy (manifesting as malabsorption and watery diarrhea), endocrinopathy (most commonly type 1 insulin-dependent diabetes mellitus), and eczematous dermatitis. In addition to these manifestations, many children have other autoimmune phenomena including cytopenias, autoimmune hepatitis, nephropathy, lymphadenopathy, splenomegaly, alopecia, arthritis, and interstitial lung disease related to immune dysregulation. Fetal presentation of IPEX syndrome includes hydrops, echogenic bowel, skin desquamation, intrauterine growth deficiency, and fetal akinesia. Without aggressive immunosuppression or hematopoietic stem cell transplantation (HSCT), the majority of affected males will die within the first one to two years of life from metabolic derangements, severe malabsorption, or sepsis. Individuals with a milder phenotype have survived into the second or third decade of life, but this is uncommon.
Diagnosis/testing.
The diagnosis is established in a male proband with typical clinical findings, absent regulatory T cells (Treg) in blood or tissues, decreased numbers of FOXP3-expressing T cells in peripheral blood determined by flow cytometry (although FOXP3 levels in Treg can be normal in some individuals), and a hemizygous pathogenic variant in FOXP3 identified by molecular genetic testing. Heterozygous females have not been reported to have clinical findings typical of IPEX syndrome.
Management.
Targeted therapies: HSCT offers the only potential cure for IPEX syndrome. T cell-directed immune suppression can include either an mTOR inhibitor (sirolimus) or calcineurin inhibitor (cyclosporin A or tacrolimus), alone or in combination with corticosteroids.
Supportive care: Total parenteral nutrition (TPN) with fluids and electrolyte support is needed until intestinal function can be established with immune suppression. Treatment of type 1 insulin-dependent diabetes mellitus with insulin and carbohydrate management is standard, as is management of autoimmune thyroid disease. Skin conditions are managed with topical therapies, which can include steroids, tacrolimus, and emollients. Autoimmune neutropenia has been successfully treated with granulocyte colony-stimulating factor; pemphigus nodularis has been treated with rituximab (anti-CD20), and rituximab has been used for other autoantibody-mediated disease. Prophylactic antibiotic therapy may be required for autoimmune neutropenia or recurrent infections with central venous access and TPN. Aggressive management of dermatitis with topical steroids and anti-inflammatory agents as needed to prevent cutaneous infections.
Surveillance: Monitor growth, nutritional intake, and stooling patterns at each visit; glucose tolerance test, hemoglobin A1c, and thyroid function tests every three to six months; skin exam at each visit; complete blood count, blood urea nitrogen, creatinine, urinalysis, and serum aspartate transaminase and alanine transaminase every three to six months.
Agents/circumstances to avoid: Withhold immunizations until after HSCT, if possible.
Evaluation of relatives at risk: It is appropriate to clarify the genetic status of at-risk males either prenatally or immediately after birth to enable early diagnosis and HSCT and/or immune suppression treatment in affected males before significant organ damage occurs.
Genetic counseling.
IPEX syndrome is inherited in an X-linked manner. The risk to sibs of the proband depends on the genetic status of the mother. If the mother of the proband has a FOXP3 pathogenic variant, the chance of transmitting the pathogenic variant in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be heterozygous (to date, IPEX syndrome has not been reported in females who are heterozygous for a FOXP3 pathogenic variant). Affected males transmit the pathogenic variant to all of their daughters and none of their sons. Once the FOXP3 pathogenic variant has been identified in an affected family member, identification of female heterozygotes and prenatal/preimplantation genetic testing are possible.
Diagnosis
The term "IPEX" is an acronym for immune dysregulation, polyendocrinopathy, enteropathy, X-linked.
Suggestive Findings
IPEX syndrome should be suspected in males with the following clinical triad, family history, and suggestive laboratory findings.
Clinical triad
- Enteropathy that manifests as chronic watery diarrhea. Onset is typically in the first months of life; villous atrophy with a mononuclear cell infiltrate (activated T cells) in the lamina propria is the most common finding in intestinal biopsy.
- Endocrinopathy, most commonly type 1 insulin-dependent diabetes mellitus with onset in the first months or years of life. Autoimmune thyroid disease leading to hypothyroidism or hyperthyroidism has also been observed [Wildin et al 2002, Gambineri et al 2003].
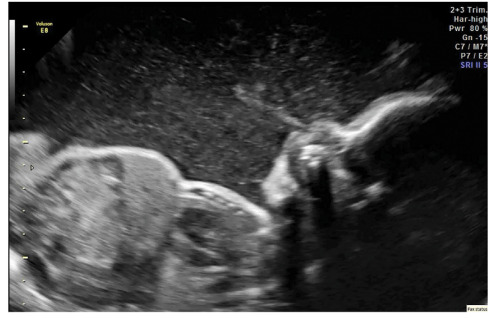
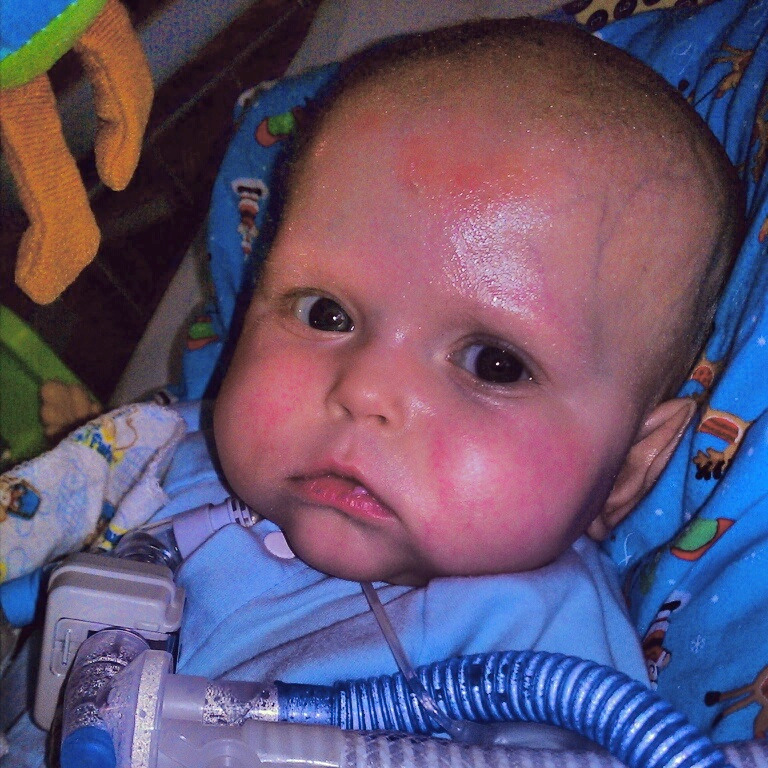
- Dermatitis, most commonly eczematous presenting within the first months of life, although prenatal skin desquamation has been reported (see Figure 1) [Louie et al 2017]. Erythroderma, exfoliative dermatitis, psoriasis-like lesions, and pemphigus nodularis have also been observed (see Figure 2) [Nieves et al 2004, McGinness et al 2006].


Figure 2.
Typical erythematous rash seen in individuals with IPEX syndrome.
Family history is consistent with X-linked inheritance (e.g., no male-to-male transmission). Absence of a known family history does not preclude the diagnosis.
Suggestive laboratory findings. No laboratory findings specifically identify affected individuals. Evidence of immune dysregulation manifesting as the following is suggestive of IPEX syndrome:
- Elevated serum concentration of immunoglobulin E (IgE), and in some individuals elevated serum concentration of IgA
- Eosinophilia
- Autoimmune anemia, thrombocytopenia, and/or neutropenia
- Autoantibodies to pancreatic islet antigens, thyroid antigens, small bowel mucosa, and other autoantigens
- Decreased numbers of FOXP3-expressing T cells in peripheral blood determined by flow cytometry – although FOXP3 levels in regulatory T cells (Treg) can be normal in some individuals
Note: Standard lymphocyte enumeration of T cells, B cells, and NK cells as well as T cell function measured by mitogen proliferation is generally normal and not helpful for diagnosis.
Establishing the Diagnosis
Male proband. The diagnosis of IPEX syndrome is established in a male proband with suggestive findings and a hemizygous pathogenic (or likely pathogenic) variant in FOXP3 identified by molecular genetic testing (see Table 1).
Female proband. Affected females have not been reported. Carrier status is determined by identification of a heterozygous pathogenic (or likely pathogenic) variant in FOXP3 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a hemizygous FOXP3 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Option 1
Single-gene testing. Sequence analysis of FOXP3 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Note: Pathogenic variants have been reported in the 5' UTR (c.-7G>T) and the 3' UTR (c.*876A>G). Since the 3' UTR variants are not typically included in sequencing assays, the assay design may need to be modified to include these variants.
A multigene panel that includes FOXP3 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in IPEX Syndrome
Clinical Characteristics
Clinical Description
Males
IPEX syndrome is generally considered to be a syndrome of neonatal enteropathy [Ruemmele et al 2004] and neonatal polyendocrinopathy [Dotta & Vendrame 2002] found in males. In a large natural history study, 95% of individuals with IPEX syndrome had disease onset in the first year of life, with 50% by age one month [Barzaghi et al 2012]. However, atypical clinical presentation has been reported with onset later in childhood [Consonni et al 2021].
Presentation. The most common presentation of IPEX syndrome is malabsorption with severe watery diarrhea, type 1 insulin-dependent diabetes mellitus, thyroiditis, and dermatitis in males younger than age one year. This disorder is frequently accompanied by other autoimmune phenomena. Males with a somewhat milder/atypical disease phenotype can present at older ages [Ge et al 2017, Hwang et al 2018]. Fetal presentation of IPEX syndrome includes hydrops, echogenic bowel, skin desquamation, intrauterine growth deficiency, and fetal akinesia. There may be a family history of pregnancy loss [Rae et al 2015, Vasiljevic et al 2015, Xavier-da-Silva et al 2015, Reichert et al 2016, Louie et al 2017, Shehab et al 2017].
Enteropathy. The enteropathy of IPEX syndrome, often the initial symptom, is present in virtually all affected individuals. Even in those with milder disease, the diarrhea typically begins in the first six to 12 months of life. Autoimmune enteropathy results in loss of intestinal villi architecture with malabsorption and watery diarrhea, which may contain mucus and blood. Malabsorption ultimately leads to growth failure and cachexia [Bacchetta et al 2018]. Small bowel biopsy is helpful in evaluating the extent of enteropathy. Histologic findings in most individuals have shown graft-vs-host-like changes with lymphocytic infiltrates with depletion of goblet cells and anti-enterocyte antibody deposition [Patey-Mariaud de Serre et al 2009]. Exocrine pancreatic insufficiency has been observed in some individuals [Gambineri et al 2008, Scaillon et al 2009], which may worsen the diarrhea. Other gastrointestinal manifestations include colitis [Lucas et al 2007] and gastritis [Gambineri et al 2008, Scaillon et al 2009]. Food allergies and intolerance are common, which can be diagnosed based on results of immunoglobulin E (IgE) testing to specific food antigens or skin prick testing [Torgerson et al 2007].
Endocrinopathy is present in the majority of affected individuals. Type 1 insulin-dependent diabetes mellitus, often with onset in the first months of life, is the most common endocrine manifestation [Gambineri et al 2008, Rubio-Cabezas et al 2009]. Thyroid disease (thyroiditis with either hypothyroidism [more common] or hyperthyroidism) is also frequently present [Wildin et al 2002, Gambineri et al 2003, Gambineri et al 2008, Rubio-Cabezas et al 2009].
Dermatitis. The dermatitis is most frequently eczematous, but psoriasiform and ichthyosiform dermatitis have been reported as well. Other dermatologic manifestations include painful chelitis and skin lesions related to food allergies. Rare cutaneous symptoms include pemphigoid nodularis and epidermolysis bullosa acquisita [Nieves et al 2004, McGinness et al 2006, Halabi-Tawil et al 2009, Bis et al 2015].
Autoimmune disorder. Most affected individuals have other autoimmune phenomena including cytopenias (autoimmune hemolytic anemia, immune thrombocytopenia, autoimmune neutropenia [Barzaghi et al 2018]), autoimmune hepatitis [López et al 2011], and nephropathy (membranous nephropathy, interstitial nephritis, and – rarely – minimal change nephrotic syndrome) [Park et al 2015, Sheikine et al 2015]. Lymphadenopathy and splenomegaly as a result of lymphoproliferation have been reported [Ochs &Torgerson 2007, Nademi et al 2014, Bacchetta et al 2018, Barzaghi et al 2018]. Alopecia and arthritis have also been observed [Barzaghi et al 2018], as well as interstitial lung disease related to immune dysregulation [Baris et al 2014].
Infectious complications. Infections of the gastrointestinal tract, skin, and airways occur in individuals with IPEX syndrome [Bacchetta et al 2018], and severe or invasive infections including sepsis, meningitis, pneumonia, and osteomyelitis affect a significant number of subjects [Gambineri et al 2008, Barzaghi et al 2012, Barzaghi et al 2018]. Common pathogens identified were Staphylococcus, Enterococcus, cytomegalovirus, and Candida [Halabi-Tawil et al 2009, Barzaghi et al 2012]. Some infections may be secondary to immunosuppressive therapy, malnutrition, and central venous access; however, many occur prior to the initiation of treatment. Serious infections in individuals with IPEX syndrome are not thought to be due to an intrinsic immune defect but instead are typically related to poor barrier function of the small intestines and skin [Bacchetta et al 2018].
Survival. The outcome of IPEX syndrome is universally poor. Many children die within the first or second year of life from metabolic derangements, severe malabsorption, or sepsis. Although improvements in immunosuppressive regimens have prolonged survival, long-term immunosuppression does not appear to prevent morbidity due to disease progression and side effects or complications in the majority of individuals [Barzaghi et al 2018].
Early hematopoietic stem cell transplantation (HSCT) can cure IPEX syndrome; some survivors are now more than ten years post transplant and doing well. If individuals develop diabetes or thyroiditis prior to HSCT, these aspects of the disorder usually persist, but the other signs of IPEX syndrome resolve. Survival and long-term outcomes are improved if HSCT occurs at an earlier age, prior to the individual developing irreversible organ damage related to the extensive, systemic autoimmunity present in virtually all individuals with IPEX syndrome [Rao et al 2007, Burroughs et al 2010, Kucuk et al 2016].
Heterozygous Females
Heterozygous females have not been reported to have IPEX syndrome.
Note: Recurrent miscarriage of male fetuses, including fetal hydrops and abnormal findings on fetal ultrasound, have been reported and are associated with fetal rather than maternal factors [Rae et al 2015, Vasiljevic et al 2015, Xavier-da-Silva et al 2015, Reichert et al 2016, Louie et al 2017, Shehab et al 2017, Carneiro-Sampaio et al 2022].
Genotype-Phenotype Correlations
There are currently no genotype-phenotype correlations. The same genotype can present with variable severity in different individuals, even within the same family [Seidel et al 2016, Bacchetta et al 2018]. Furthermore, it is difficult to correlate the type of pathogenic variant and outcome. Loss-of-function variants (frameshift) predicted to be missing the forkhead domain have been described in fetal-onset and nonviable infants, but also in individuals who survive into adolescence [Louie et al 2017, Ben-Skowronek 2021]. Within the cohort of affected individuals with extremely early onset of symptoms (<24 hours of life), the types of variants and their position within the gene vary [Reichert et al 2016].
FOXP3 missense variants can result in FOXP3 expression resulting in normal regulatory T cell (Treg) enumeration by flow cytometry but abnormal Treg function [Seghezzo et al 2017, Lin et al 2018].
Nomenclature
IPEX syndrome may also be referred to as X-linked autoimmunity-allergic dysregulation (XLAAD) syndrome or X-linked syndrome of polyendocrinopathy, immune dysfunction, and diarrhea (XPID).
Prevalence
IPEX syndrome is rare: fewer than 300 affected individuals have been identified worldwide. No accurate estimates of prevalence have been published.
Genetically Related (Allelic) Disorders
No other phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in FOXP3.
Differential Diagnosis
IPEX syndrome is classified by the International Union of Immunological Societies (IUIS) as an inborn error of immunity that results in immune dysregulation due to absent or defective regulatory T cells (Treg) [Bousfiha et al 2022]. Autoimmunity is the primary clinical manifestation. Among the ten unique disorders with Treg dysfunction, LRBA deficiency, CTLA4 haploinsufficiency, CD25 deficiency, FERMT1 deficiency, BACH2 deficiency, IKAROS GOF (gain of function), and CD122 deficiency have the most clinical overlap with IPEX syndrome [Tangye et al 2022]. Other inborn errors of immunity without Treg dysfunction, including immune dysregulation with colitis, can also mimic IPEX syndrome [Cepika et al 2018, Tangye et al 2022].
In addition to immune dysregulation disorders, monogenic forms of neonatal diabetes that are clinically evident soon after birth can present similarly to IPEX syndrome. These conditions are most commonly associated with pancreatic defects and lack autoimmune manifestations [Rubio-Cabezas et al 2011]. Similarly, intrinsic defects of the intestinal microvilli have clinical presentations consistent with enteropathy with malabsorption and diarrhea but are not immune mediated [Cai et al 2020].
See Table 2 for these and other considerations in the differential diagnosis.
Table 2.
Syndromes of Known Genetic Cause to Consider in the Differential Diagnosis of IPEX Syndrome
Syndromes of unknown genetic cause to consider in the differential diagnosis of IPEX syndrome include the following:
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with IPEX syndrome, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
IPEX Syndrome: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Targeted Therapies
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Hematopoietic stem cell transplantation (HSCT) currently offers the only potential cure for IPEX syndrome. Myeloablative conditioning regimens showed a high degree of transplant-related mortality and other complications, so most centers have used non-myeloablative conditioning regimens, resulting in better overall survival [Baud et al 2001, Burroughs et al 2007, Lucas et al 2007, Rao et al 2007]. Overall, 15-year survival is 77.5% following HSCT. Survival is similar among individuals with IPEX syndrome who receive transplants from matched related, matched unrelated, cord blood, or haploidentical donors. Individuals with more severe disease at the time of transplant have poorer outcomes. If HSCT is performed in early infancy there is evidence that early-onset diabetes and thyroiditis is reversible [Yamauchi et al 2019]. Taken together, HSCT results in better outcomes and lower overall morbidity compared with non-transplanted individuals receiving chronic immunosuppressive therapy [Barzaghi et al 2018].
Immunosuppression therapy. The most recent multicenter study examining long-term outcomes in individuals with IPEX syndrome treated with different therapeutic modalities showed that calcineurin inhibitors such as tacrolimus or mTOR inhibitors such as rapamycin were the backbone of immune suppression strategies with azathioprine, mycophenolic acid, and methotrexate used in addition to these agents [Barzaghi et al 2018]. More recently, rapamycin, dosed to achieve levels of 8-12 ng/mL, has been shown to restore regulatory T cell (Treg) function, and when used early in the course of disease it has the potential to reverse endocrinopathy [Passerini et al 2020]. Immune suppression treatment requires closing monitoring for nephrotoxicity and drug levels, and close observation for opportunistic infection. Cutaneous manifestations can be treated with topical corticosteroids and topical tacrolimus. There have been reports that improved dermatitis and diabetes is associated with the use of dupilumab [Maher et al 2021, Caruso et al 2023].
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 5 are recommended.
Table 5.
IPEX Syndrome: Recommended Surveillance
Agents/Circumstances to Avoid
Immune activation (e.g., by immunizations or severe infections) has been reported to cause worsening or exacerbation of disease symptoms [Powell et al 1982]. It is generally best practice to withhold immunizations until after HSCT, if possible.
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of at-risk males either prenatally or immediately after birth to enable early diagnosis and HSCT and/or steroid treatment in affected males before significant organ damage occurs.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
HSCT carries the risk of significant morbidity and mortality, and suitable donors are not always available. For these and other reasons, IPEX syndrome is an excellent candidate for treatment with gene therapy. However, one major hurdle is the need to regulate the expression of FOXP3 on mature T cells; thus, hematopoietic stem cells are not the ideal target for gene delivery. An alternative approach has been to convert CD4+ T cells from individuals with IPEX syndrome using lentivirus vectors that carry normal FOXP3. Lentiviral delivery of exogenous FOXP3 cDNA is accomplished under the constitutive promoter to achieve conversion of FOXP3 mutated cells to normal functioning regulatory T cells (Treg) in vivo. A potential limitation to this approach is determining how long the converted Treg will survive. A human clinical trial is now ongoing using this strategy [Borna et al 2022] (see NCT05241444).
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
IPEX syndrome is inherited in an X-linked manner.
Risk to Family Members
Parents of a male proband
- The father of an affected male will not have the disorder, nor will he be hemizygous for the FOXP3 pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected child and no other affected relatives and if the FOXP3 pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism. Maternal somatic and germline mosaicism has been reported in IPEX syndrome [Lin et al 2018].
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier), the affected male may have a de novo FOXP3 pathogenic variant (in which case the mother is not a carrier), or the mother may have somatic/germline mosaicism. The percentage of affected males who have no family history of IPEX syndrome is not known.
- Molecular genetic testing of the mother is recommended to evaluate her genetic status and inform recurrence risk assessment.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband has a FOXP3 pathogenic variant, the chance of transmitting it in each pregnancy is 50%.
- Males who inherit the pathogenic variant will be affected. Male sibs with the same FOXP3 pathogenic variant can present with variable severity (see Genotype-Phenotype Correlations).
- Females who inherit the pathogenic variant will be heterozygous (carriers). To date, IPEX syndrome has not been reported in females who are heterozygous for a FOXP3 pathogenic variant.
- If the proband represents a simplex case and if the FOXP3 pathogenic variant cannot be detected in the leukocyte DNA of the proband's mother, the recurrence risk to sibs is presumed to be low but slightly greater than that of the general population because of the possibility of maternal germline mosaicism [Lin et al 2018].
Offspring of a male proband. Affected males transmit the FOXP3 pathogenic variant to all of their daughters, who will be heterozygotes (carriers), and none of their sons.
Other family members. The proband's maternal aunts and their offspring may be at risk of being heterozygotes (carriers) for the pathogenic variant, and the aunts' offspring, depending on their sex, may be at risk of being heterozygotes for the pathogenic variant or of being affected.
Carrier Detection
Identification of female heterozygotes requires prior identification of the FOXP3 pathogenic variant in the family.
X-chromosome inactivation is skewed only in regulatory T cells [Di Nunzio et al 2009] and is random in all other lymphocyte populations [Tommasini et al 2002]; therefore, X-chromosome inactivation studies are of limited use in carrier detection.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the FOXP3 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- American Diabetes AssociationPhone: 800-DIABETES (800-342-2383)Email: [email protected]
- Diabetes UKUnited KingdomPhone: 0345 123 2399Email: [email protected]
- International Patient Organization for Primary Immunodeficiencies (IPOPI)United KingdomPhone: +44 01503 250 668Fax: +44 01503 250 668Email: [email protected]
- Jeffrey Modell Foundation/National Primary Immunodeficiency Resource CenterEmail: [email protected]
- European Society for Immunodeficiencies (ESID) RegistryEmail: [email protected]
- Latin American Society for Immunodeficiency (LASID) RegistryEmail: [email protected]
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
IPEX Syndrome: Genes and Databases
Table B.
OMIM Entries for IPEX Syndrome (View All in OMIM)
Molecular Pathogenesis
FOXP3 encodes forkhead box protein P3 (FOXP3), a forkhead DNA-binding protein that is expressed primarily in CD4+CD25+ regulatory T cells. The protein has important functional domains including:
- An N-terminal proline-rich domain, which is essential for the gene-suppressive function of FOXP3 and for interaction with other key transcription factors including RORα and RORγt [Du et al 2008, Zhou et al 2008];
- A C2H2 zinc finger and leucine zipper (both conserved structural motifs involved in protein-protein interactions) in the central portion;
- A forkhead DNA-binding domain at the C terminus, from which it derives its name (forkhead box) [Ochs et al 2005, Lopes et al 2006]. A putative nuclear localization signal is located at the C-terminal portion of the forkhead domain [Lopes et al 2006].
Proteins bearing forkhead DNA-binding motifs comprise a large family of related DNA-binding proteins that play diverse roles in enhancing or suppressing transcription from specific binding sites. Several members of this protein family are involved in patterning and development [Gajiwala & Burley 2000]. FOXP3 is expressed primarily in lymphoid tissues (thymus, spleen, and lymph nodes), particularly in CD4+CD25+ regulatory T-cell lymphocytes. In mice, it is required for the development and suppressive function of this important regulatory T cell population [Fontenot et al 2003, Hori et al 2003, Khattri et al 2003, Sakaguchi 2003]. In humans, it is not expressed at baseline in CD4+CD25− or CD8+ T cells but is expressed upon T-cell activation [Gavin et al 2006, Allan et al 2007]. The significance of this inducible expression in effector T cells is unknown.
The majority of pathogenic variants in FOXP3 are either missense variants or small in-frame amino acid deletions. Loss-of-function variants have been described both in individuals with a neonatal presentation and others with a childhood presentation; thus, haploinsufficiency of FOXP3 does not appear to be lethal. The highest concentration of variants cluster within the C-terminal forkhead DNA-binding domain. Some pathogenic variants also affect the leucine zipper and an amino-terminal proline-rich domain that is involved in interactions with other key protein partners. Clustering of variants within these key functional regions of the protein demonstrates the essential role for these domains in FOXP3 function [Chatila et al 2000, Lopes et al 2006]. Pathogenic alterations can affect mRNA stability, protein function, and intracellular localization. The FOXP3 protein is a transcription factor that regulates the expression of hundreds of targets and is necessary for proper development of Treg, a population of cells responsible for tolerance of self-antigens.
Mechanism of disease causation. Loss of function
FOXP3-specific laboratory technical considerations. FOXP3 pathogenic variants have been reported in the 5' UTR (c.-7G>T) and the 3' UTR (c.*876A>G). Since the 3' UTR variants are not typically included in sequencing assays, the assay design may need to be modified to include these variants.
Table 6.
FOXP3 Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Acknowledgments
Supported in part by an award from the Jeffrey Modell Foundation (JS).
Author History
Mark C Hannibal, MD, PhD; University of Michigan Medical School (2004-2018)
Raymond J Louie, PhD (2018-present)
John W Sleasman, MD (2018-present)
Queenie K-G Tan, MD, PhD (2018-present)
Troy Torgerson, MD, PhD; University of Washington, Seattle (2004-2018)
Revision History
- 1 February 2024 (sw) Comprehensive update posted live
- 19 July 2018 (ha) Comprehensive update posted live
- 27 January 2011 (me) Comprehensive update posted live
- 12 December 2007 (me) Comprehensive update posted live
- 19 October 2004 (me) Review posted live
- 11 February 2004 (mh) Original submission
References
Literature Cited
- Afzali B, Gronholm J, Vandrovcova J, O'Brien C, Sun HW, Vanderleyden I, Davis FP, Khoder A, Zhang Y, Hegazy AN, Villarino AV, Palmer IW, Kaufman J, Watts NR, Kazemian M, Kamenyeva O, Keith J, Sayed A, Kasperaviciute D, Mueller M, Hughes JD, Fuss IJ, Sadiyah MF, Montgomery-Recht K, McElwee J, Restifo NP, Strober W, Linterman MA, Wingfield PT, Uhlig HH, Roychoudhuri R, Aitman TJ, Kelleher P, Lenardo MJ, O'Shea JJ, Cooper N, Laurence ADJ. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat Immunol. 2017;18:813–23. [PMC free article: PMC5593426] [PubMed: 28530713]
- Allan SE, Crome SQ, Crellin NK, Passerini L, Steiner TS, Bacchetta R, Roncarolo MG, Levings MK. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–54. [PubMed: 17329235]
- Avitzur Y, Guo C, Mastropaolo LA, Bahrami E, Chen H, Zhao Z, Elkadri A, Dhillon S, Murchie R, Fattouh R, Huynh H, Walker JL, Wales PW, Cutz E, Kakuta Y, Dudley J, Kammermeier J, Powrie F, Shah N, Walz C, Nathrath M, Kotlarz D, Puchaka J, Krieger JR, Racek T, Kirchner T, Walters TD, Brumell JH, Griffiths AM, Rezaei N, Rashtian P, Najafi M, Monajemzadeh M, Pelsue S, McGovern DP, Uhlig HH, Schadt E, Klein C, Snapper SB, Muise AM. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology. 2014;146:1028–39. [PMC free article: PMC4002656] [PubMed: 24417819]
- Bacchetta R, Barzaghi F, Roncarolo MG. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. 2018;1417:5–22. [PubMed: 26918796]
- Baris S, Schulze I, Ozen A, Karakoc Aydiner E, Altuncu E, Karasu GT, Ozturk N, Lorenz M, Schwarz K, Vraetz T, Ehl S, Barlan IB. Clinical heterogeneity of immunodysregulation, polyendocrinopathy, enteropathy, X-linked: pulmonary involvement as a non-classical disease manifestation. J Clin Immunol. 2014;34:601–6. [PubMed: 24916357]
- Barzaghi F, Amaya Hernandez LC, Neven B, Ricci S, Kucuk ZY, Bleesing JJ, Nademi Z, Slatter MA, Ulloa ER, Shcherbina A, Roppelt A, Worth A, Silva J, Aiuti A, Murguia-Favela L, Speckmann C, Carneiro-Sampaio M, Fernandes JF, Baris S, Ozen A, Karakoc-Aydiner E, Kiykim A, Schulz A, Steinmann S, Notarangelo LD, Gambineri E, Lionetti P, Shearer WT, Forbes LR, Martinez C, Moshous D, Blanche S, Fisher A, Ruemmele FM, Tissandier C, Ouachee-Chardin M, Rieux-Laucat F, Cavazzana M, Qasim W, Lucarelli B, Albert MH, Kobayashi I, Alonso L, Diaz De Heredia C, Kanegane H, Lawitschka A, Seo JJ, Gonzalez-Vicent M, Diaz MA, Goyal RK, Sauer MG, Yesilipek A, Kim M, Yilmaz-Demirdag Y, Bhatia M, Khlevner J, Richmond Padilla EJ, Martino S, Montin D, Neth O, Molinos-Quintana A, Valverde-Fernandez J, Broides A, Pinsk V, Ballauf A, Haerynck F, Bordon V, Dhooge C, Garcia-Lloret ML, Bredius RG, Kałwak K, Haddad E, Seidel MG, Duckers G, Pai SY, Dvorak CC, Ehl S, Locatelli F, Goldman F, Gennery AR, Cowan MJ, Roncarolo MG, Bacchetta R; Primary Immune Deficiency Treatment Consortium (PIDTC) and the Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT). Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol. 2018;141:1036–49.e5. [PMC free article: PMC6050203] [PubMed: 29241729]
- Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211. [PMC free article: PMC3459184] [PubMed: 23060872]
- Baud O, Goulet O, Canioni D, Le Deist F, Radford I, Rieu D, Dupuis-Girod S, Cerf-Bensussan N, Cavazzana-Calvo M, Brousse N, Fischer A, Casanova JL. Treatment of the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) by allogeneic bone marrow transplantation. N Engl J Med. 2001;344:1758–62. [PubMed: 11396442]
- Ben-Skowronek I. IPEX syndrome: genetics and treatment options. Genes (Basel). 2021;12:323. [PMC free article: PMC7995986] [PubMed: 33668198]
- Bindl L, Torgerson T, Perroni L, Youssef N, Ochs HD, Goulet O, Ruemmele FM. Successful use of the new immune-suppressor sirolimus in IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome). J Pediatr. 2005;147:256–9. [PubMed: 16126062]
- Bis S, Maguiness SM, Gellis SE, Schneider LC, Lee PY, Notarangelo LD, Keles S, Chatila TA, Schmidt BA, Miller DD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome associated with neonatal epidermolysis bullosa acquisita. Pediatr Dermatol. 2015;32:e74–7. [PubMed: 25790289]
- Borna S, Lee E, Sato Y, Bacchetta R. Towards gene therapy for IPEX syndrome. Eur J Immunol. 2022;52:705-16. [PMC free article: PMC9322407] [PubMed: 35355253]
- Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, Al-Herz W, Rundles CC, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Puel A, Puck J, Seppänen MRJ, Somech R, Su HC, Sullivan KE, Torgerson TR, Meyts I. The 2022 update of IUIS phenotypical classification for human inborn errors of immunity. J Clin Immunol. 2022;42:1508-20. [PubMed: 36198931]
- Boutboul D, Kuehn HS, Van de Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, Lenoir C, Barlogis V, Farnarier C, Vely F, Yoshida N, Kojima S, Kanegane H, Hoshino A, Hauck F, Lhermitte L, Asnafi V, Roehrs P, Chen S, Verbsky JW, Calvo KR, Husami A, Zhang K, Roberts J, Amrol D, Sleasman J, Hsu AP, Holland SM, Marsh R, Fischer A, Fleisher TA, Picard C, Latour S, Rosenzweig SD. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. 2018;128:3071-87. [PMC free article: PMC6026000] [PubMed: 29889099]
- Burroughs LM, Storb R, Leisenring WM, Pulsipher MA, Loken MR, Torgerson TR, Ochs HD, Woolfrey AE. Intensive postgrafting immune suppression combined with nonmyeloablative conditioning for transplantation of HLA-identical hematopoietic cell grafts: results of a pilot study for treatment of primary immunodeficiency disorders. Bone Marrow Transplant. 2007;40:633–42. [PubMed: 17660844]
- Burroughs LM, Torgerson TR, Storb R, Carpenter PA, Rawlings DJ, Sanders MD, Scharenberg AM, Skoda-Smith S, Englund J, Ochs HD, Woolfrey AE. Stable hematopoietic cell engraftment after low-intensity nonmyeloablative conditioning in patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. J Allergy Clin Immunol. 2010;126:1000–5. [PMC free article: PMC2962731] [PubMed: 20643476]
- Cai C, Chen Y, Chen X, Ji F. Tufting enteropathy: a review of clinical and histological presentation, etiology, management, and outcome. Gastroenterol Res Pract. 2020;2020:5608069. [PMC free article: PMC7530495] [PubMed: 33029133]
- Carneiro-Sampaio M, de Jesus AA, Bando SY, Moreira-Filho CA. Inborn errors of immunity with fetal or perinatal clinical manifestations. Front Pediatr. 2022;10:891343. [PMC free article: PMC9121170] [PubMed: 35601409]
- Caruso C, Laterza L, Settanni CR, Colantuono S, Di Mario C, Tolusso B, Castrì F, Gremese E, Scaldaferri F, Armuzzi A, De Simone C, Peris K, Chiricozzi A, Gasbarrini A. Case report: dupilumab treatment improved type 2 disorders in a patient with IPEX syndrome diagnosis. Front Immunol. 2023;13:995304. [PMC free article: PMC9875030] [PubMed: 36713411]
- Cepika AM, Sato Y, Liu JM, Uyeda MJ, Bacchetta R, Roncarolo MG. Tregopathies: monogenic diseases resulting in regulatory T-cell deficiency. J Allergy Clin Immunol. 2018;142:1679-95. [PubMed: 30527062]
- Charbit-Henrion F, Jeverica AK, Begue B, Markelj G, Parlato M, Avcin SL, Callebaut I, Bras M, Parisot M, Jazbec J, Homan M, Ihan A, Rieux-Laucat F, Stolzenberg MC, Ruemmele FM, Avčin T, Cerf-Bensussan N; GENIUS Group. Deficiency in mucosa-associated lymphoid tissue lymphoma translocation 1: a novel cause of IPEX-like syndrome. J Pediatr Gastroenterol Nutr. 2017;64:378–84. [PubMed: 27253662]
- Chatila TA, Blaeser F, Ho N, Lederman HM, Voulgaropoulos C, Helms C, Bowcock AM. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic dysregulation syndrome. J Clin Invest. 2000;106:R75–81. [PMC free article: PMC387260] [PubMed: 11120765]
- Consonni F, Ciullini Mannurita S, Gambineri E. Atypical presentations of IPEX: expect the unexpected. Front Pediatr. 2021;9:643094. [PMC free article: PMC7892580] [PubMed: 33614561]
- Di Nunzio S, Cecconi M, Passerini L, McMurchy AN, Baron U, Turbachova I, Vignola S, Valencic E, Tommasini A, Junker A, Cazzola G, Olek S, Levings MK, Perroni L, Roncarolo MG, Bacchetta R. Wild-type FOXP3 is selectively active in CD4+CD25hi regulatory T cells of healthy female carriers of different FOXP3 mutations. Blood. 2009;114:4138–41. [PubMed: 19738030]
- Dotta F, Vendrame F. Neonatal syndromes of polyendocrinopathy. Endocrinol Metab Clin North Am. 2002;31:283–93. [PubMed: 12092451]
- Du J, Huang C, Zhou B, Ziegler SF. Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol. 2008;180:4785–92. [PubMed: 18354202]
- Du YT, Moore L, Poplawski NK, De Sousa SMC. Familial GATA6 mutation causing variably expressed diabetes mellitus and cardiac and renal abnormalities. Endocrinol Diabetes Metab Case Rep. 2019;2019:19-0022. [PMC free article: PMC6499914] [PubMed: 31051468]
- Engelhardt KR, Grimbacher B. IL-10 in humans: lessons from the gut, IL-10/IL-10 receptor deficiencies, and IL-10 polymorphisms. Curr Top Microbiol Immunol. 2014;380:1-18. [PubMed: 25004811]
- Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Allen HL, De Franco E, McDonald TJ, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy NP, Milenkovic T, Seppänen M, Lernmark Å, Mustjoki S, Otonkoski T, Kere J, Morgan NG, Ellard S, Hattersley AT. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat Genet. 2014;46:812–4. [PMC free article: PMC4129488] [PubMed: 25038750]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4:330–6. [PubMed: 12612578]
- Gajiwala KS, Burley SK. Winged helix proteins. Curr Opin Struct Biol. 2000;10:110–6. [PubMed: 10679470]
- Gambineri E, Perroni L, Passerini L, Bianchi L, Doglioni C, Meschi F, Bonfanti R, Sznajer Y, Tommasini A, Lawitschka A, Junker A, Dunstheimer D, Heidemann PH, Cazzola G, Cipolli M, Friedrich W, Janic D, Azzi N, Richmond E, Vignola S, Barabino A, Chiumello G, Azzari C, Roncarolo MG, Bacchetta R. Clinical and molecular profile of a new series of patients with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: inconsistent correlation between forkhead box protein 3 expression and disease severity. J Allergy Clin Immunol. 2008;122:1105–12.e1. [PubMed: 18951619]
- Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–5. [PubMed: 12819471]
- Gavin MA, Torgerson TR, Houston E, DeRoos P, Ho WY, Stray-Pedersen A, Ocheltree EL, Greenberg PD, Ochs HD, Rudensky AY. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A. 2006;103:6659–64. [PMC free article: PMC1458937] [PubMed: 16617117]
- Ge T, Wang YZ, Che YR, Xiao YM, Zhang T. Atypical late-onset immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome with intractable diarrhea: a case report. Front Pediatr. 2017;5:267. [PMC free article: PMC5732958] [PubMed: 29312905]
- Halabi-Tawil M, Ruemmele FM, Fraitag S, Rieux-Laucat F, Neven B, Brousse N, De Prost Y, Fischer A, Goulet O, Bodemer C. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br J Dermatol. 2009;160:645–51. [PubMed: 18795917]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–61. [PubMed: 12522256]
- Hwa V. Human growth disorders associated with impaired GH action: defects in STAT5B and JAK2. Mol Cell Endocrinol. 2021;519:111063. [PMC free article: PMC7736371] [PubMed: 33122102]
- Hwang JL, Park SY, Ye H, Sanyoura M, Pastore AN, Carmody D, Del Gaudio D, Wilson JF, Hanis CL, Liu X, Atzmon G, Glaser B, Philipson LH, Greeley SAW; T2D-Genes Consortium. FOXP3 mutations causing early-onset insulin-requiring diabetes but without other features of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr Diabetes. 2018;19:388–92. [PMC free article: PMC5918222] [PubMed: 29193502]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–42. [PubMed: 12612581]
- Kucuk ZY, Bleesing JJ, Marsh R, Zhang K, Davies S, Filipovich AH. A challenging undertaking: Stem cell transplantation for immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Allergy Clin Immunol. 2016;137:953–5.e4. [PubMed: 26559324]
- Lin Y, Xu A, Zeng C, Cheng J, Li N, Niu H, Liu L, Li X. Somatic and germline FOXP3 mosaicism in the mother of a boy with IPEX syndrome. Eur J Immunol. 2018;48:885–7. [PubMed: 29400909]
- Lopes JE, Torgerson TR, Schubert LA, Anover SD, Ocheltree EL, Ochs HD, Ziegler SF. Analysis of FOXP3 reveals multiple domains required for its function as a transcriptional repressor. J Immunol. 2006;177:3133–42. [PubMed: 16920951]
- López SI, Ciocca M, Oleastro M, Cuarterolo ML, Rocca A, de Dávila MT, Roy A, Fernández MC, Nievas E, Bosaleh A, Torgerson TR, Ruiz JA. Autoimmune hepatitis type 2 in a child with IPEX syndrome. J Pediatr Gastroenterol Nutr. 2011;53:690–3. [PubMed: 21629128]
- Louie RJ, Tan QK, Gilner JB, Rogers RC, Younge N, Wechsler SB, McDonald MT, Gordon B, Saski CA, Jones JR, Chapman SJ, Stevenson RE, Sleasman JW, Friez MJ. Novel pathogenic variants in FOXP3 in fetuses with echogenic bowel and skin desquamation identified by ultrasound. Am J Med Genet A. 2017;173:1219–25. [PMC free article: PMC5515470] [PubMed: 28317311]
- Lucas KG, Ungar D, Comito M, Bayerl M, Groh B. Submyeloablative cord blood transplantation corrects clinical defects seen in IPEX syndrome. Bone Marrow Transplant. 2007;39:55–6. [PubMed: 17115064]
- Maher MC, Hall EM, Horii KA. Generalized eczematous dermatitis and pruritus responsive to dupilumab in a patient with immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Pediatr Dermatol. 2021;38:1370-1. [PubMed: 34272772]
- McGinness JL, Bivens MM, Greer KE, Patterson JW, Saulsbury FT. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55:143–8. [PubMed: 16781310]
- Mustafa M, Ramdas N, Elhalik M, Faquih A. Transient neonatal diabetes mellitus with the rare association of nonsuppurative sialadenitis and genetic defects in 6q24. Case Rep Pediatr. 2021;2021:5901898. [PMC free article: PMC8376448] [PubMed: 34422424]
- Nademi Z, Slatter M, Gambineri E, Mannurita SC, Barge D, Hodges S, Bunn S, Thomas J, Haugk B, Hambleton S, Flood T, Cant A, Abinun M, Gennery A. Single centre experience of haematopoietic SCT for patients with immunodysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Bone Marrow Transplant. 2014;49:310–2. [PubMed: 24270390]
- Nieves DS, Phipps RP, Pollock SJ, Ochs HD, Zhu Q, Scott GA, Ryan CK, Kobayashi I, Rossi TM, Goldsmith LA. Dermatologic and immunologic findings in the immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Arch Dermatol. 2004;140:466–72. [PubMed: 15096376]
- Ochs HD, Torgerson TR. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked inheritance: model for autoaggression. Adv Exp Med Biol. 2007;601:27–36. [PubMed: 17712989]
- Ochs HD, Ziegler SF, Torgerson TR. FOXP3 acts as a rheostat of the immune response. Immunol Rev. 2005;203:156–64. [PubMed: 15661028]
- Park E, Chang HJ, Shin JI, Lim BJ, Jeong HJ, Lee KB, Moon KC, Kang HG, Ha IS, Cheong HI. Familial IPEX syndrome: different glomerulopathy in two siblings. Pediatr Int. 2015;57:e59–61. [PubMed: 25712815]
- Passerini L, Barzaghi F, Curto R, Sartirana C, Barera G, Tucci F, Albarello L, Mariani A, Testoni PA, Bazzigaluppi E, Bosi E, Lampasona V, Neth O, Zama D, Hoenig M, Schulz A, Seidel MG, Rabbone I, Olek S, Roncarolo MG, Cicalese MP, Aiuti A, Bacchetta R. Treatment with rapamycin can restore regulatory T-cell function in IPEX patients. J Allergy Clin Immunol. 2020;145:1262-71.e13. [PubMed: 31874182]
- Patey-Mariaud de Serre N, Canioni D, Ganousse S, Rieux-Laucat F, Goulet O, Ruemmele F, Brousse N. Digestive histopathological presentation of IPEX syndrome. Mod Pathol. 2009;22:95-102. [PubMed: 18820676]
- Powell BR, Buist NR, Stenzel P. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. J Pediatr. 1982;100:731–7. [PubMed: 7040622]
- Rae W, Gao Y, Bunyan D, Holden S, Gilmour K, Patel S, Wellesley D, Williams A. A novel FOXP3 mutation causing fetal alcinesia and recurrent male miscarriages. Clin Immunol. 2015;161:284–5. [PubMed: 26387632]
- Rao A, Kamani N, Filipovich A, Lee SM, Davies SM, Dalal J, Shenoy S. Successful bone marrow transplantation for IPEX syndrome after reduced-intensity conditioning. Blood. 2007;109:383–5. [PubMed: 16990602]
- Reichert SL, McKay EM, Moldenhauer JS. Identification of a novel nonsense mutation in the FOXP3 gene in a fetus with hydrops-expanding the phenotype of IPEX syndrome. Am J Med Genet A. 2016;170A:226–32. [PubMed: 26395338]
- Ren A, Yin W, Miller H, Westerberg LS, Candotti F, Park CS, Lee P, Gong Q, Chen Y, Liu C. Novel discoveries in immune dysregulation in inborn errors of immunity. Front Immunol. 2021;12:725587. [PMC free article: PMC8429820] [PubMed: 34512655]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rubio-Cabezas O, Klupa T, Malecki MT; CEED3 Consortium. Permanent neonatal diabetes mellitus--the importance of diabetes differential diagnosis in neonates and infants. Eur J Clin Invest. 2011;41:323-33. [PubMed: 21054355]
- Rubio-Cabezas O, Minton JA, Caswell R, Shield JP, Deiss D, Sumnik Z, Cayssials A, Herr M, Loew A, Lewis V, Ellard S, Hattersley AT. Clinical heterogeneity in patients with FOXP3 mutations presenting with permanent neonatal diabetes. Diabetes Care. 2009;32:111–6. [PMC free article: PMC2606841] [PubMed: 18931102]
- Ruemmele FM, Brousse N, Goulet O. Autoimmune enteropathy: molecular concepts. Curr Opin Gastroenterol. 2004;20:587–91. [PubMed: 15703687]
- Sakaguchi S. The origin of FOXP3-expressing CD4+ regulatory T cells: thymus or periphery. J Clin Invest. 2003;112:1310–2. [PMC free article: PMC228490] [PubMed: 14597756]
- Scaillon M, Van Biervliet S, Bontems P, Dorchy H, Hanssens L, Ferster A, Segers V, Cadranel S. Severe gastritis in an insulin-dependent child with an IPEX syndrome. J Pediatr Gastroenterol Nutr. 2009;49:368–70. [PubMed: 19633572]
- Seghezzo S, Bleesing JJ, Kucuk ZY. Persistent enteropathy in a toddler with a novel FOXP3 mutation and normal FOXP3 protein expression. J Pediatr. 2017;186:183-5. [PubMed: 28457527]
- Seidel MG, Boztug K, Haas OA. Immune dysregulation syndromes (IPEX, CD27 deficiency, and others): always doomed from the start? J Clin Immunol. 2016;36:6–7. [PubMed: 26661331]
- Shehab O, Tester DJ, Ackerman NC, Cowchock FS, Ackerman MJ. Whole genome sequencing identifies etiology of recurrent male intrauterine fetal death. Prenatal Diag. 2017;37:1040–5. [PubMed: 28833278]
- Sheikine Y, Woda CB, Lee PY, Chatila TA, Keles S, Charbonnier LM, Schmidt B, Rosen S, Rodig NM. Renal involvement in the immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) disorder. Pediatr Nephrol. 2015;30:1197–202. [PubMed: 25911531]
- Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol. 2007;178:320–9. [PubMed: 17182569]
- Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, Klein C, Morio T, Oksenhendler E, Picard C, Puel A, Puck J, Seppänen MRJ, Somech R, Su HC, Sullivan KE, Torgerson TR, Meyts I. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42:1473-507. [PMC free article: PMC9244088] [PubMed: 35748970]
- Tommasini A, Ferrari S, Moratto D, Badolato R, Boniotto M, Pirulli D, Notarangelo LD, Andolina M. X-chromosome inactivation analysis in a female carrier of FOXP3 mutation. Clin Exp Immunol. 2002;130:127–30. [PMC free article: PMC1906506] [PubMed: 12296863]
- Torgerson TR, Linane A, Moes N, Anover S, Mateo V, Rieux-Laucat F, Hermine O, Vijay S, Gambineri E, Cerf-Bensussan N, Fischer A, Ochs HD, Goulet O, Ruemmele FM. Severe food allergy as a variant of IPEX syndrome caused by a deletion in a noncoding region of the FOXP3 gene. Gastroenterology. 2007;132:1705–17. [PubMed: 17484868]
- Vasiljevic A, Poreau B, Bouvier R, Lachaux A, Arnoult C, Faure J, Cordier MP, Ray PF. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome and recurrent intrauterine fetal death. Lancet. 2015;385:2120. [PubMed: 26009232]
- Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. 2002;39:537–45. [PMC free article: PMC1735203] [PubMed: 12161590]
- Xavier-da-Silva MM, Moreira-Filho CA, Suzuki E, Patricio F, Coutinho A, Coutinho A, Carneiro-Sampaio M. Fetal-onset IPEX: report of two families and review of literature. Clin Immunol. 2015;156:131–40. [PubMed: 25546394]
- Yamauchi T, Takasawa K, Kamiya T, Kirino S, Gau M, Inoue K, Hoshino A, Kashimada K, Kanegane H, Morio T. Hematopoietic stem cell transplantation recovers insulin deficiency in type 1 diabetes mellitus associated with IPEX syndrome. Pediatr Diabetes. 2019;20:1035-40. [PubMed: 31322807]
- Yong PL, Russo P, Sullivan KE. Use of sirolimus in IPEX and IPEX-like children. J Clin Immunol. 2008;28:581–7. [PubMed: 18481161]
- Zhang Z, Gothe F, Pennamen P, James JR, McDonald D, Mata CP, Modis Y, Alazami AM, Acres M, Haller W, Bowen C, Döffinger R, Sinclair J, Brothers S, Zhang Y, Matthews HF, Naudion S, Pelluard F, Alajlan H, Yamazaki Y, Notarangelo LD, Thaventhiran JE, Engelhardt KR, Al-Mousa H, Hambleton S, Rooryck C, Smith KGC, Lenardo MJ. Human interleukin-2 receptor β mutations associated with defects in immunity and peripheral tolerance. J Exp Med. 2019;216:1311-27. [PMC free article: PMC6547869] [PubMed: 31040185]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubstov YP, Rudensky AY, Ziegler SF, Littman DR. TGFβ-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORγt function. Nature. 2008;453:236–40. [PMC free article: PMC2597437] [PubMed: 18368049]
Publication Details
Author Information and Affiliations
Rochester, Minnesota
Greenwood, South Carolina
Durham, North Carolina
Publication History
Initial Posting: October 19, 2004; Last Update: February 1, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Tan QKG, Louie RJ, Sleasman JW. IPEX Syndrome. 2004 Oct 19 [Updated 2024 Feb 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.