Summary
The goals of this overview on urea cycle disorders are the following:
Goal 1.
To define the urea cycle and to describe the clinical characteristics of urea cycle disorders
Goal 2.
To review the causes of urea cycle disorders and their prevalence
Goal 3.
To provide an evaluation strategy to identify the specific type and genetic cause of a urea cycle defect in a proband
Goal 4.
To review the differential diagnosis of urea cycle disorders
Goal 5.
To inform genetic risk assessment in family members of the proband
Goal 6.
To provide a brief summary of the acute management of a urea cycle disorder
Definition of the Urea Cycle and Clinical Characteristics of Urea Cycle Disorders
Definition
The urea cycle:
- Is the sole source of endogenous production of arginine, ornithine, and citrulline;
- Is the principal mechanism for the clearance of waste nitrogen resulting from protein turnover;
- Is the principal mechanism for the metabolism of other nitrogenous metabolic compounds such as adenosine monophosphate;
- Includes enzymes that overlap with the nitric oxide production pathway (ASS1 and ASL).
The urea cycle comprises the following (Figure 1) [Krebs & Henseleit 1932]:
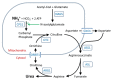
Figure 1.
The urea cycle
- Five catalytic enzymes:
- Carbamoylphosphate synthetase I (CPS1)
- Ornithine transcarbamylase (OTC)
- Argininosuccinic acid synthetase (ASS1)
- Argininosuccinic acid lyase (ASL)
- Arginase (ARG1)
- One cofactor-producing enzyme: N-acetyl glutamate synthetase (NAGS)
- Two amino acid transporters:
- Ornithine translocase (ORNT1; ornithine/citrulline carrier; solute carrier family 25, member 15)
- Citrin (aspartate/glutamate carrier; solute carrier family 25, member 13)
Urea cycle disorders (UCDs) result from inherited deficiencies in any one of the six enzymes or two transporters of the urea cycle pathway (CPS1, OTC, ASS1, ASL, ARG1, NAGS, ORNT1, or citrin).
Clinical Characteristics
Severity of the urea cycle defect is influenced by the position of the defective protein in the pathway and the severity of the defect (see Figure 1).
Severe deficiency or total absence of activity of any of the first four enzymes in the pathway (CPS1, OTC, ASS1, and ASL) or the cofactor producer (NAGS) results in the accumulation of ammonia and other precursor metabolites during the first few days of life. Because no effective secondary clearance system for ammonia exists, complete disruption of this pathway results in the rapid accumulation of ammonia and development of related symptoms.
Presentation. Individuals with complete defects normally present in the newborn period, when the immaturity of the neonatal liver accentuates defects in the urea cycle enzymes [Pearson et al 2001, Summar 2001, Summar & Tuchman 2001].
- Infants with a urea cycle disorder appear normal at birth but rapidly develop cerebral edema and the related signs of lethargy, anorexia, hyper- or hypoventilation, hypothermia, seizures, neurologic posturing, and coma.
- Because newborns are usually discharged from the hospital within one to two days after birth, the symptoms of a urea cycle disorder often develop when the child is at home and may not be recognized in a timely manner by the family and primary care physician.
The typical initial symptoms of a child with hyperammonemia are nonspecific [Summar 2001, Kölker et al 2015]:
- Failure to feed
- Loss of thermoregulation with a low core temperature
- Somnolence
Symptoms progress from somnolence to lethargy and coma.
- Abnormal posturing and encephalopathy are often related to the degree of central nervous system swelling and pressure on the brain stem [Summar 2001].
- About 50% of neonates with severe hyperammonemia may have seizures, some without overt clinical manifestations.
- Individuals with closed cranial sutures are at higher risk for rapid neurologic deterioration from the cerebral edema that results from ammonia elevation.
- Hyperventilation secondary to the effect of hyperammonemia on the brain stem, a common early finding in hyperammonemic attacks, results in respiratory alkalosis.
- Hypoventilation and respiratory arrest follow as pressure increases on the brain stem.
In milder (or partial) urea cycle enzyme deficiencies, ammonia accumulation may be triggered at almost any time of life by illness or stress (e.g., surgery, prolonged fasting, holidays, the peripartum period), resulting in multiple mild elevations of plasma ammonia concentration.
- Hyperammonemia in the milder defects is typically less severe and the symptoms more subtle than the neonatal presentation of a UCD.
- In individuals with partial enzyme deficiencies, the first recognized clinical episode may be delayed for months or years.
- Although the clinical abnormalities vary somewhat with the specific urea cycle disorder, in most the hyperammonemic episode is marked by loss of appetite, vomiting, lethargy, and behavioral abnormalities [Gardeitchik et al 2012].
- Sleep disorders, delusions, hallucinations, and psychosis may occur.
- An encephalopathic (slow-wave) EEG pattern may be observed during hyperammonemia and nonspecific brain atrophy seen subsequently on MRI.
Defects in the final enzyme in the pathway (ARG1) cause hyperargininemia, a more subtle disorder involving neurologic symptoms; however, neonatal hyperammonemia has been reported (see Arginase Deficiency).
Defects in the two amino acid transporters (ORNT1 and citrin deficiency) may both cause hyperammonemia. However, ORNT1 deficiency may also present with chronic liver dysfunction. Citrin deficiency typically only presents with hyperammonemia in adolescence or adulthood, but may present in infants with neonatal intrahepatic cholestasis, and in older children with failure to thrive.
Neurologic aspects of UCDs. Ammonia can cause brain damage through a variety of proposed mechanisms, a major component of which is cerebral edema through increased glutamine. The specific roles of ammonia, glutamate, and glutamine in cerebral edema are still under investigation [Gropman et al 2007, Lichter-Konecki 2008, Lichter-Konecki et al 2008, Albrecht et al 2010, Braissant et al 2013].
Damage resulting from acute hyperammonemia in infancy resembles that seen in hypoxic-ischemic events or stroke. The most vulnerable areas are the insular cortex, which represents deep white matter. With prolonged hyperammonemia, the parietal, occipital, and frontal regions are affected. This is best appreciated on T2-weighted MRI sequences or on diffusion tensor imaging.
Neuroimaging may be helpful in identifying affected areas of the brain. However, MRI findings may lag behind clinical changes. In fact, early imaging may be normal as some degree of injury must occur before macroscopic changes are seen on MRI.
Chronic hyperammonemia may disrupt ion-gradients and neurotransmitters, transport of metabolites, mitochondrial function, and the alpha-ketoglutarate/ glutamate/glutamine ratio.
Seizures are common in acute hyperammonemia and may result from cerebral damage. Recent findings suggest that subclinical seizures are common in acute hyperammonemic episodes, especially in neonates, and their effects on cerebral metabolism in an otherwise compromised state should be addressed (see Management, Treatment of Acute Manifestations). These seizures may be seen during the rise of glutamine even before ammonia levels are maximal [Wiwattanadittakul et al 2018].
Survival and intellectual outcome. Historically the outcome of newborns with hyperammonemia was considered poor [Brusilow 1995]. With rapid identification and current treatment strategies, survival of neonates with hyperammonemia has improved dramatically in the last few decades. See Summar [2001], Summar & Tuchman [2001], Enns et al [2007], Summar et al [2008], Tuchman et al [2008], and Krivitzky et al [2009].
More recent data from the NIH-sponsored longitudinal study on patients treated with the more recent protocols show IQ measures within a less severe range as summarized in Table 1.
Table 1.
Cognitive and Adaptive Outcome in Children with UCD Age 3-16 Years
While hyperammonemia is thought to be the main contributor to brain damage in UCDs, other factors, such as adverse effects on the nitric oxide production system [Nagamani et al 2012], may also contribute. For instance, neonates with CPS1 deficiency or OTC deficiency have more severe hyperammonemia than those with ASS1 deficiency or ASL deficiency; however, their intellectual outcomes appear similar [Ah Mew et al 2013].
In a recent study, asymptomatic female carriers of OTC deficiency demonstrated no significant differences in cognitive function compared to control participants until they were cognitively challenged with fine motor tasks, measures of executive function, and measures of cognitive flexibility [Sprouse et al 2014].
Causes of Urea Cycle Disorders (UCDs) and Prevalence
Specific Urea Cycle Disorders
N-acetylglutamate synthase deficiency (NAGS deficiency) has been described in a number of affected individuals. Symptoms mimic those of CPS1 deficiency, as CPS1 is rendered inactive in the absence of N-acetylglutamate [Caldovic et al 2003].
Carbamoylphosphate synthetase I deficiency (CPS1 deficiency) is the most severe of the urea cycle disorders. Individuals with complete CPS1 deficiency rapidly develop hyperammonemia in the newborn period. Children who are successfully rescued from crisis are chronically at risk for repeated bouts of hyperammonemia.
Ornithine transcarbamylase deficiency (OTC deficiency). Absence of OTC activity in males is as severe as CPS1 deficiency. Approximately 15% of carrier females develop hyperammonemia during their lifetime and many require chronic medical management for hyperammonemia. More recently it has been recognized that carrier females who have never had symptoms of overt hyperammonemia have deficiencies in executive function.
Citrullinemia type I (ASS1 deficiency). The hyperammonemia in this disorder can also be quite severe. Affected individuals are able to incorporate some waste nitrogen into urea cycle intermediates, which makes treatment slightly easier than in the other UCDs.
Argininosuccinic aciduria (ASL deficiency) can also present with rapid-onset hyperammonemia in the newborn period. This enzyme defect is past the point in the metabolic pathway at which all the waste nitrogen has been incorporated into the cycle. Some affected individuals develop chronic hepatic enlargement and elevation of transaminases. Liver biopsy shows enlarged hepatocytes, which may over time progress to fibrosis, the etiology of which is unclear. Affected individuals can also develop trichorrhexis nodosa, a node-like appearance of fragile hair that usually responds to arginine supplementation. Affected individuals who have never had prolonged coma nevertheless have been reported to have significant developmental disabilities [Summar 2001, Summar & Tuchman 2001, Nagamani et al 2012].
Arginase deficiency (hyperargininemia, ARG1 deficiency) is not typically characterized by rapid-onset hyperammonemia, however, some individuals present earlier with more severe symptoms [Jain-Ghai et al 2011]. Affected individuals develop progressive spasticity and can also develop tremor, ataxia, and choreoathetosis. Growth is also affected [Cederbaum et al 2004].
Ornithine translocase deficiency (ORNT1 deficiency, hyperornithinemia-hyperammonemia-homocitrullinuria syndrome) has a variable age of onset ranging from infancy to adulthood. Clinical presentation can be with chronic neurocognitive deficits, hyperammonemic crisis, or chronic liver dysfunction.
Citrin deficiency can manifest in newborns as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD), in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD), and in adults as recurrent hyperammonemia with neuropsychiatric symptoms in citrullinemia type II (CTLN2).
Prevalence of Urea Cycle Disorders
The incidence of UCDs is estimated to be at least 1:35,000 births; partial defects may make the number much higher.
Table 2.
Estimated Incidence of Individual Urea Cycle Disorders
Evaluation Strategy to Identify the Specific Type and Genetic Cause of a Urea Cycle Disorder in a Proband
The diagnosis of a urea cycle disorder (UCD) in a symptomatic individual is based on clinical, biochemical, and molecular genetic data.
Family history. A three-generation family history with attention to other relatives (particularly children) with neurologic signs and symptoms suggestive of UCD should be obtained. Documentation of relevant findings in relatives can be accomplished either through direct examination of those individuals or review of their medical records including the results of biochemical testing, molecular genetic testing, and autopsy examination. A family history consistent with X-linked inheritance suggests OTC deficiency.
Physical examination. No findings on physical examination distinguish among the eight types of urea cycle defect; however, trichorrhexis nodosa can be suggestive of ASL deficiency and progressive spasticity of the lower extremities suggestive of arginase deficiency.
Biochemical Testing
The algorithm in Figure 2 may assist with the evaluation of a newborn with hyperammonemia.
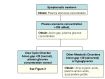
Figure 2.
Steps in the evaluation of a newborn with hyperammonemia
Plasma ammonia concentration elevation is usually the first identified laboratory abnormality in most of the urea cycle disorders. A plasma ammonia concentration of 150 μmol/L or higher associated with a normal anion gap and a normal plasma glucose concentration is a strong indication of a UCD [Summar & Tuchman 2001].
Figure 3 highlights the use of the following recommended diagnostic tests to identify the specific urea cycle disorder.
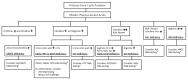
Figure 3.
Testing used in the diagnosis of urea cycle disorders * If DNA testing is not informative, enzymatic testing is available for these disorders (see Evaluation Strategy).
Quantitative plasma amino acid analysis can be used to arrive at a tentative diagnosis. (As the liver is not fully mature at birth, affected newborns often have plasma amino acid concentrations that are quite different from those in older children and adults.)
- Plasma concentration of citrulline helps discriminate between the proximal and distal urea cycle defects, as citrulline is the product of the proximal enzymes (CPS1, OTC, and NAGS) and a substrate for the distal enzymes (ASS1, ASL, ARG1).
- Plasma citrulline is either absent or present only in trace amounts in neonatal-onset CPS1 deficiency, NAGS deficiency and OTC deficiency and present in low to low-normal concentrations in late-onset disease. Plasma citrulline is also reduced in ORNT1 deficiency.
- Marked elevation in plasma citrulline concentration is seen in ASS1 deficiency.
- Moderate elevation in plasma citrulline may be observed in citrin deficiency, along with an elevated threonine/serine ratio.
- A more moderate (~2- to 5-fold) increase in plasma citrulline concentration is seen in ASL deficiency, which is also associated with high levels of argininosuccinic acid (ASA) in plasma and urine. Note: ASA is absent in unaffected individuals [Summar 2001, Summar & Tuchman 2001].
- Plasma citrulline concentration is usually normal in ARG1 deficiency.
- Plasma concentration of arginine is markedly elevated in ARG1 deficiency. It may be reduced in all other urea cycle disorders; however, in partial UCD enzyme defects, it may be normal.
- Plasma concentration of ornithine is elevated in ORNT1 deficiency, in which urine homocitrulline is also elevated. Ornithine is not elevated in OTC deficiency.
Note: Plasma concentrations of glutamine, alanine, and asparagine, which serve as storage forms of waste nitrogen, are frequently elevated.
Urinary orotic acid is measured to distinguish CPS1 deficiency or NAGS deficiency from OTC deficiency. It is normal or low in CPS1 deficiency and NAGS deficiency and significantly elevated in OTC deficiency. Note: Urinary orotic acid excretion can also be increased in argininemia (ARG1 deficiency) and citrullinemia type I (ASS1 deficiency).
Urine amino acid analysis may be used to identify the presence of urine homocitrulline, observed in ORNT1 deficiency. Additionally, ASA concentrations are higher in urine than in plasma, and therefore urine amino acid profile may be helpful when small peaks of ASA or its anhydrides are difficult to resolve on plasma amino acid analysis.
Molecular Genetic Testing
Molecular genetic testing is the primary method of diagnostic confirmation for all eight UCDs (see Table 3). Molecular testing has supplanted measurement of enzyme activity as the definitive diagnostic test. However, enzymatic testing remains available for most disorders (see Enzyme Activity), and may be helpful if DNA-based investigations are not informative.
- Serial single-gene testing can be considered if the biochemical findings indicate that mutation of a particular gene is most likely.Sequence analysis of the gene of interest is performed first, followed by gene-targeted deletion/duplication analysis if a hemizygous pathogenic variant in the case of OTC deficiency or only one or no pathogenic variant is found in the case of deficiencies of NAGS, CPS1, ASS1, ASL, ARG1, ORNT1, or citrin.
- A multigene panel that includes the eight genes discussed in this GeneReview may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene varies by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- More comprehensive genomic testing (when available) including exome sequencing and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation).
Table 3.
Urea Cycle Disorders: Molecular Genetics
Enzyme Activity
If molecular testing is uninformative, the following disorders can be diagnosed by assay of enzyme activity:
- CPS1 deficiency, NAGS deficiency, or OTC deficiency: hepatocytes
- ASL deficiency, ASS1 deficiency or ORNT1 deficiency: fibroblasts
- ARG1 deficiency: erythrocytes
Newborn Screening
Current newborn screening panels in the United States using tandem mass spectrometry detect abnormal concentrations of analytes associated with ASS1 deficiency, and ASL deficiency in all states; however, the sensitivity and specificity of screening for these disorders varies by state.
Other disorders are screened for in some states only (see www.newsteps.org for information on disorders screened for by state). For example:
- CPS1 deficiency is screened for in Florida, Maine, Massachusetts, Mississippi, New Hampshire, Pennsylvania, Rhode Island, and Vermont.
- OTC deficiency is screened for in Connecticut, Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont, and is likely to be detected in Kentucky and Utah.
- Arginase deficiency is screened for in 35 states and likely to be detected in four more.
- Citrin deficiency is screened for in 36 states and likely to be detected in 13 more.
IMPORTANT NOTE: The sensitivity and specificity of screening for these disorders varies by state, and current newborn metabolic screen cannot reliably identify all cases of these disorders. Additionally, even for UCDs detectable by newborn screening, neonates are often symptomatic prior to availability of the screening results; thus, a high level of clinical suspicion on the part of health care providers is necessary.
Differential Diagnosis of Urea Cycle Disorders
A number of other disorders that perturb the liver can result in hyperammonemia and mimic the effects of a urea cycle disorder. These include diseases of the liver and biliary tract, use of certain medications, and a number of other genetic disorders (see Table 4).
Diseases of the liver and biliary tract
- Herpes simplex virus infection
- Vascular bypass of the liver
- Biliary atresia
- Acute liver failure
Medications
- Valproic acid
- Cyclophosphamide
- 5-pentanoic acid
Table 4.
Genetic Disorders to Consider in the Differential Diagnosis of a Urea Cycle Disorder
Genetic Risk Assessment in Family Members
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
OTC deficiency is inherited in an X-linked manner.
The rest of the urea cycle disorders (deficiencies of NAGS, CPS1, ASS1, ASL, ARG1, ORNT1, and citrin) are inherited in an autosomal recessive manner.
X-Linked Inheritance – Risk to Family Members
Parents of a male proband
- The father of an affected male will not have the disorder nor will he be hemizygous for the OTC pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected child and no other affected relatives and if the OTC pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism. Germline mosaicism has been reported in OTC deficiency [Bowling et al 1999]; however, because the frequency is not known the general background risk for germline mosaicism – 3%-4% – should be used.
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier) or the affected male may have a de novo pathogenic variant, in which case the mother is not a carrier. About 26% of affected males had a de novo pathogenic variant in one study [Rüegger et al 2014].
Parents of a female proband
- A female proband may have inherited the OTC pathogenic variant from either her mother or her father, or the pathogenic variant may be de novo.
- Detailed evaluation of the parents and review of the extended family history may help distinguish probands with a de novo pathogenic variant from those with an inherited pathogenic variant. Molecular genetic testing of the mother (and possibly the father) can determine if the pathogenic variant was inherited.
Sibs of a proband. The risk to sibs depends on the genetic status of the parents:
- If the mother of the proband has an OTC pathogenic variant, the chance of transmitting it in each pregnancy is 50%.
- Males who inherit the pathogenic variant will be affected.
- Females who inherit the pathogenic variant may or may not develop clinical findings related to the disorder.
- If the father of a female proband has the OTC pathogenic variant, all of the proband's female sibs and none of the male sibs will inherit the pathogenic variant.
- If the proband represents a simplex case (i.e., a single occurrence in a family) and if the OTC pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is low, but greater than that of the general population because of the possibility of maternal germline mosaicism.
For additional specific genetic counseling considerations see OTC Deficiency: Heterozygous Females, Evaluation of Relatives at Risk, and Risk to Family Members.
Autosomal Recessive Inheritance – Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an affected individual are obligate heterozygotes (carriers) for one pathogenic variant.
Carrier detection. Carrier testing for at-risk relatives requires prior identification of the urea cycle disorder-related pathogenic variants in the family.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Connecting Families - Urea Cycle Disorders (UCD) FoundationPhone: 918-490-3055
- MedlinePlus
- MedlinePlus
- MedlinePlus
- MedlinePlus
- MedlinePlus
- MedlinePlus
- National Urea Cycle Disorders FoundationPhone: 626-578-0833
- Urea Cycle Disorders ConsortiumPhone: 202-306-6489Email: [email protected]
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- Newborn Screening in Your StateHealth Resources & Services Administration
- European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD)
- National Urea Cycle Disorders Foundation International Patient RegistryEmail: [email protected]
- Urea Cycle Disorders Consortium RegistryChildren's National Medical Center
Acute Management of a Urea Cycle Disorder
The extent of disease in an individual diagnosed with a urea cycle disorder can be estimated by the rapidity of onset of neurologic symptoms, the degree to which the brain is involved, and to a lesser extent the plasma ammonia concentration.
The NIH-funded Urea Cycle Disorders Consortium provides expert diagnosis and treatment of urea cycle disorders as well as clinical and therapeutic studies.
Once a diagnosis of a UCD is made, treatment of acute manifestations can be started.
Subsequent treatment should be tailored to the specific urea cycle disorder. See:
- Ornithine Transcarbamylase Deficiency (OTC deficiency);
- Citrullinemia Type I (ASS1 deficiency);
- Argininosuccinic Aciduria (ASL deficiency);
- Arginase Deficiency (hyperargininemia, ARG1 deficiency);
- Ornithine Translocase Deficiency (ORNT1 deficiency, hyperornithinemia-hyperammonemia-homocitrullinuria syndrome);
For NAGS deficiency see Ah Mew & Caldovic [2011].
Chronic management of CPS1 deficiency is similar to that of OTC deficiency; however, some patients with CPS1 deficiency may also benefit from therapy with oral N-carbamylglutamate [Diez-Fernandez et al 2013, Ah Mew et al 2014].
Treatment of Acute Manifestations
Care of an individual with a urea cycle disorder should be provided by a team coordinated by a metabolic specialist in a tertiary care center.
In the acute phase, the mainstays of treatment are the following:
Rapidly return plasma ammonia concentrations to normal physiologic levels. This is necessary even without a definitive diagnosis, given the toxic effect of elevated plasma ammonia concentration. The best way to reduce plasma ammonia concentration quickly is by dialysis. The faster the flow rate, the faster the clearance. The method employed depends on the affected individual's circumstances and available resources. In general, the best choice for an individual patient is whatever method the local treating team is most comfortable with and can implement most quickly. The various renal replacement modalities are reviewed by Gupta et al [2016].
- The fastest method is use of pump-driven dialysis, in which an extra corporeal membrane oxygenation (ECMO) pump is used to drive a hemodialysis machine.
- Intermittent hemofiltration (both arteriovenous and venovenous) and hemodialysis are more likely to be available than ECMO-driven dialysis.
- Continuous renal replacement therapies have lower clearance than intermittent dialysis, but are less prone to interruption and may be better tolerated in sick neonates.
- Clearance with peritoneal dialysis is substantially lower than with hemodialysis; therefore, hemodialysis (if available) is typically preferred. However, published outcome data are limited.
- Intermittent dialysis can usually be discontinued when plasma ammonia concentration falls below 150 µmol/L, but may vary based on clinical evaluation by a clinician experienced in the treatment of metabolic disease. Affected individuals often experience a "rebound" hyperammonemia that may require further dialysis. This may be attenuated with the use of continuous renal replacement therapies following intermittent HD [Gupta et al 2016].
Perform pharmacologic interventions to allow alternative pathway excretion of excessive nitrogen (see Table 5).
- Nitrogen scavenger therapy (sodium phenylacetate and sodium benzoate) is available as an intravenous infusion for acute management and an oral preparation for long-term maintenance.
- Deficient urea cycle intermediates need to be replaced depending on the diagnosis; these can include arginine (IV infusion) and/or citrulline (oral preparation).Note: Continuous arginine hydrochloride (HCl) infusion requires central access, as extravasion from a peripheral IV has on multiple occasions resulted in severe cutaneous necrosis.
- Sodium phenylacetate and sodium benzoate can be infused through a peripheral IV; however, central access is preferred.
- In persons with NAGS deficiency and in some with CPS1 deficiency, replacement of n-acetylglutamate with the analog molecule carbamyl glutamate (Carbaglu®) can improve the clinical symptoms or in NAGS deficiency can be almost curative. This compound is available in the US and should be added to the treatment regimen in a patient without a clear diagnosis at initial presentation. Dosing in adults and children is 100 mg/kg/day to 250 mg/kg/day divided into two to four doses. The only form currently available is an oral preparation; thus, administration of the medication by nasogastric/jejunal tube is necessary in the treatment of acute manifestations.
Table 5.
IV Ammonia Scavenger Therapy Protocol
Treat catabolic state with calories from glucose, fats, and essential amino acids. The introduction of nutrition support in the following manner is necessary for patients on dialysis or hemofiltration in order to resolve the catabolic state while avoiding overuse of enteral feeds.
- Complete restriction of protein should not exceed 12-24 hours because depletion of essential amino acids results in protein catabolism and nitrogen release. Frequent (often daily) quantitative assessments of plasma amino acid concentrations can help optimize nutritional management by allowing the clinician to maintain adequate levels of essential amino acids without having to provide excess nitrogen. Maintenance of appropriate levels of essential amino acids is necessary to reverse the typical catabolic state because most acutely ill patients either present with essential amino acid deficiency or become deficient quickly.
- Enteral nutrition is preferred; however, intravenous (total parenteral nutrition) is an option if the patient is so clinically unstable that adequate enteral intake is impossible. Intolerance of enteral feeding should not lead to further depletion of essential amino acids.
- The placement of a nasogastric/jejunal tube at admission is warranted for slow drip administration of solutions of essential amino acids and infant formulas and administration of cofactors like carbamyl glutamate (analog of n-acetylglutamate).
- Multiple other strategies to combat catabolism can be used, including low-dose continuous infusion of insulin with maintenance of adequate glucose delivery by high continuous delivery of carbohydrate-containing fluids; however, caution is advised since patients are often exquisitely sensitive to either the glucose or insulin.
Reduce the risk for neurologic damage
- Use intravenous fluids (≥10% dextrose with appropriate electrolytes) for physiologic stabilization.
- Use cardiac pressors as necessary while avoiding overhydration.
- Consider the use of continuous bedside EEG to detect subclinical seizures. Patients in deep coma may have subclinical seizures that are non-convulsive and therefore not apparent.
Note: In patients with prolonged hyperammonemic coma and evidence of severe neurologic damage, the relative risks versus benefits of all the treatments discussed above should be considered on an individual basis.
Chapter Notes
Author History
Nicholas Ah Mew, MD (2014-present)
Kimberly A Chapman, MD, PhD (2011-present)
Andrea Gropman, MD (2011-present)
Brendan C Lanpher, MD (2011-present)
Uta Lichter-Konecki, MD, PhD; Columbia University (2011-2014)
Kara L Simpson, MS, CGC (2014-present)
Marshall L Summar, MD (2003-present)
Mendel Tuchman, MD; Children's National Medical Center (2003-2005)
Revision History
- 22 June 2017 (ha) Comprehensive update posted live
- 11 September 2014 (me) Comprehensive update posted live
- 1 September 2011 (me) Comprehensive update posted live
- 11 August 2005 (me) Comprehensive update posted live
- 29 April 2003 (me) Overview posted live
- 29 January 2001 (mls) Original submission
References
Published Guidelines / Consensus Statements
- Alfadhel M, Mutairi FA, Makhseed N, Jasmi FA, Al-Thihli K, Al-Jishi E, AlSayed M, Al-Hassnan ZN, Al-Murshedi F, Häberle J, Ben-Omran T. Guidelines for acute management of hyperammonemia in the Middle East region. Ther Clin Risk Manag. 2016;12:479-87. [PubMed]
- Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M, Karall D, Martinelli D, Crespo PS, Santer R, Servais A, Valayannopoulos V, Lindner M, Rubio V, Dionisi-Vici C. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012;7:32. [PubMed]
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001;138:S30-9. [PubMed]
- Urea Cycle Disorders Conference Group. Consensus statement from a conference for the management of patients with urea cycle disorders. J Pediatr. 2001;138:S1-5. [PubMed]
Literature Cited
- Ah Mew N, Caldovic L. N-acetylglutamate synthase deficiency: an insight into the genetics, epidemiology, pathophysiology, and treatment. Appl Clin Genet. 2011;4:127-35. [PMC free article: PMC3681184] [PubMed: 23776373]
- Ah Mew N, Krivitzky L, McCarter R, Batshaw M, Tuchman M, et al. Clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ. J Pediatr. 2013;162:324-9.e1. [PMC free article: PMC4440324] [PubMed: 22901741]
- Ah Mew N, McCarter R, Daikhin Y, Lichter-Konecki U, Nissim I, Yudkoff M, Tuchman M. Augmenting ureagenesis in patients with partial carbamyl phosphate synthetase 1 deficiency with N-carbamyl-L-glutamate. J Pediatr. 2014;165:401-3 [PMC free article: PMC4111993] [PubMed: 24880889]
- Albrecht J, Zielińska M, Norenberg MD. Glutamine as a mediator of ammonia neurotoxicity: A critical appraisal. Biochem Pharmacol. 2010;80:1303-8. [PMC free article: PMC4714775] [PubMed: 20654582]
- Braissant O, McLin VA, Cudalbu C. Ammonia toxicity to the brain. J Inherit Metab Dis. 2013;36:595-612. [PubMed: 23109059]
- Bowling F, McGown I, McGill J, Cowley D, Tuchman M. Maternal gonadal mosaicism causing ornithine transcarbamylase deficiency. Am J Med Genet. 1999;85:452-4. [PubMed: 10405441]
- Brusilow SW. Urea cycle disorders: clinical paradigm of hyperammonemic encephalopathy. Prog Liver Dis. 1995;13:293–309. [PubMed: 9224507]
- Caldovic L, Morizono H, Panglao MG, Cheng SF, Packman S, Tuchman M. Null mutations in the N-acetylglutamate synthase gene associated with acute neonatal disease and hyperammonemia. Hum Genet. 2003;112:364–8. [PubMed: 12594532]
- Cederbaum SD, Yu H, Grody WW, Kern RM, Yoo P, Iyer RK. Arginases I and II: do their functions overlap? Mol Genet Metab. 2004;81:S38–44. [PubMed: 15050972]
- Diez-Fernandez C, Martínez AI, Pekkala S, Barcelona B, Pérez-Arellano I, Guadalajara AM, Summar M, Cervera J, Rubio V. Molecular characterization of carbamoyl-phosphate synthetase (CPS1) deficiency using human recombinant CPS1 as a key tool. Hum Mutat. 2013;34:1149-59. [PubMed: 23649895]
- Enns GM, Berry SA, Berry GT, Rhead WJ, Brusilow SW, Hamosh A. Survival after treatment with phenylacetate and benzoate for urea-cycle disorders. N Engl J Med. 2007;356:2282–92. [PubMed: 17538087]
- Gardeitchik T, Humphrey M, Nation J, Boneh A. Early clinical manifestations and eating patterns in patients with urea cycle disorders. J Pediatr. 2012;161:328-32. [PubMed: 22424941]
- Gropman AL, Summar M, Leonard JV. Neurological implications of urea cycle disorders. J Inherit Metab Dis. 2007;30:865-79 [PMC free article: PMC3758693] [PubMed: 18038189]
- Gupta S, Fenves AZ, Hootkins R. The role of RRT in hyperammonemic patients. Clin J Am Soc Nephrol. 2016;11:1872-8 [PMC free article: PMC5053785] [PubMed: 27197910]
- Jain-Ghai S, Nagamani SC, Blaser S, Siriwardena K, Feigenbaum A. Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported? Mol Genet Metab. 2011;104:107-11. [PMC free article: PMC3171515] [PubMed: 21802329]
- Kobayashi K, Iijima M, Ushikai M, Ikeda S, Saheki T. Citrin deficiency. J Jpn Pediatr Soc. 2006;110:1047–59.
- Kölker S, Garcia-Cazorla A, Valayannopoulos V, Lund AM, Burlina AB, Sykut-Cegielska J, Wijburg FA, Teles EL, Zeman J, Dionisi-Vici C, Barić I, Karall D, Augoustides-Savvopoulou P, Aksglaede L, Arnoux JB, Avram P, Baumgartner MR, Blasco-Alonso J, Chabrol B, Chakrapani A, Chapman K, I Saladelafont EC, Couce ML, de Meirleir L, Dobbelaere D, Dvorakova V, Furlan F, Gleich F, Gradowska W, Grünewald S, Jalan A, Häberle J, Haege G, Lachmann R, Laemmle A, Langereis E, de Lonlay P, Martinelli D, Matsumoto S, Mühlhausen C, de Baulny HO, Ortez C, Peña-Quintana L, Ramadža DP, Rodrigues E, Scholl-Bürgi S, Sokal E, Staufner C, Summar ML, Thompson N, Vara R, Pinera IV, Walter JH, Williams M, Burgard P. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J Inherit Metab Dis. 2015;38:1041-57 [PubMed: 25875215]
- Krebs HA, Henseleit K. Untersuchungen uber die harnstoffbildung im tierkorper. Hoppe-Seyler's Z Physiol Chem. 1932;210:325-32.
- Krivitzky L, Babikian T, Lee HS, Thomas NH, Burk-Paull KL, Batshaw ML. Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders. Pediatr Res. 2009;66:96–101. [PMC free article: PMC2746951] [PubMed: 19287347]
- Lee HC, Mak CM, Lam CW, Yuen YP, Chan AO, Shek CC, Siu TS, Lai CK, Ching CK, Siu WK, Chen SP, Law CY, Tai HL, Tam S, Chan AY. Analysis of inborn errors of metabolism: disease spectrum for expanded newborn screening in Hong Kong. Chin Med J (Engl). 2011;124:983–9. [PubMed: 21542954]
- Lichter-Konecki U. Profiling of astrocyte properties in the hyperammonemic brain: Shedding new light on the pathophysiology of the brain damage in hyperammonemia. J Inherit Metab Dis. 2008;31:492–502. [PubMed: 18683079]
- Lichter-Konecki U, Mangin JM, Gordish-Dressman H, Hoffman EP, Gallo V. Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo. Glia. 2008;56:365–77. [PMC free article: PMC4116685] [PubMed: 18186079]
- Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y, Liu TT, Chiang SH, Hsiao KJ, Lau YL, Tsui LC, Lee DH, Saheki T. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency. J Hum Genet. 2005;50:338–46. [PubMed: 16059747]
- Nagamani SC, Campeau PM, Shchelochkov OA, Premkumar MH, Guse K, Brunetti-Pierri N, Chen Y, Sun Q, Tang Y, Palmer D, Reddy AK, Li L, Slesnick TC, Feig DI, Caudle S, Harrison D, Salviati L, Marini JC, Bryan NS, Erez A, Lee B. Nitric-oxide supplementation for treatment of long-term complications in argininosuccinic aciduria. Am J Hum Genet. 2012;90:836-46. [PMC free article: PMC3376491] [PubMed: 22541557]
- Pearson DL, Dawling S, Walsh WF, Haines JL, Christman BW, Bazyk A, Scott N, Summar ML. Neonatal pulmonary hypertension--urea-cycle intermediates, nitric oxide production, and carbamoyl-phosphate synthetase function. N Engl J Med. 2001;344:1832–8. [PubMed: 11407344]
- Rüegger CM, Lindner M, Ballhausen D, Baumgartner MR, Beblo S, Das A, Gautschi M, Glahn EM, Grünert SC, Hennermann J, Hochuli M, Huemer M, Karall D, Kölker S, Lachmann RH, Lotz-Havla A, Möslinger D, Nuoffer JM, Plecko B, Rutsch F, Santer R, Spiekerkoetter U, Staufner C, Stricker T, Wijburg FA, Williams M, Burgard P, Häberle J. Cross-sectional observational study of 208 patients with non-classical urea cycle disorders. J Inherit Metab Dis. 2014;37:21–30 [PMC free article: PMC3889631] [PubMed: 23780642]
- Sprouse C, King J, Helman G, Pacheco-Colón I, Shattuck K, Breeden A, Seltzer R, VanMeter JW, Gropman AL. Investigating neurological deficits in carriers and affected patients with ornithine transcarbamylase deficiency. Mol Genet Metab. 2014;113:136-41. [PMC free article: PMC4458385] [PubMed: 24881970]
- Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr. 2001;138:S30–9. [PubMed: 11148547]
- Summar M, Tuchman M. Proceedings of a consensus conference for the management of patients with urea cycle disorders. J Pediatr. 2001;138:S6–10. [PubMed: 11148544]
- Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year, multicentre study of acute hyperammonaemic episodes. Acta Paediatr. 2008;97:1420–5. [PMC free article: PMC2675643] [PubMed: 18647279]
- Summar ML, Koelker S, Freedenberg D, Le Mons C, Ha J, Lee HS, Kirmse B; European Registry and Network for Intoxication Type Metabolic Diseases (E-IMD). The incidence of urea cycle disorders. Mol Genet Metab. 2013;110:179-80. [PMC free article: PMC4364413] [PubMed: 23972786]
- Tabata A, Sheng J-S, Ushikai M, Song Y-Z, Gao H-Z, Lu Y-B, Okumura F, Iijima M, Mutoh K, Kishida S, Saheki T, Kobayashi K. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency. J Hum Genet. 2008;53:534–45. [PubMed: 18392553]
- Tuchman M, Lee B, Lichter-Konecki U, Summar ML, Yudkoff M, Cederbaum SD, Kerr DS, Diaz GA, Seashore MR, Lee HS, McCarter RJ, Krischer JP, Batshaw ML. Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network; Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol Genet Metab. 2008;94:397–402. [PMC free article: PMC2640937] [PubMed: 18562231]
- Wiwattanadittakul N, Prust M, Gaillard WD, Massaro A, Vezina G, Tsuchida TN, Gropman AL. The utility of EEG monitoring in neonates with hyperammonemia due to inborn errors of metabolism. Mol Genet Metab. 2018;125:235-40. [PubMed: 30197275]
Publication Details
Author Information and Affiliations
Children’s National Health System
Washington, DC
Children’s National Health System
Washington, DC
Division of Neurology
Children’s National Health System
Washington, DC
Children’s National Health System
Washington, DC
Children’s National Health System
Washington, DC
Publication History
Initial Posting: April 29, 2003; Last Update: June 22, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Ah Mew N, Simpson KL, Gropman AL, et al. Urea Cycle Disorders Overview. 2003 Apr 29 [Updated 2017 Jun 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


