X-Linked Congenital Stationary Night Blindness
Synonym: X-Linked CSNB
Ian M MacDonald, MD, CM
Departments of Ophthalmology and Medical Genetics
University of Alberta
Edmonton, Alberta, Canada
Stephanie Hoang, MSc
Alberta Health Services
Edmonton, Alberta, Canada
Sari Tuupanen, PhD
Blueprint Genetics
Helsinki, Finland
Initial Posting: January 16, 2008; Last Update: July 3, 2019.
Estimated reading time: 18 minutes
Summary
Clinical characteristics.
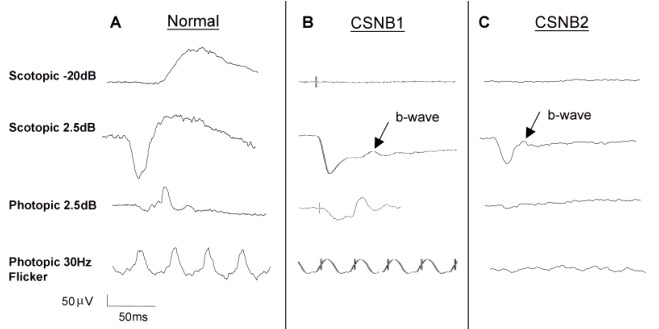
X-linked congenital stationary night blindness (CSNB) is characterized by non-progressive retinal findings of reduced visual acuity ranging from 20/30 to 20/200; defective dark adaptation; refractive error, most typically myopia ranging from low (-0.25 diopters [D] to -4.75 D) to high (≥-10.00 D) but occasionally hyperopia; nystagmus; strabismus; normal color vision; and normal fundus examination. Characteristic ERG findings can help distinguish between complete X-linked CSNB and incomplete X-linked CSNB.
Diagnosis/testing.
The diagnosis of X-linked CSNB is established in a male proband with characteristic clinical and electroretinogram (ERG) findings and a family history consistent with X-linked inheritance. Identification of a hemizygous pathogenic variant in CACNA1F or NYX by molecular genetic testing can confirm the diagnosis if clinical features are inconclusive. The diagnosis of X-linked CSNB may be established in a female proband with ERG findings suggestive of X-linked CSNB and identification of a heterozygous or biallelic pathogenic variant in CACNA1F or NYX by molecular genetic testing.
Management.
Treatment of manifestations: Glasses or contact lenses to treat refractive error (myopia or hyperopia); conventional strabismus surgery may be required to improve binocularity or head posture.
Surveillance: At a young age yearly eye examinations with refraction to identify and treat myopia as early as possible.
Agents/circumstances to avoid: Reduced visual acuity and difficulties seeing at night may preclude driving a car or restrict the class of driving license.
Genetic counseling.
By definition, X-linked CSNB is inherited in an X-linked manner. The father of an affected male will not have X-linked CSNB nor will he be hemizygous for the pathogenic variant. If the mother of the proband is a carrier, the chance of transmitting the pathogenic variant in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be carriers and will usually not be affected. Males with X-linked CSNB will pass the pathogenic variant to all of their daughters and none of their sons. Carrier testing for at-risk relatives and prenatal testing for a pregnancy at increased risk are possible for families in which the pathogenic variant has been identified.
Diagnosis
Suggestive Findings
Males. X-linked congenital stationary night blindness (CSNB) should be suspected in a male
proband with the following characteristic clinical and electroretinogram (ERG) findings characteristicec of complete X-linked CSNB or incomplete X-linked CSNB (see Table 1):
Characteristic clinical findings:
Reduced visual acuity
Night blindness
Myopia
Nystagmus (not universal) and strabismus (50%-70%)
Normal color vision
Normal fundus examination
Family history consistent with X-linked inheritance
Characteristic findings on ERG examination:
Table 1.
Electroretinogram Findings in Complete and Incomplete X-Linked Congenital Stationary Night Blindness
View in own window
| ERG Finding | Complete (NYX X-linked CSNB) | Incomplete (CACNA1F X-linked CSNB) |
|---|
| Scotopic rod b-wave | Severely reduced or absent | Reduced |
| Mixed scotopic a-wave | Normal | Slightly reduced |
| Mixed scotopic b-wave | Reduced | Reduced |
| Scotopic OP | Absent | Slightly reduced |
| Photopic a-wave | Normal, slightly reduced, sawtooth (square) shaped | Reduced |
| Photopic b-wave | Slightly reduced | Reduced |
| Photopic OP | Lost, except for OP4 | All OPs are lost. |
| 30-Hz flicker | Normal / slightly reduced | Reduced w/double peak |
OP = oscillatory potential
Note: Pupillary responses have been described in the literature and in textbooks as "paradoxic" (i.e., miosis of pupils when lights are turned off, as opposed to dilation). This description predates genotyping. In 17 individuals with incomplete X-linked CSNB ages five to 51 years examined by one of the authors, none clearly demonstrated a paradoxic pupillary response. Further clarification of the presence or absence of this phenomenon in individuals with X-linked CSNB may require measurement with pupillometry.
Heterozygous females. X-linked CSNB should be suspected in a female
proband with the following ERG findings (observed in some heterozygous females):
Establishing the Diagnosis
Male proband. The diagnosis of X-linked CSNB is established in a male
proband with the characteristic clinical and ERG findings described in Suggestive Findings and a family history consistent with X-linked inheritance. Identification of a hemizygous pathogenic variant in CACNA1F or NYX by molecular genetic testing can confirm the diagnosis if clinical features are inconclusive (see Table 2).
Female proband. The diagnosis of X-linked CSNB may be established in a female
proband with ERG findings suggestive of X-linked CSNB and a heterozygous or biallelic pathogenic variant in CACNA1F or NYX identified by molecular genetic testing (see Table 2).
Molecular Genetic Testing
Approaches can include serial single-gene testing (recommended in individuals with a clear family history consistent with X-linked inheritance) or a multigene panel (recommended in individuals without a clear family history consistent with X-linked inheritance).
Serial single-gene testing. For individuals with a clear family history consistent with X-linked inheritance, ERG findings can be used to direct molecular genetic testing to the appropriate gene (see Table 1).
Sequence analysis of NYX should be performed first in individuals with ERG findings consistent with complete X-linked CSNB to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
Sequence analysis of CACNA1F should be performed first in individuals with ERG findings consistent with incomplete X-linked CSNB to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. If no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
Note: Targeted analysis for the CACNA1F founder variant c.3167_3168dupC can be performed first in individuals of Dutch-German Mennonite ancestry [Bech-Hansen et al 1998, Boycott et al 2000].
Multigene panel. For individuals without a clear family history consistent with X-linked inheritance, a CSNB multigene panel that includes CACNA1F, NYX, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 2).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Table 2.
Molecular Genetic Testing Used in X-Linked Congenital Stationary Night Blindness
View in own window
| Gene 1, 2 | Proportion of X-Linked CSNB Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants 3 Detectable by Method |
|---|
Sequence
analysis 4 | Gene-targeted deletion/duplication analysis 5 |
|---|
|
CACNA1F
| 55% 6, 7 | >98% 6, 7 | 5 reported 8 |
|
NYX
| 45% 6, 9 | >99% 6, 9, 10 | 4 reported 10 |
- 1.
Genes are listed in alphabetic order.
- 2.
- 3.
- 4.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
- 7.
- 8.
- 9.
- 10.
Clinical Characteristics
Clinical Description
X-linked congenital stationary night blindness (CSNB) is a congenital non-progressive retinal disorder characterized by defective night vision, reduced visual acuity, myopia, nystagmus, and strabismus that primarily affects males.
Males
Reduced visual acuity. Vision is reduced in all affected males in the range of 20/30 (6/9; log MAR 0.1) to 20/200 (6/60; log MAR 1.0).
Defective dark adaptation. Night blindness is a subjective finding. Individuals with NYX X-linked CSNB generally report severe night blindness. Individuals with CACNA1F X-linked CSNB do not uniformly report severe night blindness.
Myopia may range from low (-0.25 diopters [D] to -4.75 D) to high (≥ -10.00 D) [Boycott et al 2000, Allen et al 2003]. A few affected individuals have hyperopia.
Nystagmus and strabismus are reported in 50%-70% of affected individuals [Boycott et al 2000, Allen et al 2003]. Transient head posture with nystagmus was noted in the first two years of life in eight individuals with CACNA1F X-linked CSNB and one with NYX X-linked CSNB [Simonsz et al 2009].
In a large Mennonite cohort with incomplete (i.e., CACNA1F) X-linked CSNB, at least one of the following was not present in 72% of individuals: myopia, nystagmus, or night blindness [Boycott et al 2000].
Normal color vision is present in most individuals. Individuals with a severe X-linked CSNB may show mild color vision deficits.
Normal fundus examination is present in most individuals, although those with high myopia may show myopic degeneration.
Females
Phenotype Correlations by Gene
NYX pathogenic variants are associated with the complete form of X-linked CSNB (see Table 1) [Bech-Hansen et al 2000, Pusch et al 2000]. Individuals with NYX X-linked CSNB generally report severe night blindness.
CACNA1F pathogenic variants are associated with the incomplete form of X-linked CSNB [Bech-Hansen et al 1998, Strom et al 1998] (see Table 1). Individuals with CACNA1F X-linked CSNB do not uniformly report severe night blindness.
Genotype-Phenotype Correlations
No genotype-phenotype correlations are known.
Penetrance
Penetrance of X-linked CSNB is probably 100%, but expressivity is variable [Boycott et al 2000]; individuals with mild presentations may be missed if electroretinography is not performed.
Nomenclature
X-linked CSNB has in the past been referred to as Schubert-Bornschein CSNB, which is a reference to the characteristic "negative" waveform (a-wave larger than the b-wave in response to a bright flash in the scotopic state) of the ERG seen in both X-linked forms of CSNB [Schubert & Bornschein 1952].
The terms "CSNB1" and "CSNB2" are sometimes used as abbreviations for complete and incomplete CSNB irrespective of the mode of inheritance; originally the terms referred to the two X-linked entities of CSNB.
Differential Diagnosis
Only a few conditions may initially be confused with the X-linked form of congenital stationary night blindness (CSNB).
Table 4.
Disorders to Consider in the Differential Diagnosis of X-Linked Congenital Stationary Night Blindness
View in own window
| Fundus 1 | DiffDx Disorder | Gene(s) | MOI | Clinical Features of DiffDx Disorder |
|---|
| Overlapping w/X-linked CSNB | Distinguishing from X-linked CSNB |
|---|
|
Normal fundus
| CSNB (non-X-linked) | See footnote 2. | AR
AD | Most autosomal CSNB is clinically identical, w/exception of AD CSNB, Nougaret type. |
|
| Blue cone monochromacy (OMIM 303700) |
OPN1LW
OPN1MW
| XL |
|
|
|
FRMD7-related infantile nystagmus
|
FRMD7
| XL |
|
Normal ERG Normal VEP Normal foveal contour
|
|
Abnormal fundus
| Ocular albinism type I (OMIM 300500) |
OA1
| XL |
|
Iris transillumination Foveal hypoplasia Heterozygous females have fundus signs (hypopigmentation of retinal pigment epithelium). 4 Absence of selective ↓ in amplitude of b-wave on ERG VEP responses show propensity for more crossing fibers than expected at level of chiasm.
|
|
X-linked juvenile retinoschisis
|
RS1
| XL |
| Fundus exam shows foveal schisis or foveal findings in virtually all affected males & ~50% have areas of peripheral retinoschisis. |
| Oguchi disease 5 (OMIM 258100, 613411) |
SAG
GRK1
| AR | Non-progressive | Fundus has abnormal color that becomes normal w/prolonged dark adaptation (Mizuo phenomenon). 6 |
| Fundus albipunctatus 7 (OMIM 136880) |
RDH5
RLBP1
| AR
AD | Non-progressive |
Fundus shows discretely scattered white retinal dots. ERG, when recorded under standard conditions, shows selective ↓ in b-wave that normalizes w/prolonged dark adaptation. 6
|
AD = autosomal dominant; AR = autosomal recessive; DiffDx = differential diagnosis; ERG = electroretinogram; MOI = mode of inheritance; VEP = visual evoked potential; XL = X-linked
- 1.
X-linked CSNB is characterized by a normal fundus.
- 2.
- 3.
- 4.
- 5.
Oguchi disease is a form of CSNB reported in the Japanese.
- 6.
- 7.
Fundus albipunctatus is a form of CSNB.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with X-linked congenital stationary night blindness (CSNB), the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Ophthalmologic examination
Electroretinography
Dark adaptation (optional)
Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Coincident high myopia or hyperopia can be managed with glasses or contact lenses.
Occasionally, a boy with X-linked CSNB may adopt a cosmetically unacceptable or functionally awkward head posture to dampen the degree of nystagmus in a particular position of gaze (the so-called "null point"). In some instances the position of gaze for the null point may be shifted to a better functional range by carefully planned strabismus surgery.
Surveillance
Regular (yearly) eye examinations are recommended with refraction at a young age to monitor for the development of myopia.
Agents/Circumstances to Avoid
Reduced visual acuity and difficulties seeing at night may preclude driving a car or restrict the class of driving license.
Evaluation of Relatives at Risk
For infants identified with high myopia, unusual head posture, or nystagmus and a family history of CSNB, ophthalmic examination and molecular genetic testing may confirm the diagnosis of CSNB, obviating the need for neuroimaging or clinical electrophysiologic testing under sedation or general anesthesia.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
By definition, X-linked congenital stationary night blindness (CSNB) is inherited in an X-linked manner.
Risk to Family Members
Parents of a proband
The father of an affected male will not have X-linked CSNB nor will he be hemizygous for the CACNA1F or NYX pathogenic variant; therefore, he does not require further evaluation/testing.
In a family with more than one affected male, the mother of an affected male is an obligate heterozygote (carrier). Note: If a woman has more than one affected child and no other affected relatives and if the CACNA1F or NYX pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism.
If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote (carrier) or the affected male may have a de novo
CACNA1F or NYX pathogenic variant, in which case the mother is not a carrier.
If the proband is female and has biallelic pathogenic variants (rare), both the mother and the father may have X-linked CSNB-causing pathogenic variants (i.e., the mother may be a carrier and the father may be affected) [
Bech-Hansen et al 1998].
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
If the mother of the proband has an CACNA1F or NYX pathogenic variant, the chance of transmitting it in each pregnancy is 50%. Males who inherit the pathogenic variant will be affected; females who inherit the variant will be heterozygous and will usually not be affected.
If the proband represents a simplex case (i.e., a single occurrence in a family) and if the CACNA1F or NYX pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population because of the possibility of maternal germline mosaicism.
Offspring of a male proband. Affected males transmit the X-linked CSNB-causing pathogenic variant to:
Other family members. The proband's maternal aunts may be at risk of being heterozygotes (carriers) for the pathogenic variant, and the aunts' offspring, depending on their sex, may be at risk of being carriers or of being affected.
Carrier (Heterozygote) Detection
Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the pathogenic variant has been identified in the proband.
Note: (1) Females who are heterozygous (carriers) for this X-linked disorder will usually not be affected. (2) Identification of female heterozygotes requires either (a) prior identification of the CACNA1F or NYX pathogenic variant in the family or, (b) if an affected male is not available for testing, molecular genetic testing first by sequence analysis, and if no pathogenic variant is identified, by gene-targeted deletion/duplication analysis.
Prenatal Testing and Preimplantation Genetic Testing
Once the CACNA1F or NYX pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for X-linked CSNB are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
Fighting Blindness Canada
890 Yonge Street
12th Floor
Toronto Ontario M4W 3P4
Canada
Phone: 800-461-3331 (toll-free); 416-360-4200
Fax: 416-360-0060
Foundation Fighting Blindness
Phone: 800-683-5555
National Eye Institute
Phone: 301-496-5248
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
X-Linked Congenital Stationary Night Blindness: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
OMIM Entries for X-Linked Congenital Stationary Night Blindness (View All in OMIM)
View in own window
|
300071 | NIGHT BLINDNESS, CONGENITAL STATIONARY, TYPE 2A; CSNB2A |
|
300110 | CALCIUM CHANNEL, VOLTAGE-DEPENDENT, ALPHA-1F SUBUNIT; CACNA1F |
|
300278 | NYCTALOPIN; NYX |
|
310500 | NIGHT BLINDNESS, CONGENITAL STATIONARY, TYPE 1A; CSNB1A |
Molecular Pathogenesis
Genes associated with X-linked congenital stationary night blindness (X-linked CSNB) encode proteins that are specifically expressed in the retina: nyctalopin and voltage-dependent L-type calcium channel subunit alpha-1F (Cav1.4/α1F) for complete and incomplete CSNB, respectively. Pathogenic variants identified in these genes impinge on synaptic transmission from photoreceptors (rods and cones) to inner retinal cells.
Pathogenic variants in NYX are predicted to cause defects in nyctalopin, including alterations in its conformation, loss of the GPI anchor, and deletions of a portion or all of the protein [Zeitz 2007].
Expression studies have shown that some (not all) CACNA1F pathogenic missense variants alter the channel activation properties of the Cav1.4 calcium channel [McRory et al 2004, Hemara-Wahanui et al 2005, Hoda et al 2005]; other missense variants may affect the assembly or expression of the presynaptic ribbon complex [Hoda et al 2006]. Pathogenic nonsense and frameshift variants are predicted to cause loss of channel function or/and photoreceptor synapses.
Mechanism of disease causation. Loss of function
Table 5.
X-Linked Congenital Stationary Night Blindness: Notable Pathogenic Variants by Gene
View in own window
| Gene | Reference Sequences | DNA Nucleotide Change (Alias 1) | Predicted Protein Change (Alias 1) | Comment [Reference] |
|---|
|
CACNA1F
|
NM_005183.2
| c.3166dupC
(c.3167_3168dupC) | p.Leu1056ProfsTer11
(Leu991insC) | Founder variant reported in persons of Dutch-German Mennonite descent [Boycott et al 2000] |
|
NYX
|
NM_022567.2
NP_072089.1
| c.85_108del | p.Arg_Ala36del | Founder variant identified in the US [Bech-Hansen et al 2000] |
| c.856delG | p.Asp286ThrfsTer62 | Identified in Flemish persons from Belgium [Leroy et al 2009] |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
Chapter Notes
Acknowledgments
The authors would like to thank Linda MacLaren and Karen McElligott for years of service to the Mennonite community affected with CSNB2A.
Author History
N Torben Bech-Hansen, PhD; University of Calgary (2007-2012)
Kym M Boycott, PhD, MD; University of Ottawa, Canada (2007-2019)
Stephanie Hoang, MSc (2019-present)
Ian M MacDonald, MD, CM (2007-present)
Yves Sauvé, PhD; University of Alberta (2007-2019)
Sari Tuupanen, PhD (2019-present)
Revision History
3 July 2019 (sw) Comprehensive update posted live
26 April 2012 (me) Comprehensive update posted live
16 January 2008 (me) Review posted live
9 August 2007 (im) Original submission