This publication is provided for historical reference only and the information may be out of date.
Scope
It is increasingly easy to determine where an individual’s nucleotide sequence may differ from a reference standard. It is much more difficult to determine which if any of those sequence variants has an effect on health. ClinVar has been developed to facilitate the evaluation of variation-phenotype relationships by archiving submitted interpretations of these relationships with supporting evidence, by aggregating data from multiple groups such as laboratories to determine if there is a consensus about the interpretation, and by making summary data freely available. ClinVar differs from NCBI’s variation archives, namely dbSNP and dbVar, which have the responsibility of maintaining information about the types and locations of all sequence variation. In contrast, ClinVar provides a curated layer on top of these resources, focusing on the subset of all variation that may be medically relevant.
ClinVar integrates and cross-references data from multiple databases at NCBI. In addition to dbSNP and dbVar, ClinVar depends on MedGen to represent phenotype, Gene to represent genes, and on human RefSeqs to represent the location of sequence variation.
History
As a public database, ClinVar is young, having been fully released for the first time in April, 2013. However, ClinVar has been in development for several years, growing out of discussions of the Variome project about the benefits of centralizing information about rare human variation and its relationship to health. In 2008 dbSNP launched several tools to make it easier to submit such data, http://www.ncbi.nlm.nih.gov/SNP/tranSNP/tranSNP.cgi for single alleles and http://www.ncbi.nlm.nih.gov/SNP/tranSNP/vsub.cgi for spreadsheets. An application was developed to provide gene-specific views of such submissions (VarView); the single record display indicated if the location was submitted via the clinical channel; and our sequence displays provided a Clinical channel track. Several locus-specific databases used this functionality to submit data about rare human variation.
In addition to submissions from external groups, the RefSeqGene staff shepherded data from GeneReviews and OMIM into dbSNP to augment the connections among the published literature, other databases, and the variation archives. Based on this foundation, and NCBI’s maintenance first of GeneTests, and now the NIH Genetic Testing Registry (GTR), NCBI was approached by several stakeholders to develop what is now called ClinVar. The genetic testing community was seeking a comprehensive, up-to-date, freely accessible resource in which to share data and pool resources to evaluate human variation.
Data Model
ClinVar’s data model is based on five major categories of content: submitter data for attribution, definition of the variation, characterization of the phenotype, evidence about the effect of variation on health, and interpretation of that evidence. Whenever possible, the content is highly structured rather than free text, and is harmonized to controlled vocabularies or other data standards.
ClinVar is a submitter-driven resource that maintains an archive of what has been received and processed. Data from submitters is assigned an accession of the format SCV123456789 (SCV) and data from multiple submitters about the same variation/phenotype combination is aggregated and assigned an accession of the format RCV123456789 (RCV). Content is versioned, i.e., the first submission is assigned version 1 and any updates to a submission is represented as an incremented version of the same accession. The RCV record also includes content added by NCBI, such as accessions from other databases, standard terminology, and analysis of related submissions.
Submitter
ClinVar represents submitters as both organizations and individuals. The infrastructure supporting this content is shared with the NIH Genetic Testing Registry (GTR), dbSNP, and dbVar. Submitters have the right to request anonymity, although to date no submitter has requested this option. Summary data about submissions are provided on the website (http://www.ncbi.nlm.nih.gov/clinvar/submitters/).
Variation
Variation is a key component of ClinVar’s data model, especially to be able to represent variation’s relationship to phenotype. Variation is thus represented both as the sequence at a particular location, or a combination of sequence changes. In other words, ClinVar can represent the interpretation of a single allele, compound heterozygotes, haplotypes, and combinations of alleles in different genes. Variation is modeled in the database as a set of varations, but currently most sets have only one member. The goal is to represent each variation on a reference sequence, but the data flow from some submitters is not amenable to establishing this immediately. Thus free text is accepted.
Variations submitted to ClinVar are compared to variations accessioned by dbSNP or dbVar. If known, ClinVar adds the rs# (dbSNP) or variant call identifier (dbVar) to the RCV record. If novel, the information is submitted to the appropriate variation database to be accessioned, so that the identifiers can be added to ClinVar. In other words, ClinVar does not create new identifiers for locations of variation. Also the archival databases do not note the number of submitters that have contributed information to ClinVar about a variation.That said, to support internal data flows and some public reports ClinVar does assign an internal unique identifier to the sequence change at each location, which is reported in the XML and tab-delimited exports as an integer identifier (Table 1).
Table 1.
Identifiers used by ClinVar
ClinVar reports multiple types of attributes for each variant. HGVS expressions are reported based on the current reference assembly, RefSeqGenes, cDNAs and proteins as appropriate. When there are multiple transcripts for a gene, ClinVar selects one HGVS expression to display as the preferred name. By default, this selection is based on the first reference standard transcript identified by the RefSeqGene/Locus Reference Genomic (LRG) collaboration, but can be overridden upon request.
Some of the data ClinVar reports related to variation are values added by NCBI. These are reported only as part of the RCV record (because the SCV accession is what the submitter provides), and can include alternate HGVS expressions, allele frequencies from the 1000 Genomes project or GO-ESP, identifiers from dbSNP or dbVar, molecular consequences (e.g., nonsense/missense/frameshift), location data (splice site, UTR’s, cytogenetic band, genes), and confidence in variation calls at that location.
Phenotype
ClinVar represents phenotype as concepts identified in MedGen. Similar to management of variation, these concepts can be single or sets of multiple values. Sets are used primarily to report a combination of clinical features; single values are used to represent diagnostic terms or indications. Submitters are encouraged to submit phenotypic information via identifier, e.g., MIM number, MeSH term, or identifier from the Human Phenotype Ontology (HPO). Free text is accepted and ClinVar staff will work with submitters to determine if that text can be mapped to current standardized concepts. If not, ClinVar establishes a new identifier to be represented in MedGen and adds that MedGen identifier to the RCV record.
Interpretation
All interpretations of the relationship between variation and phenotype in ClinVar are supplied by submitters. ClinVar reports clinical significance, the date that clinical significance was last interpreted by the submitter, and functional significance. To support interpretation, mode of inheritance of a variation relative to a disorder and qualification of severity of phenotype are also represented. Terms for clinical significance are those recommend by the American College of Medical Genetics (ACMG). If submitters disagree on the interpretation of the clinical significance of any variation, that record is marked in the aggregate report as having conflicts. If one submitter does not provide this information, and another does, that is not marked as conflicting.
Comparison of clinical signficiance provided by multiple submitters is computed by two methods. One is a strict interpretation, per RCV accession, of any difference. In other words, pathogenic and likely pathogenic are reported as being in conflict. The second is more relaxed, and based only on the variation and not the variation as related to a specific phenotype. In this mode, the conflicts are reported only at the extremes, i.e., differences between pathogenic/likely pathogenic, benign/likely benign, and uncertain significance.
Evidence
Evidence that supports an interpretation of the variation-phenotype relationship can be highly structured and/or a free-text summary discussing how the evidence was evaluated. When structured, content includes the description of how the variants were called and in what context (genetic testing, family studies, comparison of tumor/normal tissue, animal models, etc.) Based on that context, the results can be represented as number of independent observations per person or chromosome, number of segregations observed, number of times other rare variations were identified in the same gene or other genes, etc. At present, most structured data are reports of number of individuals in which non-somatic variation was observed, sometimes with indication of number of families.
Dataflow
Initial records
The major data flows for ClinVar are diagrammed in Figure 1. Direct submissions are validated, converted to XML, and accessioned. If any content does not validate, submitters are contacted and corrections are requested. When valid, the records are assigned accessions (SCV) and processed. Submitters are provided reports including the accessions assigned to their data and indications as to whether any of their data conflicted with current public submissions.

Figure 1.
Overview of the flow of information through ClinVar. ClinVar validates content and looks for differences relative to previous submissions and returns reports to the submitter before the data are released to the public.
Data that NCBI processes from OMIM or GeneReviews are managed slightly differently. Data from OMIM are updated daily from automatic feeds, and bypass the validation assigned to direct submissions. If possible, novel variations are converted to sequence coordinates by testing possible reference sequences and determining if the data in the text of OMIM’s description of the allele are consistent with reported sequence changes. As resources permit, NCBI staff reviews recent records from OMIM that cannot be processed automatically. Data from GeneReviews are extracted from the tables embedded in the GeneReview, as well as attached tables provided by the submitter. Any questions that arise in processing data from GeneReviews are reported to GeneReviews staff for review.
Updates
Submitters may update their submissions at any time. With an update, the accession is assigned a new version. Thus if a unit record in a submission were assigned the accession SCV000000001, with an update the version would be incremented, in this case to 2 (SCV000000001.2).
RCV-specific processing
Data associated with an RCV accession can change in one of two ways. One is represented by an increment of a version. Again, if there are multiple submissions about the same variation-phenotype relationship, these are aggregated into one RCV accession and versioned. The version of an RCV accession is incremented if a new submission is received for the same variation-phenotype relationship (i.e., a new SCV accession is added to the set represented by the RCV accession), or if any SCV accession in the set is itself updated and assigned a new version.
The content of an RCV accession can also change without that being reflected in a new version. If a genomic assembly changes, if genomic coordinates are established for a variation for the first time, if database identifiers such as rs#, nsv#, or PubMed ids are added, if preferred terms are redefined, then the content will be updated without assigning a new version, but with a new unique identifier. These snapshots of content are calculated weekly, and the unique integer identifer is detected when accessing ClinVar via E-Utilities.
Access
Web
ClinVar’s website, http://www.ncbi.nlm.nih.gov/clinvar, is part of NCBI’s Entrez system and thus is searchable with the standard query interface and Advanced query options. ClinVar supports retrieval by any text in the RCV record, including descriptions of variation (HGVS expression, rs, nsv, nssv, OMIM allelic variant identifier, identifier used in a locus-specific database or LSDB), genes (symbol or full name), disease (names and identifiers), submitter names, and clinical significance. To facilitate a common search strategy, a query that is detected to be a human gene symbol displays a link to make it easier to limit your query results by that symbol. The default result set is a table of 20 rows, but that can be altered using Display Settings (Figure 2). When multiple results are returned from a query, filters are provided at the left that reflect the content of the retrieval set (values and counts of each). Clicking on one of those options removes all but the selection from the display, a restriction that can be reversed by using the Clear option.
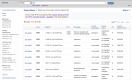
Figure 2.
Tabular results of a ClinVar search.
The full record is accessed by clicking on See details in the first column of the tabular display, or the title row if the summary display option is used. At present, the detailed display corresponds to content of an RCV accession (Figure 3). The Clinical significance, Allele description, Condition(s) sections, and the Genome view report aggregate data; the Clinical Assertions are submitter-specific, and the Evidence (not shown) is provided both in aggregate and submitter-specific sections.
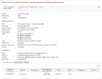
Figure 3.
Detailed display of an RCV record. This is currently the default display.
Before the end of 2013, a new display will be provided via See details, quite similar to the RCV report but aggregated per single variation rather than variation-phenotype combination. This new display allows users to see all data for a variation even when submitters’ representation of phenotype differs.
Data in ClinVar can also be discovered via other NCBI databases, based on the links that are built when content is shared. Examples include dbSNP, dbVar, Gene, MedGen, Nucleotide, and PubMed. Locations of variation represented in ClinVar are annotated on RefSeqs and are visible in the graphical sequence displays (e.g., http://www.ncbi.nlm.nih.gov/nuccore/125662814?report=graph), and browsers such as 1000 Genomes (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). ClinVar also provides specialized pages for certain types of access. One is the list of genes and disorders for which ACMG recommends that incidental findings be reported (1) (http://www.ncbi.nlm.nih.gov/clinvar/docs/acmg/); another is the listing of submitters and all their submissions (http://www.ncbi.nlm.nih.gov/clinvar/submitters/).
FTP
Data from ClinVar are reported from several directories at NCBI and in several formats. The README file (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/README.txt) provides a comprehensive list. Current content includes the file converting MIM numbers, GeneIDs, and MedGen concepts ids on Gene’s FTP site (mim2gene_medgen), the listing of standard terms used by ClinVar at GTR’s FTP site, and the tab-delimited, XML, and VCF files from ClinVar. The VCF files are available from dbSNP (with the symbolic link from ClinVar).
E-Utilities
ClinVar supports programmatic access via E-Utilities as esearch, esummary, and elink. E-fetch is not enabled. Please note that esearch (e.g., http://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=clinvar&term=brca1[gene]&retmax=1000)
returns the unique identifiers for an RCV record, which does not correspond 1:1 with an accession.version. The unique identifiers represent an instance of that record, which may change without a version change if NCBI adds data to the record such as an rs# or a ConceptUID from MedGen. A record retrieved by an outdated ID provides a link to the current record.
Related Tools
The data for which ClinVar is responsible, namely the archive of interpretations of clinical significance, is integrated into the various tools NCBI maintains to manage recalculation of sequence coordinates (Clinical Remap) and to report what is known about human variation at a genomic location (1000 Genomes Browser, Variation Reporter, Variation View). These data are integrated monthly on the first Thursday.
References
- 1.
- Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, McGuire AL, Nussbaum RL, O'Daniel JM, Ormond KE, Rehm HL, Watson MS, Williams MS, Biesecker LG. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013Jul 15(7):565–74. [PMC free article: PMC3727274] [PubMed: 23788249]
Publication Details
Author Information and Affiliations
Authors
Melissa Landrum, PhD, Jennifer Lee, PhD, George Riley, PhD, Wonhee Jang, PhD, Wendy Rubinstein, MD, PhD, Deanna Church, PhD, and Donna Maglott, PhD.Publication History
Created: November 21, 2013.
Copyright
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Landrum M, Lee J, Riley G, et al. ClinVar. 2013 Nov 21. In: The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.


