Summary
Clinical characteristics.
EPB42-related hereditary spherocytosis (EPB42-HS) is a chronic nonimmune hemolytic anemia that is usually of mild-to-moderate severity. EPB42-HS can present with jaundice as early as the first 24 hours of life or can present later in childhood with anemia resulting from a hemolytic crisis or aplastic crisis (usually associated with a viral infection). In addition to the hematologic manifestations, serious complications include splenomegaly, which can become evident in early childhood, and cholelithiasis, which usually becomes evident in the second or third decade of life.
Typical laboratory findings in EPB42-HS include anemia (decreased hemoglobin [Hgb] level) and reticulocytosis (increased percentage of reticulocytes), with high mean corpuscular Hgb concentration, presence of spherocytes in the peripheral blood smear, significantly decreased or absent haptoglobin, mildly increased osmotic fragility in osmotic fragility assay, increased Omin (osmolality at which 50% of red blood cells hemolyze), and decreased maximal elongation index (EImax) in osmotic gradient ektacytometry.
Diagnosis/testing.
The diagnosis of EPB42-HS is established by identification of biallelic pathogenic variants in EPB42.
Management.
Treatment of manifestations: Treatment of hyperbilirubinemia as needed; folic acid supplementation; red blood cell transfusion as needed for a hemolytic or aplastic crisis; routine immunizations; iron chelation therapy as needed. Prior to splenectomy, immunizations for S pneumoniae, N meningitidis, and H influenzae. Although splenectomy is rarely indicated in EPB42-HS because disease severity is usually mild or moderate, partial or total splenectomy may be recommended in those with moderately severe EPB42-HS who are older than age five years when quality of life is compromised. Following splenectomy, booster immunizations for S pneumoniae and N meningitidis, prophylactic antibiotics, and prompt antibiotics for fever are recommended. Cholecystectomy in those with signs and/or symptoms of cholelithiasis; affected individuals with a history of cholelithiasis should have cholecystectomy at the time of splenectomy.
Surveillance: Monitor serum bilirubin concentration in neonates during the first week of life and Hgb in infants during the first two to four months of life. Those dependent on frequent transfusions and those receiving iron chelation therapy require monitoring of serum ferritin concentration. Monitor efficacy of chelation via T2*-weighted liver MRI and adjust appropriately. Abdominal ultrasound examination to evaluate for cholelithiasis either when symptoms are present or, when hemolysis is significant, every five to ten years beginning at age ten to 12 years.
Agents/circumstances to avoid: Avoid supplements containing iron unless iron studies have documented iron deficiency. If so, treatment with supplemental iron must be closely monitored and then discontinued when iron stores have been repleted. Avoidance of contact sports is recommended in those with splenomegaly; of note, acute or excessive splenomegaly is a greater risk than chronic mild splenomegaly.
Evaluation of relatives at risk: When EPB42-HS has been diagnosed in a family member, the following is recommended for at-risk sibs: (1) Neonates at risk require monitoring of serum bilirubin concentration during the first week of life so that treatment for hyperbilirubinemia can be instituted promptly; (2) Infants at risk require monitoring in the first two to four months of life for significant anemia, which may require red blood cell transfusion and initiation of folic acid supplementation. Laboratory evaluation (CBC and reticulocyte count, blood smear, osmotic fragility or ektacytometry) and/or molecular genetic testing for the EPB42 pathogenic variants in the family (if known) is appropriate for at-risk relatives.
Pregnancy management: Folic acid supplementation (800-1,000 µg daily); monitor for exacerbation of anemia with CBC and reticulocyte count.
Genetic counseling.
EPB42-HS is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an EPB42 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial EPB42 pathogenic variants. Once the EPB42 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
EPB42-related hereditary spherocytosis (EPB42-HS) should be suspected in individuals with any of the following clinical and laboratory findings.
Clinical findings
- Pallor and/or fatigue due to anemia, which is usually of mild-to-moderate severity
- Jaundice
- Usually intermittent and caused by unconjugated hyperbilirubinemia resulting from exacerbated hemolysis
- In rare instances, caused by conjugated hyperbilirubinemia resulting from biliary obstruction
- Splenomegaly
- Cholelithiasis in the second or third decade of life
Laboratory findings
- Complete blood count consistent with:
- Chronic, nonimmune hemolytic anemia (decreased hemoglobin [Hgb] with reticulocytosis), usually of mild-to-moderate severity
- Decreased Hgb level (See Table 1 for Hgb levels that define the severity of hereditary spherocytosis.)Note: Hgb values in EPB42-HS may also vary depending on the clinical status of the affected individual (baseline or during a hemolytic or aplastic crisis).
- Increased percent of reticulocytes as well as increased absolute reticulocyte count (See Table 1 for percent of reticulocytes that define the severity of hereditary spherocytosis.)Note: Percent of reticulocytes may vary (depending on baseline or crisis status) from 2.5% to greater than 10% (or even normal or low when in aplastic crisis).
- High mean corpuscular Hgb concentration. Normal values are typically 31-37 g/dL. Values in individuals with hereditary spherocytosis are usually 35.5-37.5 g/dL.
- Negative (i.e., normal) direct anti-globulin test (DAT)Note: DAT should always be evaluated in a person with newly diagnosed hemolytic anemia to evaluate for an acute immune-mediated (acquired) hemolytic anemia.
- Peripheral blood smear demonstrating presence of spherocytes and occasionally a few ovalocytes and elliptocytesNote: The term "spherocyte" refers to the sphere-shaped red blood cells (with a decreased surface-to-volume ratio) that characterize the red blood cell membrane skeleton disorders (see Differential Diagnosis).
- Significantly decreased or absent haptoglobin. After age six months normal haptoglobin values are 16-200 mg/dL. In hereditary spherocytosis, haptoglobin is typically undetectable; however, haptoglobin can be normal in the presence of concurrent inflammation (as it is an acute phase reactant).
- Mildly increased osmotic fragility (as in Figure 1B of Hammill et al [2011]; see full text)
- Decreased maximal deformability index (DImax; also known as elongation index, or EImax) and increased Omin (osmolality at which 50% of red blood cells hemolyze) measured by osmotic gradient ektacytometry, giving a typical hereditary spherocytosis curve [Clark et al 1983, Hammill et al 2011]
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Table 1.
Severity of Hereditary Spherocytosis
Establishing the Diagnosis
The diagnosis of EPB42-HS is established in a proband by the identification of biallelic pathogenic variants in EPB42 (see Table 2).
Molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of EPB42 to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. To date, exon-level deletions/duplications have not been identified in individuals with EPB42-HS.
- A multigene panel that includes EPB42 and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 2.
Molecular Genetic Testing Used in EPB42-Related Hereditary Spherocytosis
Clinical Characteristics
Clinical Description
Children with EPB42-related hereditary spherocytosis (EPB42-HS) frequently present within the first 24 hours of life with jaundice that requires treatment with phototherapy or, rarely, exchange transfusion to prevent kernicterus. They may also present later in childhood with anemia resulting from a hemolytic crisis or aplastic crisis usually associated with a viral infection.
Toddlers with hereditary spherocytosis are occasionally found to have age-related iron deficiency anemia; however, the anemia fails to completely resolve with iron supplementation and reticulocytosis persists [Hammill et al 2011].
As with all forms of mild or moderate hereditary spherocytosis (see Table 1 for definitions), EPB42-HS can be more severe in the first four to six months of life, requiring regular red blood cell transfusions. Thus, frequent transfusions during the first few months of life do not necessarily correlate with disease severity later on.
EPB42-HS, if not recognized during infancy or early childhood, may be diagnosed later in life as a mild (hemoglobin [Hgb] = 11-15 g/dL) to moderate (Hgb = 8-11.5 g/dL) chronic hemolytic anemia (see Table 1), with jaundice, splenomegaly, and cholelithiasis at a relatively young age [Eber & Lux 2004].
Genotype-Phenotype Correlations
Homozygosity for p.Ala142Thr has been found most commonly in individuals of Japanese descent and was reported to lead to moderately severe hereditary spherocytosis, with Hgb as low as 6.1 g/dL [Bouhassira et al 1992]. An Italian individual with the same genotype was reported with moderate hemolytic anemia from birth and splenomegaly [Perrotta et al 1999].
Homozygosity for p.Ala142Thr or homozygosity for p.Asp175Tyr results in atypical hereditary spherocytosis with the presence of ovalostomatocytes in addition to few spherocytes in the blood smear. Hemolysis may be mild to moderately severe and improves after splenectomy [Bouhassira et al 1992, Kanzaki et al 1995].
Compound heterozygosity for p.Ala142Thr with another EPB42 pathogenic variant causes typical hereditary spherocytosis with microspherocytes in the blood smear and increased osmotic fragility [Takaoka et al 1994, Kanzaki et al 1995]. Case reports of individuals with other EPB42 pathogenic variants also indicate mild-to-moderate hereditary spherocytosis with only occasional need for blood transfusion [Hayette et al 1995, van den Akker et al 2010, Hammill et al 2011].
One individual homozygous for the null c.950delG variant (resulting in premature termination of the transcript and lack of production of any viable erythrocyte membrane protein 4.2) developed a strong antibody response against protein 4.2 after multiple red blood cell transfusions for gastrointestinal bleeding, causing alloimmune hemolytic anemia [Beauchamp-Nicoud et al 2000]. To date, antibody development has not been described following red blood cell transfusion in persons with other EPB42 pathogenic variants, although in most individuals with EPB42-HS no protein 4.2 is detectable in the RBC membrane [Satchwell et al 2009].
Prevalence
Hereditary spherocytosis is the most common inherited anemia in individuals of northern European ancestry and is present worldwide, with a prevalence of 1:1,000-1:2,500 [Risinger & Kalfa 2020].
EPB42-HS is responsible for 40%-50% of hereditary spherocytosis in Japan, where the carrier frequency of p.Ala142Thr among healthy persons is as high as 3% [Yawata 1994, Yawata et al 2000].
In other populations, EPB42-HS accounts for 5% or less of hereditary spherocytosis [Eber & Lux 2004, Perrotta et al 2008].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in EPB42.
Differential Diagnosis
The initial evaluation of a person with hemolytic anemia typically includes:
- Complete blood count and reticulocyte count;
- Blood smear review;
- Direct and indirect anti-globulin test (DAT and IAT, traditionally called direct and indirect Coombs, respectively) to evaluate for autoimmune (or, in an infant, alloimmune) hemolytic anemia;
- Hemoglobin electrophoresis;
- G6PD enzyme activity (especially in males).
Osmotic fragility testing and/or ektacytometry can identify erythrocyte membrane disorders. Figure 1 demonstrates ektacytometry results typical of hereditary spherocytosis.
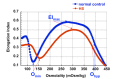
For the individual with nonimmune hemolytic anemia, the differential diagnosis includes other forms of hereditary spherocytosis (see Table 3a) and other causes of hereditary hemolytic anemia (see Table 3b).
Table 3a.
Genes Associated With Hereditary Spherocytosis
Table 3b.
Other Causes of Hereditary Hemolytic Anemia
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with EPB42-related hereditary spherocytosis (EPB42-HS), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with EPB42-Related Hereditary Spherocytosis
Treatment of Manifestations
Detailed management guidelines for hereditary spherocytosis have been published [Eber & Lux 2004, Bolton-Maggs et al 2012]. However, increased information on genotype-phenotype correlation along with the current wider availability of partial splenectomy calls for a new consensus statement of experts in the field on updated management guidelines [Iolascon et al 2017, Rothman et al 2020, Kalfa 2021].
Table 5.
Treatment of Manifestations in Individuals with EPB42-Related Hereditary Spherocytosis
Surveillance
Table 6.
Recommended Surveillance for Individuals with EPB42-Related Hereditary Spherocytosis
Agents/Circumstances to Avoid
Any preparations containing iron should be avoided except in those with iron deficiency documented with appropriate studies (see Treatment of Manifestations).
Contact sports are not advisable in those with splenomegaly; of note, acute or excessive splenomegaly is a greater risk than chronic mild splenomegaly.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger sibs of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of treatment and preventive measures. Evaluations include:
- Laboratory evaluation of the phenotype (complete blood count and reticulocyte count, blood smear, osmotic fragility, or ektacytometry);
- Molecular genetic testing for the EPB42 pathogenic variants in the family.
Neonates require monitoring of serum bilirubin concentration during the first week of life so that treatment for hyperbilirubinemia can be instituted promptly to avoid complications such as kernicterus. Infants require monitoring in the first two to four months of life for significant anemia, which may require red blood cell transfusion. Initiation of folate supplementation should be considered in individuals with chronic hemolytic anemia with significant reticulocytosis.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Folic acid supplementation (800-1,000 µg daily) is recommended in pregnant women with EPB42-HS.
Monitoring for exacerbation of anemia with complete blood count and reticulocyte count is recommended in pregnant women, as hemolytic crisis and persistent anemia have been reported during pregnancy in women with hemolytic anemia, especially in women who have not undergone splenectomy [Pajor et al 1993].
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
EPB42-related hereditary spherocytosis (EPB42-HS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are presumed to be heterozygous for an EPB42 pathogenic variant.
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an EPB42 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an EPB42 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of inheriting neither of the familial EPB42 pathogenic variants.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with EPB42-HS are obligate heterozygotes (carriers) for an EPB42 pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an EPB42 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the EPB42 pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the EPB42 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for EPB42-HS are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
EPB42-Related Hereditary Spherocytosis: Genes and Databases
Table B.
OMIM Entries for EPB42-Related Hereditary Spherocytosis (View All in OMIM)
Molecular Pathogenesis
Erythrocyte membrane protein band 4.2 (also known as protein 4.2), encoded by EPB42, is a major component of the red blood cell (RBC) cytoskeleton and maintains the stability and flexibility of red blood cells through interactions with other key RBC proteins, many of which (in their pathogenic forms) also cause hereditary spherocytosis.
Protein 4.2 is a part of the ankyrin-band 3 complex, connecting band 3 anion transport protein (encoded by SLC4A1; see Table 3a) with the CD47 and Rhesus protein complex antigens. Protein 4.2 supports physical associations between the cytoskeleton and the membrane lipid bilayer [Bruce et al 2003]. Protein 4.2 interacts with spectrin, a tetramer comprising an alpha subunit and a beta subunit (see Table 3a), which is the largest protein in the RBC cytoskeleton [Mandal et al 2002, Korsgren et al 2010].
Mechanism of disease causation. Loss of function
Table 7.
Notable EPB42 Pathogenic Variants
Chapter Notes
Acknowledgments
The authors' work related to this review was supported by the National Institutes of Health, National Center for Advancing Translational Sciences (award 1UL1TR001425-01), and National Heart, Lung, and Blood Institute grant R01 HL152099. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author History
Amber H Begtrup, PhD, FACMG (2014-present)
Jessica A Connor, MS; Counsyl, Inc (2014-2022)
Theodosia A Kalfa, MD, PhD (2014-present)
Revision History
- 7 April 2022 (sw) Comprehensive update posted live
- 10 November 2016 (ma) Comprehensive update posted live
- 13 March 2014 (me) Review posted live
- 20 October 2013 (tk) Original submission
References
Published Guidelines / Consensus Statements
- Iolascon A, Andolfo I, Barcellini W, Corcione F, Garçon L, De Franceschi L, Pignata C, Graziadei G, Pospisilova D, Rees DC, de Montalembert M, Rivella S, Gambale A, Russo R, Ribeiro L, Vives-Corrons J, Martinez PA, Kattamis A, Gulbis B, Cappellini MD, Roberts I, Tamary H, et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Available online. 2017. Accessed 3-30-22.
Literature Cited
- American Academy of Pediatrics Subcommittee on Hyperbilirubinemia. Management of hyperbilirubinemia in the newborn infant 35 or more weeks of gestation. Pediatrics. 2004;114:297–316. [PubMed: 15231951]
- Beauchamp-Nicoud A, Morle L, Lutz HU, Stammler P, Agulles O, Petermann-Khder R, Iolascon A, Perrotta S, Cynober T, Tchernia G, Delaunay J, Baudin-Creuza V. Heavy transfusions and presence of an anti-protein 4.2 antibody in 4. 2(-) hereditary spherocytosis (949delG). Haematologica. 2000;85:19–24. [PubMed: 10629586]
- Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012;156:37–49. [PubMed: 22055020]
- Bouhassira EE, Schwartz RS, Yawata Y, Ata K, Kanzaki A, Qiu JJ, Nagel RL, Rybicki AC. An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood. 1992;79:1846–54. [PubMed: 1558976]
- Bruce LJ, Beckmann R, Ribeiro ML, Peters LL, Chasis JA, Delaunay J, Mohandas N, Anstee DJ, Tanner MJ. A band 3-based macrocomplex of integral and peripheral proteins in the RBC membrane. Blood. 2003;101:4180–8. [PubMed: 12531814]
- Casale M, Perrotta S. Splenectomy for hereditary spherocytosis: complete, partial or not at all? Expert Rev Hematol. 2011;4:627–35. [PubMed: 22077527]
- Clark MR, Mohandas N, Shohet SB. Osmotic gradient ektacytometry: comprehensive characterization of red cell volume and surface maintenance. Blood. 1983;61:899–910. [PubMed: 6831052]
- Cohn AC, MacNeil JR, Clark TA, Ortega-Sanchez IR, Briere EZ, Meissner HC, Baker CJ, Messonnier NE, et al. Prevention and control of meningococcal disease: recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2013;62:1–28. [PubMed: 23515099]
- Committee on Infectious Diseases. Meningococcal conjugate vaccines policy update: booster dose recommendations. Pediatrics. 2011;128:1213–8. [PubMed: 22123893]
- Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41:118–41. [PubMed: 15071790]
- Hammill AM, Risinger MA, Joiner CH, Keddache M, Kalfa TA. Compound heterozygosity for two novel mutations in the erythrocyte protein 4.2 gene causing spherocytosis in a Caucasian patient. Br J Haematol. 2011;152:780–3. [PMC free article: PMC3105174] [PubMed: 21275958]
- Hayette S, Dhermy D, dos Santos ME, Bozon M, Drenckhahn D, Alloisio N, Texier P, Delaunay J, Morlé L. A deletional frameshift mutation in protein 4.2 gene (allele 4.2 Lisboa) associated with hereditary hemolytic anemia. Blood. 1995;85:250–6. [PubMed: 7803799]
- Iolascon A, Andolfo I, Barcellini W, Corcione F, Garçon L, De Franceschi L, Pignata C, Graziadei G, Pospisilova D, Rees DC, de Montalembert M, Rivella S, Gambale A, Russo R, Ribeiro L, Vives-Corrons J, Martinez PA, Kattamis A, Gulbis B, Cappellini MD, Roberts I, Tamary H, et al. Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica. 2017;102:1304–13. [PMC free article: PMC5541865] [PubMed: 28550188]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Kalfa TA. Diagnosis and clinical management of red cell membrane disorders. Hematology Am Soc Hematol Educ Program. 2021;2021:331–40. [PMC free article: PMC8791164] [PubMed: 34889366]
- Kanzaki A, Hayette S, Morle L, Inoue F, Matsuyama R, Inoue T, Yawata A, Wada H, Vallier A, Alloisio N, Yawata Y, Delaunay J. Total absence of protein 4.2 and partial deficiency of band 3 in hereditary spherocytosis. Br J Haematol. 1997;99:522–30. [PubMed: 9401060]
- Kanzaki A, Yawata Y, Yawata A, Inoue T, Okamoto N, Wada H, Harano T, Harano K, Wilmotte R, Hayette S. Band 4.2 Komatsu: 523 GAT-->TAT (175 Asp-->Tyr) in exon 4 of the band 4.2 gene associated with total deficiency of band 4.2, hemolytic anemia with ovalostomatocytosis and marked disruption of the cytoskeletal network. Int J Hematol. 1995;61:165–78. [PubMed: 8547605]
- Korsgren C, Peters LL, Lux SE. Protein 4.2 binds to the carboxyl-terminal EF-hands of erythroid alpha-spectrin in a calcium- and calmodulin-dependent manner. J Biol Chem. 2010;285:4757–70. [PMC free article: PMC2836081] [PubMed: 20007969]
- Mandal D, Moitra PK, Basu J. Mapping of a spectrin-binding domain of human erythrocyte membrane protein 4.2. Biochem J. 2002;364:841–7. [PMC free article: PMC1222634] [PubMed: 12049649]
- Mizukawa B, George A, Pushkaran S, Weckbach L, Kalinyak K, Heubi JE, Kalfa TA. Cooperating G6PD mutations associated with severe neonatal hyperbilirubinemia and cholestasis. Pediatr Blood Cancer. 2011;56:840–2. [PMC free article: PMC3023834] [PubMed: 20949590]
- Mohandas N, Clark MR, Health BP, Rossi M, Wolfe LC, Lux SE, Shohet SB. A technique to detect reduced mechanical stability of red cell membranes: relevance to elliptocytic disorders. Blood. 1982;59:768–74. [PubMed: 7059678]
- Nuorti JP, Whitney CG, et al. Prevention of pneumococcal disease among infants and children--use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2010;59:1–18. [PubMed: 21150868]
- Pajor A, Lehoczky D, Szakacs Z. Pregnancy and hereditary spherocytosis. Report of 8 patients and a review. Arch Gynecol Obstet. 1993;253:37–42. [PubMed: 8328819]
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372:1411–26. [PubMed: 18940465]
- Perrotta S, Iolascon A, Polito R, d'Urzo G, Conte ML, Miraglia del Giudice E. 4.2 Nippon mutation in a non-Japanese patient with hereditary spherocytosis. Haematologica. 1999;84:660–2. [PubMed: 10406914]
- Risinger M, Kalfa TA. Red cell membrane disorders: structure meets function. Blood. 2020;136:1250–61. [PMC free article: PMC7483429] [PubMed: 32702754]
- Rothman JA, Stevens JL, Gray FL, Kalfa TA. How I approach hereditary hemolytic anemia and splenectomy. Pediatr Blood Cancer. 2020;67:e28337. [PubMed: 32391969]
- Ruparel RK, Bogert JN, Moir CR, Ishitani MB, Khan SP, Rodriguez V, Zarroug AE. Synchronous splenectomy during cholecystectomy for hereditary spherocytosis: is it really necessary? J Pediatr Surg. 2014;49:433–5. [PubMed: 24650472]
- Satchwell TJ, Shoemark DK, Sessions RB, Toye AM. Protein 4.2: a complex linker. Blood Cells Mol Dis. 2009;42:201–10. [PubMed: 19269200]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Takaoka Y, Ideguchi H, Matsuda M, Sakamoto N, Takeuchi T, Fukumaki Y. A novel mutation in the erythrocyte protein 4.2 gene of Japanese patients with hereditary spherocytosis (protein 4.2 Fukuoka). Br J Haematol. 1994;88:527–33. [PubMed: 7819064]
- Toye AM, Williamson RC, Khanfar M, Bader-Meunier B, Cynober T, Thibault M, Tchernia G, Dechaux M, Delaunay J, Bruce LJ. Band 3 Courcouronnes (Ser667Phe): a trafficking mutant differentially rescued by wild-type band 3 and glycophorin A. Blood. 2008;111:5380–9. [PMC free article: PMC2605348] [PubMed: 18174378]
- van den Akker E, Satchwell TJ, Pellegrin S, Flatt JF, Maigre M, Daniels G, Delaunay J, Bruce LJ, Toye AM. Investigating the key membrane protein changes during in vitro erythropoiesis of protein 4.2 (-) cells (mutations Chartres 1 and 2). Haematologica. 2010;95:1278–86. [PMC free article: PMC2913075] [PubMed: 20179084]
- Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, Eber SW. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol. 2000;71:118–35. [PubMed: 10745622]
- Yawata Y. Red cell membrane protein band 4.2: phenotypic, genetic and electron microscopic aspects. Biochim Biophys Acta. 1994;1204:131–48. [PubMed: 8142452]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: March 13, 2014; Last Update: April 7, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Kalfa TA, Begtrup AH. EPB42-Related Hereditary Spherocytosis. 2014 Mar 13 [Updated 2022 Apr 7]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
