Clinical Description
Schmid metaphyseal chondrodysplasia (SMCD) is typically diagnosed in early childhood and is the most common and least severe metaphyseal chondrodysplasia [Al Kaissi et al 2018]. It results from disrupted calcification of metaphyseal cartilage and consequent restricted longitudinal growth of bones with preservation of the epiphyses. The clinical and radiographic features are usually not present at birth, but manifest in early childhood with limb shortening, genu varum, and a waddling gait [Bateman et al 2005]. The pattern of radiographic features is generally similar across individuals with SMCD, but clinical severity varies considerably [Mäkitie et al 2005]. There are no extraskeletal manifestations.
A comprehensive review of the published reports and clinical databases identified at least 150 published unrelated individuals with SMCD and a confirmed pathogenic variant in COL10A1. The following description of the phenotypic features associated with this condition is based on these reports and author observations in a multidisciplinary skeletal dysplasia clinic.
Growth. Most neonates with SMCD have normal growth parameters. Progressive growth failure typically begins in the second year of life, with individuals coming to medical attention after age two years with short-limbed short stature and bowed legs [Mäkitie et al 2005]. Adult height is typically more than 3.5 standard deviations (SD) below the mean, although a wide spectrum that overlaps normal height has been reported [Mäkitie et al 2005]. No standardized growth curves for SMCD are available.
Musculoskeletal. Most individuals with SMCD have genu varum (outward bowing at the knees), although valgus deformity has been reported. Waddling gait due to coxa vara is common by age two years and may require surgical correction. Joint pain in the knees and hips is common in children and adults with SMCD and may limit physical activity. Chronic joint pain may develop and limit mobility for some individuals. Lumbar lordosis is common, with typical onset by age three to five years. Scoliosis is less commonly reported [Park et al 2015, Cammarata-Scalisi et al 2019].
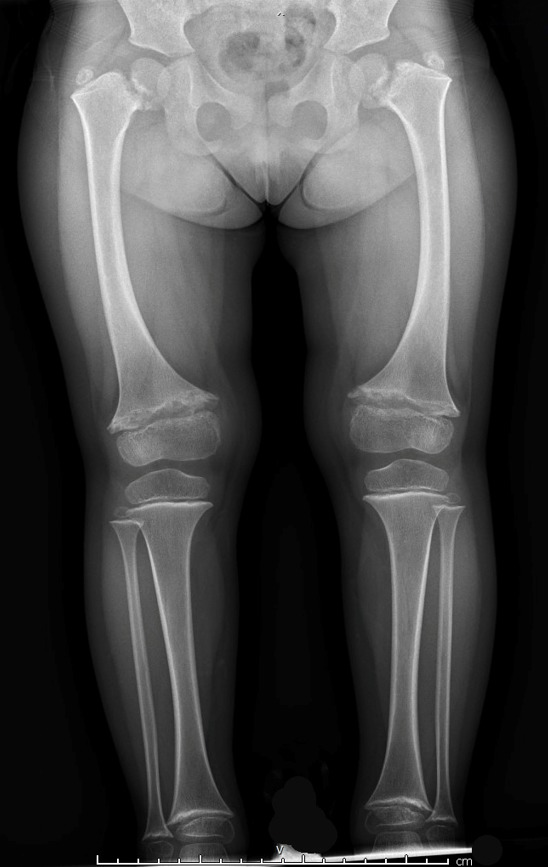
Radiographic findings. The metaphyses of the long bones become flared or widened. The proximal and distal femoral and proximal tibial metaphyses are consistently affected (ragged, cupped, sclerotic, or splayed) with widening of the growth plates. Enlarged capital femoral epiphyses are commonly reported and typically resolve between age 11 and 14 years [Tüysüz et al 2023]. Coxa vara (reduced angle to <120 degrees between the head and the shaft of the femur) is seen in a majority of individuals and is more prominent after age five years [Tüysüz et al 2023]. Coxa vara distinguishes SMCD from other forms of metaphyseal chondrodysplasia. Tibial and femoral bowing is typical. Metaphyseal irregularities of the distal ulnae and radii, and proximal and distal humeral metaphyses are less commonly reported [Lachman et al 1988, Al Kaissi et al 2018, de França et al 2020]. The anterior rib ends are often cupped or sclerotic [Bateman et al 2005].
Hand involvement is present in fewer than half of individuals and is usually mild. Metaphyseal cupping of the distal metacarpals and proximal phalanges and shortening of the phalanges may be seen and are more pronounced in the fifth ray. Hand features may become less apparent with age [Elliott et al 2005].
Vertebral involvement is less common and, when present, is usually mild. Reported findings include platyspondyly and vertebral end plate anomalies (e.g., rounding of the anterior aspects of the vertebral bodies, superior and inferior indentations of the vertebral bodies, posterior wedging of the vertebrae) [Hasegawa et al 1994, Savarirayan et al 2000]. Vertebral changes may become less apparent with age.
Obesity. Limited mobility due to chronic joint pain may contribute to the development of obesity and associated comorbidities.
Craniofacial. The craniofacial bones and facial appearance are normal [Bateman et al 2005].
Neurodevelopment. Intelligence is normal. Attainment of early motor milestones is usually preserved in infancy; however, mild gross and fine motor delays may accompany orthopedic complications in early childhood.
Other. Hypothyroidism, growth hormone deficiency, celiac disease, hypospadias, and mild microcephaly have been rarely reported [de França et al 2020, Chreitah et al 2023]; however, these may represent coincidental occurrences and may be unrelated to SMCD.
Homozygotes. Biallelic pathogenic variants in COL10A1 have been associated with a more severe phenotype [Ain et al 2018, Tüysüz et al 2023] and in some instances embryonic lethality [Zhang et al 2018].
Nomenclature
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], SMCD is referred to as metaphyseal dysplasia Schmid (MCS), COL10A1-related.
Spondylometaphyseal dysplasia (SMD) Japanese type. Affected individuals from one reported family had bowed legs, short stature, coxa vara, metaphyseal changes, and mild platyspondyly [Hasegawa et al 1994]. A heterozygous COL10A1 pathogenic variant was identified in affected individuals [Ikegawa et al 1998]. Expansion of the clinical phenotype of SMCD to include spinal changes led to the conclusion that SMD Japanese type and SMCD are the same disorder [Savarirayan et al 2000].