Clinical Description
Salih myopathy is characterized by muscle weakness manifest during the neonatal period or in early infancy, and delayed motor development. Children acquire independent walking between ages 20 months and four years. In the first decade of life, global motor performance is stable or tends to improve. During this period skeletal muscle involvement mainly manifests as difficulty in running, climbing stairs, and rising from a sitting position. Those who survive childhood remain ambulant, with or without support, and maintain normal cognitive function.
Musculoskeletal. Moderate joint and neck contractures and spinal rigidity may appear in the first decade but become more obvious in the second decade. Respiratory function tests show a moderate restrictive pattern. Scoliosis develops after age 11 years.
Cardiac dysfunction manifests between ages five and 16 years and progresses rapidly, leading to death between ages eight and 20 years. Heart rhythm disturbances are the major cause of sudden death and their frequency and severity suggest primary involvement of the conduction system.
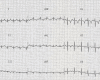
Electrocardiography (EKG) is very helpful in signaling the occurrence of cardiac involvement. Left axis deviation (left anterior fascicular block) can be seen as early as age four years (). With the onset of dilated cardiomyopathy (between ages 5 and 16 years), major rhythm disturbances become evident on EKG and Holter monitoring, including polymorphic premature ventricular complexes, bigeminism and trigeminism, couplets, triplets, atrioventricular heart block, atrioventricular nodal reentrant tachycardia, premature atrial complexes, premature ventricular complexes, and ventricular tachycardia.
Echocardiogram reveals, at the onset of cardiomyopathy, reduced function of the left ventricle and dilatation and global hypokinesia without wall hypertrophy. Later, dilatation involves the left atrium and ventricle, subsequently affecting all chambers and leading to congestive heart failure.
Radionuclide angiography using MUGA (multi-gated acquisition) scan reveals the deteriorating ventricular function with reduction of the left ventricular ejection fraction followed by reduction of the right ventricle ejection fraction.
Heart muscle biopsies (taken from 2 individuals) showed increased interstitial fibrosis compatible with dilated cardiomyopathy [Carmignac et al 2007]. Oxidative staining was normal without focal oxidative defects or significant disarray of the cardiomyocyte structure, in contrast to the classic observation in hypertrophic cardiomyopathy.
Heterozygotes. In contrast to individuals with heterozygous pathogenic variants in TTN associated with Udd distal myopathy, the heterozygous parents of individuals with Salih myopathy remain asymptomatic with no cardiac or muscle disorder ().
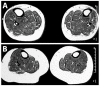
Mid-calf muscle MRI of parents of a proband at age (A) 55 years and (B) 44 years were normal and showed no fatty degeneration of the anterior tibial muscles.