Clinical Description
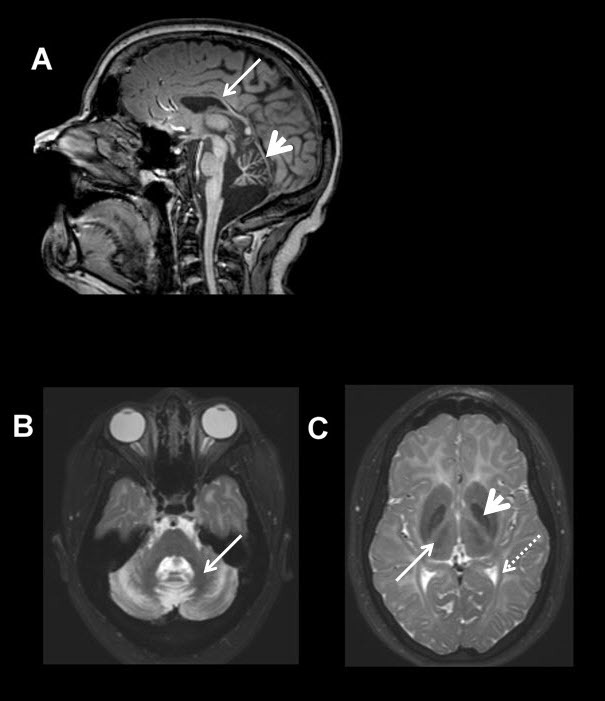
POLR3-related leukodystrophy is a hypomyelinating leukodystrophy characterized by neurologic (cerebellar, extrapyramidal, pyramidal, and cognitive) and non-neurologic (dental, endocrine, and ocular) features.
Before the identification of the involved genes, five overlapping clinical phenotypes were described and are now all recognized as part of the spectrum of POLR3-related leukodystrophy:
Age of onset is typically in early childhood but later-onset cases have also been reported.
Neurologic dysfunction includes prominent cerebellar features leading to progressive gait abnormalities, dysmetria, dysdiadochokinesis, dysarthria, and extraocular movement abnormalities including progressive loss of pursuits (saccadic ocular pursuits), abnormal saccades (i.e., hypo- or hypermetric), and gaze-evoked nystagmus. Progressive vertical more than horizontal gaze restriction has also been reported. Hypersalivation and dysphagia are also commonly seen, typically later in the disease course.
Individuals may also develop extrapyramidal features – almost always dystonia.
Mild pyramidal features are typically present but are not problematic in the vast majority of individuals.
An upper-extremity tremor (usually cerebellar in nature) with or without a dystonic component may be a presenting sign.
Cognitive decline typically occurs later, except in late-onset cases.
Approximately 10% of individuals have later onset and slower progression; they typically present with academic difficulties and cognitive decline which manifests as a plateau in learning at school. Those with later onset may have also behavioral abnormalities.
Dental abnormalities include delayed dentition, hypodontia, oligodontia, and eruption of abnormally placed or shaped teeth, among others [Wolff et al 2010]. Dental abnormalities may be subtle and have not been seen in all individuals.
Endocrine abnormalities. Hypogonadotropic hypogonadism (HH), the most typical endocrine characteristic, is note seen in all individuals. When reported, HH typically presents with delayed or arrested puberty or absence of early pubertal changes. Other reported endocrine abnormalities include short stature (in 50% of individuals [Wolf et al 2014]) and growth hormone deficiency [Potic et al 2012, Billington et al 2015, Potic et al 2015].
Ocular abnormality. Myopia, typically progressive over several years and eventually leading to severe myopia, is the most common non-neurologic feature.
Course and life span. The course of POLR3-related leukodystrophy is invariably progressive. Life span depends on the supportive measures put in place to prevent secondary complications. POLR3-related leukodystrophy is considered to be a life-limiting condition; those with earlier onset are at a higher risk for mortality in young adulthood. Individuals with later onset or with a slower progressive disease course could survive later into adulthood (i.e., 4th-5th decade).
Additional features seen less frequently include the following [Wolf et al 2005, Timmons et al 2006, Vázquez-López et al 2008, Bekiesinska-Figatowska et al 2010, Orcesi et al 2010, Wolff et al 2010, Sato et al 2011, Potic et al 2012]:
Neurophysiologic investigations. All affected individuals undergoing EMG and nerve conduction studies had normal results [Author, personal observation].
Wiedemann-Rautenstrauch syndrome (neonatal progeroid syndrome). A newborn with features of this disorder (including extreme small size, sparse scalp hair, broad abnormally shaped skull, triangular face with midface retraction, natal teeth, decreased subcutaneous fat, and limb contractures) was recently reported to have biallelic truncating variants in POLR3A on exome sequencing that were predicted to result in little residual POLR3 function. Confirmation of this as a very severe form of POLR3-related leukodystrophy awaits replication in other individuals with a clinical diagnosis of Wiedemann-Rautenstrauch syndrome [Jay et al 2016].