Summary
Clinical characteristics.
Cytochrome P450 oxidoreductase deficiency (PORD) is a disorder of steroidogenesis with a broad phenotypic spectrum including cortisol deficiency, altered sex steroid synthesis, disorders of sex development (DSD), and skeletal malformations of the Antley-Bixler syndrome (ABS) phenotype. Cortisol deficiency is usually partial, with some baseline cortisol production but failure to mount an adequate cortisol response in stress. Mild mineralocorticoid excess can be present and causes arterial hypertension, usually presenting in young adulthood. Manifestations of altered sex steroid synthesis include ambiguous genitalia/DSD in both males and females, large ovarian cysts in females, poor masculinization and delayed puberty in males, and maternal virilization during pregnancy with an affected fetus. Skeletal malformations can manifest as craniosynostosis, mid-face retrusion with proptosis and choanal stenosis or atresia, low-set dysplastic ears with stenotic external auditory canals, hydrocephalus, radiohumeral synostosis, neonatal fractures, congenital bowing of the long bones, joint contractures, arachnodactyly, and clubfeet; other anomalies observed include urinary tract anomalies (renal pelvic dilatation, vesicoureteral reflux). Cognitive impairment is of minor concern and likely associated with the severity of malformations; studies of developmental outcomes are lacking.
Diagnosis/testing.
The diagnosis of PORD can be established by urinary steroid profiling using gas chromatography / mass spectrometry (GC/MS), which documents combined impairment of 17α-hydroxylase (CYP17A1) and 21-hydroxylase (CYP21A2) enzymatic activity located at key branch points of cortisol, aldosterone, and sex steroid synthesis. Identification of biallelic POR pathogenic variants on molecular genetic testing confirms the diagnosis. Molecular genetic testing is desirable for all individuals affected by PORD to confirm the diagnosis, but is mandatory if clinical and laboratory features are inconclusive.
Management.
Treatment of manifestations: Glucocorticoid replacement therapy for cortisol deficiency including stress-dose cover in intercurrent illness; surgery as needed for craniosynostosis, hypospadias, and cryptorchidism in males and clitoromegaly and vaginal hypoplasia in females; dihydrotestosterone treatment has been successful in some males with micropenis; testosterone replacement in males in whom testosterone levels remain relatively low after onset of puberty; females with absent pubertal development may require estrogen replacement therapy; treatment with estradiol to reduce the size of ovarian cysts; endotracheal intubation, nasal stints or tracheotomy, and tracheostomy as needed; physical and occupational therapy for joint contractures and help with fine and gross motor skills.
Prevention of secondary complications: Supplementation with appropriate steroid hormones in individuals who are deficient has helped alleviate adrenal crisis, lack of or poor pubertal development in males and females, sleepiness, and fatigue. Early intervention services may improve the outcome for individuals at risk for developmental delays and learning difficulties.
Surveillance: Evaluations with a specialist tertiary pediatric endocrine service throughout childhood to closely monitor development and adjust steroid supplementation. Periodic formal developmental assessments in centers with expertise and experience in developmental testing.
Evaluation of relatives at risk: It is appropriate to evaluate apparently asymptomatic older and younger sibs of a proband in order to identify as early as possible those who would benefit from initiation of treatment and preventive measures.
Genetic counseling.
PORD is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk family members and prenatal genetic testing for pregnancies at increased risk are possible if the POR pathogenic variants have been identified in the family. In addition, noninvasive testing of maternal urine steroid excretion by GC/MS can indicate whether the unborn child is affected by PORD from gestational week 12 onwards.
GeneReview Scope
Table
Antley-Bixler syndrome Congenital adrenal hyperplasia due to apparent combined CYP17A1 and CYP21A2 enzymatic deficiency
Diagnosis
Suggestive Findings
Cytochrome P450 oxidoreductase deficiency (PORD) is an autosomal recessive disorder with a broad phenotypic spectrum including skeletal malformations resembling the Antley-Bixler syndrome (ABS) phenotype and abnormalities in adrenal steroid biosynthesis resulting in congenital adrenal hyperplasia (CAH).
Clinical Findings
Skeletal abnormalities. PORD should be suspected in individuals with features of ABS. Affected individuals may present with the following congenital craniofacial and skeletal anomalies:
- Midface retrusion
- Craniosynostosis (i.e. brachycephaly or turricephaly)
- Hand and feet malformations (arachnodactyly, clinodactyly, camptodactyly, metacarpal and metatarsal synostoses, wrist deviation, rocker-bottom feet, talipes)
- Large joint synostosis, predominantly radiohumeral or radioulnar synostosis, in severely affected individuals. Other large joints (e.g., knees, ankles) can also be affected.
- Femoral bowing
Ambiguous genitalia at birth. The majority of individuals with PORD have disordered sex development (DSD) and present with ambiguous genitalia at birth. DSD can occur in both sexes:
- Females may present with masculinized genitalia (46,XX DSD; e.g., enlarged clitoris, labial fusion).
- Males can present undermasculinized (46,XY DSD; e.g., hypospadias, micropenis).
Laboratory Findings
Because POR is required for normal enzymatic function at various steps within the cholesterol and steroid synthesis pathways, individuals with PORD exhibit characteristic abnormalities in both sterol and steroid metabolism (see Figures 1, 2, and 3).
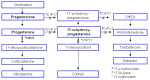


Serum steroid abnormalities
- ACTH plasma concentration is normal or elevated at baseline.
- Cortisol serum concentration is normal or low at baseline, and may not increase as expected following ACTH stimulation.
- Pregnenolone, progesterone, 17-OH pregnenolone, and 17-OH progesterone serum concentrations are often elevated at baseline and/or after ACTH stimulation.
- Dehyroepiandrosterone (DHEA), dehydroepiandrosterone sulfate (DHEAS), and androstenedione serum concentrations are normal or decreased before and/or after ACTH stimulation.
- Androgen serum concentration may be low and unresponsive to ACTH or hCG stimulation.
Urinary steroid anomalies detected by gas chromatography / mass spectrometry (GC/MS). Steroid abnormalities in individuals with PORD are consistent with attenuated activity of 21-hydroxylase (encoded by CYP21A2) and 17α-hydroxlyase/17,20 lyase activities (encoded by CYP17A1) [Krone et al 2012] and are characterized by:
- Increased concentration of pregnenediol (metabolite of pregnenolone) and pregnanediol (metabolite of progesterone);
- Significantly elevated ratio of metabolites associated with:
- Deficiency of 17α-hydroxylase (5α-tetrahydrocorticosterone, tetrahydrocorticosterone, and 11-dehydro metabolites);
- Deficiency of 21-hydroxylase (17α-hydroxypregnanolone, pregnanetriol, and pregnanetriolone).
Note: The term "apparent pregnene hydroxylation deficiency (APHD)" refers to individuals with this unique urinary steroid profile [Shackleton & Malunowicz 2003]. Despite sharing common characteristics, steroid profiles vary somewhat among affected individuals, presumably because of differences in how the various POR pathogenic variants affect different enzymatic reactions [Pandey et al 2007, Huang et al 2008, Dhir et al 2009, Miller et al 2009].
Evidence of steroid anomalies during pregnancy. Low or undetectable maternal serum unconjugated estriol (uE3) and/or failure of urinary E3 excretion to increase have been noted during pregnancies in which fetuses have PORD [Cragun et al 2004, Shackleton et al 2004]. Prenatal testing for PORD by maternal urinary steroid profiling (GC/MS) has been developed as a sensitive tool to establish the diagnosis in the fetus [Reisch et al 2013] (see Prenatal Testing and Preimplantation Genetic Testing).
Newborn screening for congenital adrenal hyperplasia. In some individuals with PORD, newborn screening for CAH may be positive with moderately elevated serum 17-OH progesterone [Fukami et al 2005]. However, newborn screening does not appear to be sensitive enough to detect all individuals with PORD.
Cholesterol abnormalities. Subtle sterol abnormalities consistent with a partial block in cholesterol synthesis at the level of CYP51 may be present (Figure 3) [Kelley et al 2002, Cragun et al 2004, Fukami et al 2005]. CYP51 catalyzes the conversion of lanosterol into principal intermediates of the distal portion of the cholesterol biosynthesis pathway. Although serum concentrations of cholesterol are grossly normal in individuals with ABS [Fukami et al 2005], lanosterol and dihydrolanosterol accumulate when cells from affected individuals are grown in cholesterol-depleted medium. Sterol profiling of amniotic fluid in an affected pregnancy may reveal di- and trimethylated sterols, but this finding is not unique to PORD [Chevy et al 2005].
Establishing the Diagnosis
The diagnosis of PORD is established in a proband with the characteristic urinary steroid profile:
- Increased concentration of metabolites of pregnenolone (pregnenediol) and progesterone (pregnanediol)
- Significantly elevated metabolites associated with:
- Deficiency of 17α-hydroxylase (5α-tetrahydrocorticosterone, tetrahydrocorticosterone, and 11 dehydrometabolites)
- Deficiency of 21-hydroxylase (17α-hydroxypregnanolone, pregnanetriol, and pregnanetriolone)
Identification of biallelic pathogenic (or likely pathogenic) variants in POR on molecular genetic testing confirms the diagnosis (see Table 1). Molecular genetic testing is desirable for all individuals affected by PORD to confirm the diagnosis, but mandatory if clinical and laboratory features are inconclusive.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic POR variants of uncertain significance (or of one known POR pathogenic variant and one POR variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular testing approaches can include single-gene testing, use of a multigene panel, and more comprehensive genomic testing:
- Single-gene testing. Sequence analysis of POR is performed first and followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
- A multigene panel that includes POR and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
- More comprehensive genomic testing (when available) including exome sequencing, mitochondrial sequencing, and genome sequencing may be considered. Such testing may provide or suggest a diagnosis not previously considered (e.g., mutation of a different gene or genes that results in a similar clinical presentation). For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Cytochrome P450 Oxidoreductase Deficiency
Clinical Characteristics
Clinical Description
The natural history of cytochrome P450 oxidoreductase deficiency (PORD) varies because it encompasses a wide phenotypic spectrum. However, steroid abnormalities, which occur in all individuals with PORD, can be associated with a number of characteristics.
The summary of clinical characteristics is based on 26 studies on 140 individuals with molecularly confirmed PORD published to date (June 2017).
Cortisol deficiency found in PORD varies, but is present in the majority of individuals.
Based on ACTH stimulation tests, Krone et al [2012] reported severe cortisol deficiency (requiring permanent hydrocortisone replacement) in 43% of individuals, and partial cortisol deficiency (requiring glucocorticoid replacement during stress only) in 40%; no replacement was required in 10% of the cohort.
Mineralocorticoid excess due to inhibition of 17α-hydroxylase activity can result in hypertension, which typically manifests in young adulthood.
Disorders of sex development (DSD) occur in approximately 75% of individuals with molecularly confirmed PORD. DSD can occur in both sexes: 46,XY DSD (e.g., small penis, undescended testes) and 46,XX DSD (e.g., enlarged clitoris, fused and hypoplastic labia). The unusual finding that both sexes can present with DSD (e.g., virilized genitalia in 46,XX and undermasculinized genitalia in 46,XY) was suggested to be caused by the presence of an alternative pathway to dihydrotestosterone [Arlt et al 2004] (see Molecular Genetics). The manifestation of DSD is related to genotype, with some pathogenic variants leading to normal male virilization and 46,XX DSD in girls while other pathogenic variants result in normal female genital appearance and 46,XY DSD in boys (see Genotype-Phenotype Correlations).
Primary amenorrhea was the presenting feature in at least one woman with PORD (milder phenotype) [Scott et al 2007].
Large ovarian cysts with a tendency to spontaneous rupture are present in a number of adolescent and adult females with PORD [Scott et al 2007, Fukami et al 2009, Idkowiak et al 2011].
Poor masculinization and delayed puberty have been reported in some males, but spontaneous progression during puberty has also been observed [Fukami et al 2005, Hershkovitz et al 2008, Idkowiak et al 2011]. Hypospermatogenesis was documented on testicular biopsy in a male with PORD [Fukami et al 2005].
Fertility may be a concern. No reports describe reproduction in individuals with PORD; thus, the prevalence of infertility among individuals with PORD remains uncertain.
Signs of maternal virilization during pregnancy with an affected fetus, including hirsutism, enlargement of the nose and lips, deepening of the voice, and acne, have been reported in women during pregnancies in which fetuses were later found to have PORD [Fukami et al 2009, Krone et al 2012, Reisch et al 2013].
Skeletal abnormalities of the Antley-Bixler syndrome (ABS) phenotype are frequently observed in individuals with PORD. The severity of malformations varies from mild to moderate and severe. Functional studies are currently lacking but individuals with milder skeletal features similar to those in classic ABS probably have moderate PORD. Those with mild PORD tend to have few if any notable physical characteristics. Krone et al have introduced a clinical scoring system rating the severity of the skeletal and craniofacial malformations in PORD (Table 2) [Krone et al 2012], which is useful for systematic assessment of malformations in PORD.
- Skeletal malformations occur in approximately 85% of individuals with molecularly confirmed PORD. Elbow ankylosis, often from radiohumeral synostosis, causes fixation of the elbow in a flexed position. Elbow extension may be restricted in the absence of radiohumeral synostosis. Neonatal fractures and congenital bowing of the long bones (especially the femurs) are common. Other common malformations of the limbs include long palms, camptodactyly, other joint contractures, arachnodactyly, clubfeet, irregularly positioned toes, and rocker-bottom feet. Vertebral and rib anomalies, hypoplasia of the scapula, scoliosis, and narrow chest and/or pelvis have been reported. Other skeletal anomalies reported in individuals with PORD include diastases of the radioulnar joint, ulnar deviation of the wrists, marfanoid habitus, flattened metacarpal epiphyses, cubitus valgus, brachymetacarpia, and brachytelephalangy.
- Craniofacial anomalies. Craniosynostosis is usually severe and most commonly involves the coronal and lambdoid sutures, resulting in turricephaly; synostosis of other cranial sutures has also been reported. Other craniofacial anomalies include frontal bossing, enlarged anterior fontanelle, severe midface retrusion, choanal stenosis or atresia, short bulbous nose, depressed nasal bridge, narrow mouth, high arched narrow palate, and dysplastic ears that may be low-set with stenotic external auditory canals. In milder forms, the craniofacial features – if present – may not be as easily identified at birth and/or tend to be less severe than those in individuals with severe disease. Although craniosynostosis and/or brachycephaly may be observed, surgical treatment may not require as many procedures. Individuals may have conductive hearing loss.
- Hydrocephalus requiring ventriculoperitoneal shunt insertion has been reported in a number of affected individuals [Krone et al 2012].
Table 2.
PORD Skeletal Malformation Scoring System Classifying the Major Features of the Antley-Bixler Syndrome Phenotype
Associated anomalies, presumably not related to the disruption of sterol or steroid synthesis, are rare in individuals with PORD. These include urinary tract anomalies including several individuals with renal pelvic dilatation and vesicoureteral reflux [Krone et al 2012, Bonamichi et al 2016] and one individual with unilateral renal agenesis [Krone et al 2012]. Gastrointestinal conditions are reported in one individual with PORD who developed severe gastroesophageal reflux and constipation [Williamson et al 2006] and another individual with an anteriorly placed anus [Krone et al 2012]. Other observations include a two vessel umbilical cord in two individuals with PORD; Arnold-Chiari malformation and a frontal capillary hemangioma have each been reported in one individual [Krone et al 2012].
Cognitive function and development. There is a paucity of data on cognitive function and developmental outcomes in individuals with PORD. Developmental delays have been reported in a number of children with PORD, mainly delayed speech and language development and fine motor skills, presumably secondary to conductive hearing loss, skeletal abnormalities, multiple surgical procedures with anesthesia, and prolonged hospitalization with immobility [Williamson et al 2006, Sahakitrungruang et al 2009]. Early and effective management of upper airway obstruction, craniosynostosis, hydrocephalus, and hearing loss appear to be a prerequisite for good cognitive development.
Prognosis is primarily determined by the severity of the skeletal and craniofacial malformations. Stillbirth has been reported in infants with very severe skeletal malformations [Krone et al 2012, Reisch et al 2013]. In individuals with mild to moderate clinical features, the prognosis is guarded in infancy and improves with age. Early death caused by respiratory complications is a concern. However, with careful airway management, many children with ABS survive and the prognosis may be reasonably good. Prospective follow-up studies describing the natural course of PORD are currently lacking.
Genotype-Phenotype Correlations
While individuals with the same pathogenic variants (even sibs) can show phenotypic variations, some commonalities are observed among individuals with the same genotype. The broad phenotypic spectrum of PORD may be caused by the effect of various POR pathogenic variants on different enzymatic reactions [Huang et al 2005, Dhir et al 2007, Pandey et al 2007, Huang et al 2008, Burkhard et al 2017].
Three studies report the investigation of genotype-phenotype correlations in larger cohorts of individuals with PORD [Huang et al 2005, Fukami et al 2009, Krone et al 2012]. The observations from these studies include the following.
Skeletal malformations
- The overall reported frequency of skeletal malformations in genetically confirmed PORD is about 85%.
- Homozygotes for p.Arg457His tend to have less severe skeletal abnormalities.
- Compound heterozygosity for p.Arg457His and a severe loss-of-function pathogenic variant on the other allele is associated with a more severe skeletal phenotype.
- Homozygotes for p.Ala287Pro have moderate skeletal malformations.
- Compound heterozygosity for p.Ala287Pro and a severe loss-of-function pathogenic variant on the other allele is associated with a severe skeletal phenotype.
Disorders of sex development (DSD). Homozygosity for either pathogenic variant p.Arg457His or p.Ala287Pro causes 46,XX DSD (virilized females at birth) but undermasculinization occurs less frequently in 46,XY individuals.
Adrenal steroid biosynthesis
- An abnormal urinary steroid pattern has been found in all persons with genetically confirmed PORD investigated; however, not all reported patients have had a urinary steroid profile as part of their work-up.
- Adrenal insufficiency is present in most persons with PORD: based on ACTH stimulation tests, Krone et al [2012] reported hydrocortisone replacement in 43%, stress dose cover only in 40%, and no replacement in 10% of his cohort; Fukami et al [2009] found the prevalence of adrenal crisis was increased in individuals with pathogenic variant p.Arg457His combined with a severe loss-of-function pathogenic variant.
- However, a prediction of the overall severity of glucocorticoid deficiency based on genotype is not possible.
Nomenclature
Peterson et al [1985] first reported a male with ambiguous genitalia and apparent combined partial 21-hydroxylase (P450c21, CYP21A2) deficiency and partial 17-hydroxylase (P450c17, CYP17A1) deficiency, and referred to this condition as "mixed oxidase disease" [Peterson et al 1985]. This condition, also known as congenital adrenal hyperplasia due to apparent combined CYP21A1 and CYP17A1 deficiency, has been shown to be caused by biallelic POR pathogenic variants [Arlt et al 2004, Flück et al 2004].
Prevalence
The prevalence of PORD has yet to be determined, however it is a very rare condition. Since POR pathogenic variants were first reported in 2004, approximately 140 individuals with PORD have been reported in the literature (June 2017).
The most prevalent pathogenic variant in individuals with PORD of Japanese ancestry is p.Arg457His. In individuals of European background with PORD, p.Ala287Pro is the most prevalent pathogenic variant.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in POR.
Differential Diagnosis
Congenital adrenal hyperplasia (CAH) is a heterogeneous group of autosomal recessive conditions that result in impaired synthesis of cortisol, mineralocorticoids, and/or sex steroids. Based on this definition, the term CAH can be used to describe cytochrome P450 oxidoreductase deficiency (PORD). PORD and the following etiologies of CAH may be distinguished by differences in urinary steroid profiles, molecular genetic testing, and/or the presence of skeletal anomalies, as skeletal anomalies are never found in other forms of CAH, but may occur in PORD. POR acts as an electron donor to two major steroidogenic enzymes, CYP21A2 and CYP17A1. Therefore, individuals with POR show biochemical features of both 21-hydroxylase and 17α-hydroxylase deficiency.
- 21-hydroxylase deficiency (21-OHD, CYP21A2 deficiency), the most common form of CAH, is associated with glucocorticoid and, in most cases, mineralocorticoid deficiency as well as with sex steroid excess resulting in masculinization of the external genitalia in females (46,XX DSD). Unlike PORD, 21-OHD is characterized by increased circulating androgens and progressive virilization of females after birth.
- 17α-hydroxylase/17,20-lyase deficiency (CYP17A1 deficiency) (OMIM 202110) is associated with mineralocorticoid excess, glucocorticoid deficiency and sex steroid deficiency, which is clinically associated with ambiguous genitalia in males (46,XY DSD) and lack of pubertal progression in both sexes. Milder pathogenic variants on the other end of a broad phenotypic spectrum might affect sex steroid production only ("isolated 17,20-lyase deficiency").
- 11β-hydroxylase deficiency (OMIM 202010) is associated with glucocorticoid deficiency but excess of both mineralocorticoids and sex steroids – therefore, with hypertension and ambiguous genitalia in females (46,XX DSD).
- 3β-hydroxysteroid dehydrogenase deficiency (OMIM 201810) is associated with glucocorticoid and mineralocorticoid deficiency and leads to ambiguous genitalia in males (46,XY DSD), but in rare cases also in females (46,XX DSD).
Antley-Bixler syndrome (ABS) without genital anomalies or disordered steroidogenesis (OMIM 207410) is characterized by the skeletal features of ABS including craniosynostosis, radioulnar and radiohumeral synostosis, midface hypoplasia with narrowing of the upper airway, frontal bossing, arachnodactyly and camptodactyly, femoral bowing, irregular positioned toes, and rocker-bottom feet. Pathogenic variants in FGFR2 are causative. Inheritance is autosomal dominant.
Autosomal dominant craniosynostosis syndromes. Craniosynostosis occurs in the autosomal dominant FGFR-related craniosynostosis syndromes that include Pfeiffer syndrome, Crouzon syndrome, Jackson-Weiss syndrome, Apert syndrome, and Beare-Stevenson syndrome (see FGFR-Related Craniosynostosis).
- Pfeiffer and Crouzon syndromes. Some individuals with severe FGFR-related craniosynostosis have clinical features that overlap ABS. Individuals with FGFR pathogenic variants may have more severe proptosis compared to those with PORD. Although common in PORD, a short bulbous nose, low-set and dysplastic ears, arachnodactyly, and rocker-bottom feet are not usually described in FGFR-related craniosynostosis syndromes. In Pfeiffer syndrome, limb deformities are part of the spectrum (e.g., broad thumbs and toes, brachydactyly, tarsal fusion). FGFR1 and FGFR2 pathogenic variants are causative.
- Apert syndrome can usually be distinguished from ABS by the presence of the typical syndactyly. FGFR2 pathogenic variants are causative.
- Muenke syndrome is caused by FGFR3 pathogenic variant p.Pro250Arg. Muenke syndrome is characterized by uni-or bilateral synostosis of the coronal suture. Deformities of the hand and feet (e.g., broad toes, carpal/tarsal fusions) can be part of the spectrum.
- Saethre-Chotzen syndrome is characterized by uni-or bilateral coronal synostosis, facial asymmetry, ptosis, ear anomalies, and syndactyly in some individuals. Pathogenic variants of TWIST1 are causative.
Cytochrome P450 26B1 (CYP26B1) deficiency (OMIM 614416) is characterized by craniosynostosis, oligodactyly, femoral bowing, radiohumeral synostosis, narrow thorax, and small pelvic bones [Laue et al 2011, Morton et al 2016]. The spectrum of severity in the small number of individuals reported has included perinatal lethal, early infant demise, and a milder phenotype in a female age 22 years, which resembled ABS and Pfeiffer syndrome [Morton et al 2016]. Individuals with cytochrome P450 26B1 deficiency are not reported to have features consistent with abnormal steroid metabolism. Biallelic CYP26B1 pathogenic variants are causative.
Thanatophoric dysplasia. The combination of femoral bowing and craniosynostosis may be seen in thanatophoric dysplasia, but genital anomalies do not occur in this condition. Unlike ABS, thanatophoric dysplasia is characterized by severe rhizomelia. Brain malformations and severe intellectual disability are also universal features of thanatophoric dysplasia but are not reported as part of PORD [Wang et al 2014, Weaver et al 2014]. Thanatophoric dysplasia is caused by pathogenic variants in FGFR3 and is inherited in an autosomal dominant manner; the majority of probands have a de novo pathogenic variant.
Shprintzen-Goldberg syndrome (SGS) overlaps with PORD skeletal malformations in that both can have camptodactyly, arachnodactyly, femoral bowing, craniosynostosis, and a marfanoid habitus. However, individuals with SGS do not have ambiguous genitalia or the distinctive facial features of PORD. SGS is associated with significant cognitive disability and brain malformations. At least two individuals with PORD without ABS-like malformations were originally classified as having SGS. Pathogenic variants in SKI are causative.
Bent-bone dysplasias, which include campomelic dysplasia, kyphomelic dysplasia (OMIM 211350), and Stüve-Wiedemann syndrome (OMIM 601559), are commonly associated with long-bone bowing, primarily of the femora. These conditions are distinguished from PORD by the lack of craniosynostosis or radiohumeral synostosis. Long-bone fractures occur in campomelic dysplasia, but usually after the neonatal period, whereas fractures in PORD usually occur during the neonatal period. Campomelic dysplasia is often associated with abnormal sexual development (e.g., hypospadias to phenotypic female with a 46,XY karyotype) and is caused by pathogenic variants in SOX9. Campomelic dysplasia is inherited in an autosomal dominant manner but is most commonly the result of a de novo pathogenic variant.
Osteogenesis imperfecta (OI; see COLA1/2-Related Osteogenesis Imperfecta) is associated with neonatal fractures, but lacks the characteristic craniofacial, limb, and urogenital anomalies of PORD. Unlike OI, PORD is not associated with osteoporosis or wormian bones.
Teratogen exposure. Early prenatal exposure to oral, high-dose fluconazole has resulted in an ABS-like phenotype in five reported individuals [Aleck & Bartley 1997, Lopez-Rangel & Van Allen 2005]. However, frontal bossing, choanal stenosis/atresia, genital abnormalities, and camptodactyly were not observed. Pregnancy history is important in identifying this exposure.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with PORD, the following evaluations are recommended:
- Evaluations by appropriate specialists in endocrinology, clinical genetics, neurosurgery, otolaryngology, and cardiology
- Assessment for airway problems in individuals with skeletal malformations
- Functional adrenal studies (cosyntropin test) to assess glucocorticoid deficiency, regardless of the presence or absence of genital abnormalities
- Additional studies that may be indicated:
- Cranial CT scan and/or MRI to determine the degree of craniosynostosis, hydrocephaly, choanal stenosis, and orbital depth
- Radiographs to identify long-bone fractures and/or bowing, bony synostoses, and/or joint contractures
- Echocardiogram if a heart defect is suspected
- Abdominal and pelvic ultrasound examination to identify internal sex organs, detect any renal anomalies, and detect and monitor ovarian cysts in adolescent girls.
Treatment of Manifestations
Cortisol deficiency
- Regular hydrocortisone replacement therapy is indicated if baseline serum cortisol concentrations are low.
- Stress-dose steroids should be provided perioperatively and during times of physiologic stress in individuals in whom cortisol response to ACTH stimulation (cosyntropin test) is below normal [Krone et al 2012].
Genital abnormalities
- Hypospadias and cryptorchidism may be corrected with surgery.
- When clitoromegaly is severe, surgical reduction and plastic reconstruction of the clitoris may be considered.
- Vaginal reconstruction may be performed in females with vaginal hypoplasia.
- Dihydrotestosterone treatment has been successful in some males with micropenis [Fukami et al 2005].
- Testosterone replacement has been initiated in males in whom testosterone levels remained relatively low after onset of puberty [Hershkovitz et al 2008, Idkowiak et al 2011]. Similarly, females with absent pubertal development may require estrogen replacement therapy.
Ovarian cysts. Treatment with estradiol appeared to successfully reduce the size of ovarian cysts in females with PORD [Fukami et al 2009, Idkowiak et al 2011]. Ovarian cysts in females can be a significant problem as they tend to be large and prone to spontaneous rupture; cases of girls treated with GnRH agonists and potent steroids have been reported [Idkowiak et al 2011].
Craniosynostosis. Treatment for craniosynostosis is similar to that for other syndromes associated with premature fusion of cranial sutures. Although surgical correction can be performed at any age, it is generally believed that earlier surgical correction results in better cognitive outcome.
Airway management is often a primary concern in individuals with ABS as a result of choanal stenosis or atresia, small chest, narrow trachea, and/or shortening of the larynx.
- Endotracheal intubation is often required in the first minutes after delivery.
- Nasal stints or tracheotomy may be required.
- Tracheostomy may be necessary until age three to five years when the pharyngeal encroachment can be corrected.
Hydrocephalus. If present, hydrocephalus may be treated by surgical placement of a ventriculoperitoneal shunt.
Joint contractures and elbow synostosis. Physical and occupational therapy can help individuals with contractures and elbow synostosis develop fine and gross motor skills.
Prevention of Secondary Complications
Supplementation with appropriate steroid hormones in individuals who are deficient has helped alleviate:
- Adrenal crisis
- Lack of or poor pubertal development in males and females
- Sleepiness and fatigue
Early intervention services may improve the outcome for individuals at risk for developmental delays and learning difficulties.
Surveillance
Individuals with PORD should be seen by a specialist tertiary pediatric endocrine service throughout childhood to closely monitor their development and adjust steroid supplementation.
Because of the presence of developmental delays in many individuals with ABS, periodic formal developmental assessments may be indicated. However, interpretation of these assessments may be complicated by the physical limitations of the disorder. Screening evaluations are likely to underestimate cognitive abilities. Therefore, evaluations should be done in centers with expertise and experience in developmental testing.
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger sibs of a proband in order to identify as early as possible those who would benefit from initiation of treatment and preventive measures. Evaluations can include:
- Molecular genetic testing if the pathogenic variants in the family are known;
- Urinary steroid profiling using gas chromatography / mass spectrometry (GC/MS) can be done if the pathogenic variants in the family are not known. The characteristic urinary steroid profile:
- Increased coancentration of metabolites of pregnenolone (pregnenediol) and progesterone (pregnanediol)
- Significantly elevated metabolites associated with:
- Deficiency of 17α-hydroxylase (5α-tetrahydrocorticosterone, tetrahydrocorticosterone, and 11 dehydrometabolites)
- Deficiency of 21-hydroxylase (17α-hydroxypregnanolone, pregnanetriol, and pregnanetriolone)
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Fertility may be a concern. No reports describe reproduction in individuals with PORD; thus, the prevalence of infertility among individuals with PORD remains uncertain.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Hepatic drug metabolism. NADPH--cytochrome P450 reductase (POR) plays an important role in the metabolism of various drugs and endogenous metabolites/hormones by hepatic microsomal (type 2) P450 cytochromes. Initial studies using bacterially expressed POR pathogenic variants and two hepatic P450 enzymes suggest that pathogenic variants in POR may alter drug metabolism [Miller et al 2009]. In these studies different pathogenic variants resulted in different effects, ranging from no apparent activity to elevated activity of the hepatic P450 enzymes; these activities were not correlated with their activity in CYP17A1 assays.
In vitro activity assays on major drug-metabolizing enzymes and in vivo investigations to assess the impact of various pathogenic and non-pathogenic variants of POR have been performed by various groups [Agrawal et al 2008, Hart & Zhong 2008, Kranendonk et al 2008, Gomes et al 2009, Oneda et al 2009, Agrawal et al 2010, Flück et al 2010, Tomalik-Scharte et al 2010]. For comprehensive reviews on the pharmacogenetics of POR see Pandey & Sproll [2014] and Burkhard et al [2017].
In brief, the different pathogenic variations have different effects on microsomal P450 drug metabolizing enzymes. The common pathogenic variant p.Ala287Pro reduces the activity of the major P450 cytochrome CYP3A4 (which metabolizes ~50% of clinically used drugs) by more than 75% [Nicolo et al 2010]. In vivo cocktail phenotyping in an individual homozygous for the p.Ala287Pro pathogenic variant confirmed altered hepatic detoxification of a variety of drugs due to impaired activity of various hepatic CYP enzymes, including CYP3A4, CYP1A1, CYP2C9, and CYP2D6; the heterozygous mother of this individual also showed impaired activities of CYP1A1 and CYP2C9 [Tomalik-Scharte et al 2010]. In addition, in vitro assays suggest that some variants cause a substrate-specific modulation of CYP3A4 [Agrawal et al 2010].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Cytochrome P450 oxidoreductase deficiency (PORD) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one POR pathogenic variant).
- Heterozygotes (carriers) are typically asymptomatic but may have altered hepatic drug metabolism [Tomalik-Scharte et al 2010] (see Management, Other).
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are typically asymptomatic but may have altered hepatic drug metabolism [Tomalik-Scharte et al 2010] (see Management, Other).
Offspring of a proband. The offspring of an individual with PORD would be obligate heterozygotes (carriers) for a pathogenic variant in POR. However, no reports describe reproduction in individuals with PORD.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a POR pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the POR pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Maternal serum screening. Low or undetectable levels of maternal serum unconjugated estriol (uE3) have been noted in women carrying a fetus with PORD. Low uE3, however, is not specific and frequently found in other conditions affecting steroid and sterol synthesis, such as Smith-Lemli-Opitz syndrome, STS deficiency, aromatase deficiency, and some forms of severe congenital adrenal hypoplasia.
Maternal urine steroid profiling. PORD can be established by urinary steroid profiling by gas chromatography / mass spectrometry (GC/MS) [Reisch et al 2013]: a distinctive abnormal und uniform steroid profile was observed from week 12 onwards in a prospective study of genetically confirmed fetuses/babies with PORD. Maternal urine shows elevated levels of the pregnenolone metabolite epiallopregnanediol and androgen metabolites of fetal origin with low levels of estriol.
Fetal ultrasound examination. Prenatal diagnosis of PORD by mid-trimester ultrasound examination is possible and would detect skeletal malformations in severely affected fetuses. Fixed flexion of the elbows is an important finding; other supportive findings include bowing of the long bones, midface retrusion, depressed nasal bridge, brachycephaly, and rocker-bottom feet. Individuals with less severe features are unlikely to be identified on ultrasound examination: in a cohort of 20 pregnancies examined, 19 had overt malformations at birth but only five had detectable malformations with fetal ultrasound; the detection of fetal malformations on antenatal ultrasound predicts a very severe phenotype [Reisch et al 2013].
Molecular genetic testing. Once the POR pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Congenital Adrenal Hyperplasia Research, Education and Support (CARES) Foundation2414 Morris AveSuite 110Union NJ 07083Phone: 866-227-3737Email: [email protected]
- Living with CAHCLIMB Congenital Adrenal Hyperplasia Support Group
- Children's Craniofacial AssociationPhone: 800-535-3643Email: [email protected]
- Face Equality InternationalUnited Kingdom
- FACES: National Craniofacial AssociationPhone: 800-332-2373; 423-266-1632Email: [email protected]
- National Institute of Neurological Disorders and Stroke (NINDS)Phone: 800-352-9424
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Cytochrome P450 Oxidoreductase Deficiency: Genes and Databases
Table B.
OMIM Entries for Cytochrome P450 Oxidoreductase Deficiency (View All in OMIM)
Molecular Pathogenesis
Gene structure. POR consists of 16 exons; exon 1 is a non-coding exon upstream of the translation initiation site (NM_000941.2). For a detailed summary of gene and protein information, see Table A, Gene.
Benign variants and population genetics. Huang et al [2008] sequenced POR in 842 healthy Americans from different ethnic backgrounds (northern European, African, Chinese, and Mexican). Some of the missense variants were associated with decreased catalytic activity during in vitro studies, however the majority maintained more than 40% of wild type activity in most in vitro assays employed. The most common benign sequence variant, NM_000941.2:c.1508C>T (p.Ala503Val), found in 20%-35% of the ethnic groups studied, showed decreased catalytic activity (50%-60%) for CYP17A1, but no decreased activity for CYP1A2 or 2C19 [Miller et al 2009]. Recently, Burkhard et al [2017] reported POR variants from the 1000 genomes project.
Pathogenic variants. More than 75 pathogenic variants have been reported in approximately 140 individuals with PORD. They include missense/nonsense variants, splice site variants, small deletions, insertions, and duplications, and large deletions or insertions (see Table 1 and Table A, Locus Specific and HGMD).
Two pathogenic variants, p.Ala287Pro and p.Arg457His, are common in distinct ethnic groups: p.Ala287Pro accounts for about 40% of pathogenic variants among individuals of European ancestry and p.Arg457His accounts for about 60% of pathogenic variants among individuals of Japanese ancestry, although it is important to note that p.Arg457His has also been reported in individuals of European and African ancestry [Scott & Miller 2008].
Table 3.
POR Pathogenic Variants Discussed in This GeneReview
Normal gene product. NADPH--cytochrome P450 reductase (POR) comprises 680 amino acids and is required for the activity of all 50 of the known human microsomal (type II) P450 enzymes, which play an important role in steroid and sterol synthesis as well as hepatic drug metabolism. POR binds NADPH and accepts a pair of electrons through its FAD moiety. Electrons are then transferred to the FMN moiety and then directly to P450 enzymes.
Abnormal gene product. Expression of recombinant mutated proteins with amino acid substitutions in yeast or bacteria revealed a deficient or reduced capacity for various POR mutants to oxidize cytochrome c, NADPH, CYP17A1, CYP21A2, CYP19A1, CYP3A4, CYP2D6, CYP1A2, and CYP2C19 [Arlt et al 2004, Flück et al 2004, Huang et al 2005, Huang et al 2006, Dhir et al 2007, Pandey et al 2007, Kranendonk et al 2008]. The diminished enzyme activity is believed to be caused by the effects of the amino acid substitution on steric conformation, charge, and/or FAD-binding affinity. POR pathogenic variants impair the activity of each P450 enzyme to different degrees.
Pathogenic variants that create a premature stop codon are predicted to result in a truncated nonfunctional protein [Adachi et al 2004, Arlt et al 2004, Flück et al 2004, Fukami et al 2005].
A murine knockout of Por results in embryonic lethality [Shen et al 2002, Otto et al 2003], while a liver-specific knockout results in a normal morphologic and reproductive phenotype [Gu et al 2003].
References
Literature Cited
- Adachi M, Tachibana K, Asakura Y, Yamamoto T, Hanaki K, Oka A. Compound heterozygous mutations of cytochrome P450 oxidoreductase gene (POR) in two patients with Antley-Bixler syndrome. Am J Med Genet A. 2004;128A:333–9. [PubMed: 15264278]
- Agrawal V, Choi JH, Giacomini KM, Miller WL. Substrate-specific modulation of CYP3A4 activity by genetic variants of cytochrome P450 oxidoreductase. Pharmacogenet Genomics. 2010;20:611–8. [PMC free article: PMC2940949] [PubMed: 20697309]
- Agrawal V, Huang N, Miller WL. Pharmacogenetics of P450 oxidoreductase: effect of sequence variants on activities of CYP1A2 and CYP2C19. Pharmacogenet Genomics. 2008;18:569–76. [PubMed: 18551037]
- Aleck KA, Bartley DL. Multiple malformation syndrome following fluconazole use in pregnancy: report of an additional patient. Am J Med Genet. 1997;72:253–6. [PubMed: 9332650]
- Arlt W, Walker EA, Draper N, Ivison HE, Ride JP, Hammer F, Chalder SM, Borucka-Mankiewicz M, Hauffa BP, Malunowicz EM, Stewart PM, Shackleton CHL. Congenital adrenal hyperplasia caused by mutant P450 oxidoreductase and human androgen synthesis: analytical study. Lancet. 2004;363:2128–35. [PubMed: 15220035]
- Bonamichi BDSF, Santiago SLM, Bertola DR, Kim CA, Alonso N, Mendonca BB, Bachega TASS, Gomes LG. Long-term follow-up of a female with congenital adrenal hyperplasia due to P450-oxidoreductase deficiency. Arch Endocrinol Metab. 2016;60:500–4. [PMC free article: PMC10118638] [PubMed: 27737328]
- Burkhard FZ, Parween S, Udhane SS, Flück CE, Pandey AV. P450 oxidoreductase deficiency: analysis of mutations and polymorphisms. J Steroid Biochem Mol Biol. 2017;165:38–50. [PubMed: 27068427]
- Chevy F, Humbert L, Wolf C. Sterol profiling of amniotic fluid: a routine method for the detection of distal cholesterol synthesis deficit. Prenat. Diagn. 2005;25:1000–6. [PubMed: 16231320]
- Cragun DL, Trumpy SK, Shackleton CHL, Kelley RI, Leslie ND, Mulrooney NP, Hopkin RJ. Undetectable maternal serum uE3 and postnatal abnormal sterol and steroid metabolism in Antley-Bixler syndrome. Am J Med Genet A. 2004;129A:1–7. [PubMed: 15266606]
- Dhir V, Ivison HE, Krone N, Shackleton CHL, Doherty AJ, Stewart PM, Arlt W. Differential inhibition of CYP17A1 and CYP21A2 activities by the P450 oxidoreductase mutant A287P. Mol Endocrinol. 2007;21:1958–68. [PubMed: 17505056]
- Dhir V, Reisch N, Bleicken CM, Lebl J, Kamrath C, Schwarz H-P, Grötzinger J, Sippell WG, Riepe FG, Arlt W, Krone N. Steroid 17alpha-hydroxylase deficiency: functional characterization of four mutations (A174E, V178D, R440C, L465P) in the CYP17A1 gene. J Clin Endocrinol Metab. 2009;94:3058–64. [PubMed: 19454579]
- Flück CE, Mullis PE, Pandey AV. Reduction in hepatic drug metabolizing CYP3A4 activities caused by P450 oxidoreductase mutations identified in patients with disordered steroid metabolism. Biochem Biophys Res Commun. 2010;401:149–53. [PubMed: 20849814]
- Flück CE, Tajima T, Pandey AV, Arlt W, Okuhara K, Verge CF, Jabs EW, Mendonça BB, Fujieda K, Miller WL. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat Genet. 2004;36:228–30. [PubMed: 14758361]
- Fukami M, Horikawa R, Nagai T, Tanaka T, Naiki Y, Sato N, Okuyama T, Nakai H, Soneda S, Tachibana K, Matsuo N, Sato S, Homma K, Nishimura G, Hasegawa T, Ogata T. Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: molecular and clinical studies in 10 patients. J Clin Endocrinol Metab. 2005;90:414–26. [PubMed: 15483095]
- Fukami M, Nishimura G, Homma K, Nagai T, Hanaki K, Uematsu A, Ishii T, Numakura C, Sawada H, Nakacho M, Kowase T, Motomura K, Haruna H, Nakamura M, Ohishi A, Adachi M, Tajima T, Hasegawa Y, Hasegawa T, Horikawa R, Fujieda K, Ogata T. Cytochrome P450 oxidoreductase deficiency: identification and characterization of biallelic mutations and genotype-phenotype correlations in 35 Japanese patients. J Clin Endocrinol Metab. 2009;94:1723–31. [PubMed: 19258400]
- Gomes LG, Huang N, Agrawal V, Mendonca BB, Bachega TASS, Miller WL. Extraadrenal 21-hydroxylation by CYP2C19 and CYP3A4: effect on 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2009;94:89–95. [PMC free article: PMC2630875] [PubMed: 18957504]
- Gu J, Weng Y, Zhang Q-Y, Cui H, Behr M, Wu L, Yang W, Zhang L, Ding X. Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase. J Biol Chem. 2003;278:25895–901. [PubMed: 12697746]
- Hart SN, Zhong X-B. P450 oxidoreductase: genetic polymorphisms and implications for drug metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2008;4:439–52. [PubMed: 18433346]
- Hershkovitz E, Parvari R, Wudy SA, Hartmann MF, Gomes LG, Loewental N, Miller WL. Homozygous mutation G539R in the gene for P450 oxidoreductase in a family previously diagnosed as having 17,20-lyase deficiency. J Clin Endocrinol Metab. 2008;93:3584–8. [PMC free article: PMC2567854] [PubMed: 18559916]
- Huang N, Agrawal V, Giacomini KM, Miller WL. Genetics of P450 oxidoreductase: sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proc Natl Acad Sci U S A. 2008;105:1733–8. [PMC free article: PMC2234213] [PubMed: 18230729]
- Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, Jabs EW, van Vliet G, Sack J, Flück CE, Miller WL. Diversity and function of mutations in p450 oxidoreductase in patients with Antley-Bixler syndrome and disordered steroidogenesis. Am J Hum Genet. 2005;76:729–49. [PMC free article: PMC1199364] [PubMed: 15793702]
- Huang N, Shoichet B, Irwin J. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–801. [PMC free article: PMC3383317] [PubMed: 17154509]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Idkowiak J, O'Riordan S, Reisch N, Malunowicz EM, Collins F, Kerstens MN, Köhler B, Graul-Neumann LM, Szarras-Czapnik M, Dattani M, Silink M, Shackleton CHL, Maiter D, Krone N, Arlt W. Pubertal presentation in seven patients with congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2011;96:E453–62. [PMC free article: PMC3124345] [PubMed: 21190981]
- Kelley RI, Kratz LE, Glaser RL, Netzloff ML, Wolf LM, Jabs EW. Abnormal sterol metabolism in a patient with Antley-Bixler syndrome and ambiguous genitalia. Am J Med Genet. 2002;110:95–102. [PubMed: 12116245]
- Kranendonk M, Marohnic CC, Panda SP, Duarte MP, Oliveira JS, Masters BSS, Rueff J. Impairment of human CYP1A2-mediated xenobiotic metabolism by Antley-Bixler syndrome variants of cytochrome P450 oxidoreductase. Arch Biochem Biophys. 2008;475:93–9. [PMC free article: PMC4269338] [PubMed: 18455494]
- Krone N, Reisch N, Idkowiak J, Dhir V, Ivison HE, Hughes BA, Rose IT, O'Neil DM, Vijzelaar R, Smith MJ, Macdonald F, Cole TR, Adolphs N, Barton JS, Blair EM, Braddock SR, Collins F, Cragun DL, Dattani MT, Day R, Dougan S, Feist M, Gottschalk ME, Gregory JW, Haim M, Harrison R, Olney AH, Hauffa BP, Hindmarsh PC, Hopkin RJ, Jira PE, Kempers M, Kerstens MN, Khalifa MM, Köhler B, Maiter D, Nielsen S, O'Riordan SM, Roth CL, Shane KP, Silink M, Stikkelbroeck NMML, Sweeney E, Szarras-Czapnik M, Waterson JR, Williamson L, Hartmann MF, Taylor NF, Wudy SA, Malunowicz EM, Shackleton CHL, Arlt W. Genotype-phenotype analysis in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2012;97:E257–67. [PMC free article: PMC3380101] [PubMed: 22162478]
- Laue K, Pogoda H-M, Daniel PB, van Haeringen A, Alanay Y. Ameln von S, Rachwalski M, Morgan T, Gray MJ, Breuning MH, Sawyer GM, Sutherland-Smith AJ, Nikkels PG, Kubisch C, Bloch W, Wollnik B, Hammerschmidt M, Robertson SP. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011;89:595–606. [PMC free article: PMC3213388] [PubMed: 22019272]
- Lopez-Rangel E, Van Allen MI. Prenatal exposure to fluconazole: an identifiable dysmorphic phenotype. Birth Defects Res A Clin Mol Teratol. 2005;73:919–23. [PubMed: 16265639]
- Miller WL, Huang N, Agrawal V, Giacomini KM. Genetic variation in human P450 oxidoreductase. Mol Cell Endocrinol. 2009;300:180–4. [PubMed: 18930113]
- Morton JEV, Frentz S, Morgan T, Sutherland-Smith AJ, Robertson SP. Biallelic mutations in CYP26B1: A differential diagnosis for Pfeiffer and Antley-Bixler syndromes. Hennekam RCM, Biesecker LG, eds. Am J Med Genet A 2016;170:2706–10. [PubMed: 27410456]
- Nicolo C, Flück CE, Mullis PE, Pandey AV. Restoration of mutant cytochrome P450 reductase activity by external flavin. Mol Cell Endocrinol. 2010;321:245–52. [PubMed: 20188793]
- Oneda B, Crettol S, Jaquenoud Sirot E, Bochud M, Ansermot N, Eap CB. The P450 oxidoreductase genotype is associated with CYP3A activity in vivo as measured by the midazolam phenotyping test. Pharmacogenet Genomics. 2009;19:877–83. [PubMed: 19801957]
- Otto DME, Henderson CJ, Carrie D, Davey M, Gundersen TE, Blomhoff R, Adams RH, Tickle C, Wolf CR. Identification of novel roles of the cytochrome p450 system in early embryogenesis: effects on vasculogenesis and retinoic Acid homeostasis. Mol Cell Biol. 2003;23:6103–16. [PMC free article: PMC180925] [PubMed: 12917333]
- Pandey AV, Kempná P, Hofer G, Mullis PE, Flück CE. Modulation of human CYP19A1 activity by mutant NADPH P450 oxidoreductase. Molecular Endocrinology. 2007;21:2579–95. [PubMed: 17595315]
- Pandey AV, Sproll P. Pharmacogenomics of human P450 oxidoreductase. Front Pharmacol. 2014;5:103. [PMC free article: PMC4023047] [PubMed: 24847272]
- Peterson RE, Imperato-McGinley J, Gautier T, Shackleton C. Male Pseudohermaphroditism Due to Multiple Defects in Steroid-Biosynthetic Microsomal Mixed-Function Oxidases. N Engl J Med. 1985;313:1182–91. [PubMed: 2932643]
- Reisch N, Idkowiak J, Hughes BA, Ivison HE, Abdul-Rahman OA, Hendon LG, Olney AH, Nielsen S, Harrison R, Blair EM, Dhir V, Krone N, Shackleton CHL, Arlt W. Prenatal diagnosis of congenital adrenal hyperplasia caused by P450 oxidoreductase deficiency. J Clin Endocrinol Metab. 2013;98:E528–36. [PMC free article: PMC3708032] [PubMed: 23365120]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Sahakitrungruang T, Huang N, Tee MK, Agrawal V, Russell WE, Crock P, Murphy N, Migeon CJ, Miller WL. Clinical, genetic, and enzymatic characterization of P450 oxidoreductase deficiency in four patients. J Clin Endocrinol Metab. 2009;94:4992–5000. [PMC free article: PMC2795645] [PubMed: 19837910]
- Scott RR, Gomes L, Huang N, Van Vliet G, Miller W. Apparent manifesting heterozygosity in P450 oxidoreductase deficiency and its effect on coexisting 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:2318–22. [PubMed: 17389698]
- Scott RR, Miller WL. Genetic and clinical features of p450 oxidoreductase deficiency. Horm Res. 2008;69:266–75. [PubMed: 18259105]
- Shackleton C, Malunowicz E. Apparent pregnene hydroxylation deficiency (APHD): seeking the parentage of an orphan metabolome. Steroids. 2003;68:707–17. [PubMed: 14625002]
- Shackleton C, Marcos J, Arlt W, Hauffa B. Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): a disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype. Am J Med Genet A. 2004;129A:105–12. [PubMed: 15316970]
- Shen AL, O'Leary KA, Kasper CB. Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2002;277:6536–41. [PubMed: 11742006]
- Soneda S, Yazawa T, Fukami M, Adachi M, Mizota M, Fujieda K, Miyamoto K, Ogata T. Proximal promoter of the cytochrome P450 oxidoreductase gene: identification of microdeletions involving the untranslated exon 1 and critical function of the SP1 binding sites. J Clin Endocrinol Metab. 2011;96:E1881–7. [PubMed: 21900384]
- Tomalik-Scharte D, Maiter D, Kirchheiner J, Ivison HE, Fuhr U, Arlt W. Impaired hepatic drug and steroid metabolism in congenital adrenal hyperplasia due to P450 oxidoreductase deficiency. Eur J Endocrinol. 2010;163:919–24. [PMC free article: PMC2977993] [PubMed: 20844025]
- Wang DC, Shannon P, Toi A, Chitayat D, Mohan U, Barkova E, Keating S, Tomlinson G, Glanc P. Temporal lobe dysplasia: a characteristic sonographic finding in thanatophoric dysplasia. Ultrasound Obstet Gynecol. 2014;44:588–94. [PubMed: 24585534]
- Weaver KN, Johnson J, Kline-Fath B, Zhang X, Lim F-Y, Tinkle B, Saal HM, Hopkin RJ. Predictive value of fetal lung volume in prenatally diagnosed skeletal dysplasia. Prenat Diagn. 2014;34:1326–31. [PubMed: 25102973]
- Williamson L, Arlt W, Shackleton CH, Kelley R, Braddock S. Linking Antley-Bixler syndrome and congenital adrenal hyperplasia: a novel case of P450 oxidoreductase deficiency. Am J Med Genet A. 2006;140A:1797–803. [PubMed: 16906539]
Chapter Notes
Acknowledgments
The authors would like to thank Dr Cedric Shackleton and Dr Richard Kelley for providing information and inspiration for this project.
Revision History
- 3 August 2017 (sw) Comprehensive update posted live
- 18 August 2009 (me) Comprehensive update posted live
- 8 September 2005 (me) Review posted live
- 9 September 2004 (dlc) Original submission
Publication Details
Author Information and Affiliations
University of Birmingham
Birmingham, United Kingdom
Tampa, Florida
Cincinnati, Ohio
University of Birmingham
Birmingham, United Kingdom
Publication History
Initial Posting: September 8, 2005; Last Update: August 3, 2017.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Idkowiak J, Cragun D, Hopkin RJ, et al. Cytochrome P450 Oxidoreductase Deficiency. 2005 Sep 8 [Updated 2017 Aug 3]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


