Summary
Clinical characteristics.
The spectrum of ANO5 muscle disease is a continuum that ranges from asymptomatic hyperCKemia and exercise-induced myalgia to proximal and/or distal muscle weakness. The most typical presentation is limb-girdle muscular dystrophy type 2L (LGMD2L) with late-onset proximal lower-limb weakness in the fourth or fifth decade (range 15-70 years). Less common is Miyoshi-like disease (Miyoshi muscular dystrophy 3) with early-adult-onset calf distal myopathy (around age 20 years). Incidental hyperCKemia may be present even earlier. Initial symptoms are walking difficulties, reduced sports performance, and difficulties in standing on toes as well as nonspecific exercise myalgia and/or burning sensation in the calf muscles. Muscle weakness and atrophy are frequently asymmetric. Cardiac findings can include cardiomyopathy and arrhythmias and/or left ventricular dysfunction. Bulbar or respiratory symptoms have not been reported. Females have milder disease manifestations than males. Disease progression is slow in both the LGMD and distal forms; ambulation is preserved until very late in the disease course. Life span is normal.
Diagnosis/testing.
The diagnosis of ANO5 muscle disease is established in a proband with identification of biallelic pathogenic variants in ANO5 on molecular genetic testing.
Management.
Treatment of manifestations: No definitive treatments for the limb-girdle muscular dystrophies exist. Management is tailored to the individual. To assist with decreased mobility, the following are suggested: weight control to avoid obesity, physical therapy to promote mobility and prevent contractures, and use of mechanical aids to help ambulation and mobility.
Surveillance: Evaluate muscle strength and functional status every six to 12 months.
Agents/circumstances to avoid: Heavy muscle force training of weak muscles. The use of statins, which can induce muscle pain and worsen muscle weakness should be avoided, but if absolutely necessary for the health of the individual, use requires extra monitoring of clinical status especially at the beginning of treatment.
Genetic counseling.
ANO5 muscle disease is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk family members, prenatal diagnosis for a pregnancy at increased risk and preimplantation testing are possible if the pathogenic variants in the family have been identified.
GeneReview Scope
Table
Limb-girdle muscular dystrophy type 2L Miyoshi muscular dystrophy 3
Diagnosis
Suggestive Findings
ANO5 muscle disease should be suspected in individuals with any of the following presentations:
- Late-adult-onset proximal lower-limb weakness (known as limb-girdle muscular dystrophy type 2L (LGMD2L)
- Mean onset age 35 years; range 15-70 years
- Asymmetric muscle weakness and atrophy, especially in thigh muscles
- Early-adult-onset (age 20-25 years) calf distal myopathy (known as Miyoshi muscular dystrophy 3)
- Asymptomatic hyperCKemia and exercise-induced myalgia
In all presentations:
- EMG shows myopathic changes with scattered necrotic fibers or may be normal in mildly affected individuals
- CT and MRI show fatty degeneration of the gastrocnemius medialis muscle in most affected individuals with involvement of the soleus and posterior thigh muscles with disease progression (see Figure 2).
- Serum CK concentration is markedly elevated (≥2-3x – and usually 10-50x – the upper limit of normal)
- Muscle biopsy shows scattered necrotic fibers or nonspecific myopathic or dystrophic findings
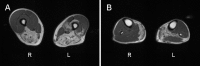
Figure 2
A. Fatty degenerative changes in posterior thigh muscles in the left vastus medialis and intermedius muscles B. Fatty degenerative changes in the medial gastrocnemius (left greater than right) and in the left soleus muscle
Establishing the Diagnosis
The diagnosis of ANO5 muscle disease is established in a proband with biallelic pathogenic variants in ANO5 identified on molecular genetic testing (see Table 1).
Because the phenotype of ANO5 muscle disease is indistinguishable from many other inherited muscle disorders, recommended molecular genetic testing approaches include use of a multigene panel or comprehensive genomic testing.
Note: Single-gene testing (sequence analysis of ANO5, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
- A muscle disease multigene panel that includes ANO5 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1). Note: To date such variants have not been identified as a cause of ANO5 muscle disease.
- Comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is another good option. Exome sequencing is most commonly used; genome sequencing is also possible.If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis. Note: To date such variants have not been identified as a cause of ANO5 muscle disease.
Table 1.
Molecular Genetic Testing Used in ANO5 Muscle Disease
Clinical Characteristics
Clinical Description
The spectrum of ANO5 muscle disease is a continuum ranging from asymptomatic hyperCKemia and exercise-induced myalgia to proximal and/or distal muscle weakness. The most typical presentation is late-onset proximal lower-limb weakness (also called limb-girdle muscular dystrophy type 2L or LGMD2) [Bolduc et al 2010, Hicks et al 2011, Penttilä et al 2012] (Table 2). Less common is early-adult-onset calf distal myopathy (also called Miyoshi muscular dystrophy 3) [Mahjneh et al 2010, Hicks et al 2011, Penttilä et al 2012].
Presentation. Onset of the proximal weakness in the limb-girdle muscular dystrophy (LGMD) form is in the fourth or fifth decade (age range 15-70 years); onset of calf weakness in the distal form is around age 20 years. Incidental hyperCKemia may be present even earlier. Initial symptoms are walking difficulties, reduced sports performance, and difficulties in standing on toes as well as nonspecific exercise myalgia and/or burning sensation in the calf muscles [Bolduc et al 2010, Hicks et al 2011, Penttilä et al 2012, Papadopoulos et al 2017, Ten Dam et al 2019].
- In the limb-girdle muscular dystrophy form, muscle weakness is mainly proximal and more pronounced in the lower limbs; however, many affected individuals also have proximal upper-limb involvement.
- In the distal myopathy form, calf hypertrophy and exercise myalgia can occur before apparent weakness and later calf atrophy. Clinical manifestations can be mild or subjectively nonexistent even with clear changes observed on muscle imaging. Individuals with distal onset may have proximal lower-limb weakness in the later stages of the disease. Distal upper-limb weakness has not been reported.
- Muscle weakness and atrophy are frequently asymmetric (see Figure 1).
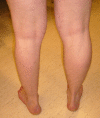
Figure 1.
Asymmetric atrophy of the muscles of the left calf in an individual with an ANO5 pathogenic variant
Table 2.
The LGMD2L Phenotype of the Frst 20 Reported Individuals
Cardiac findings. In 19 individuals with ANO5 muscle disease who underwent systematic cardiac investigations, two had dilated cardiomyopathy at age ≤42 years and three had left ventricular dilatation [Wahbi et al 2013]. Rarely, other individuals have been reported with arrhythmias [Witting et al 2013, Ten Dam et al 2019], hypertrophic cardiomyopathy [van der Kooi et al 2013], or left ventricular dysfunction [Ten Dam et al 2019].
Other. Bulbar or respiratory symptoms have not been reported.
Sex differences. Females tend to have milder disease manifestations than males [Bolduc et al 2010, Hicks et al 2011, Magri et al 2012, Penttilä et al 2012, Sarkozy et al 2013, van der Kooi et al 2013]. It is possible females are underrepresented in the reports due to milder disease course, which may be only hyperCKemia with or without myalgia or mild muscle weakness.
Progression and life span. Disease progression is slow in both the LGMD and distal form; ambulation is preserved until very late in the disease course. Most affected individuals remain ambulatory without assistance for decades. Life span appears to be normal.
Genotype-Phenotype Correlations
No genotype-phenotype correlations for ANO5 have been identified [Penttilä et al 2012].
Nomenclature
Miyoshi muscular dystrophy 3 may also be referred to as non-dysferlin Miyoshi muscular dystrophy or MMD3.
Prevalence
ANO5 muscle disease has been estimated to be one of the most common causes of limb-girdle muscular dystrophy. Prevalence in Finland is as high as 2:100,000 [Penttilä et al 2012]; in the North of England it has been estimated at 0.26:100,000 [Hicks et al 2011]. In Europe, ANO5 muscle disease has been estimated to be the third most prevalent LGMD subtype (26%) although this may be an underestimation due to the less severe phenotype [Ten Dam et al 2019]. In a large cohort study of 4656 individuals with clinically suspected LGMD across the US, ANO5 was the fourth most prevalent LGMD subtype (7%) [Nallamilli et al 2018].
Genetically Related (Allelic) Disorders
Dominant pathogenic variants in ANO5 are known to cause gnathodiaphyseal dysplasia (OMIM 166260), a generalized skeletal syndrome characterized by cemento-osseus lesions of the jawbones, in conjunction with bone fragility, bowing/cortical thickening of tubular bones, and diaphyseal sclerosis of long bones. The functional difference between dominant and recessive ANO5 pathogenic variants is not understood. However, dominant ANO5 variants appear to locate in an extracellular domain following the first transmembrane domain or in the fourth putative transmembrane domain [Jin et al 2017]. Recessive ANO5 variants have been shown to decrease the ANO5 protein expression [Vihola et al 2018].
Differential Diagnosis
Limb-girdle muscular dystrophy (LGMD). The differential diagnosis of ANO5 muscle disease includes all the LGMDs (see OMIM phenotypic series: LGMD, autosomal recessive and LGMD, autosomal dominant). Of particular note is LGMD2B with primarily proximal weakness. LGMD2B is a dysferlinopathy characterized by early weakness and atrophy of the pelvic and shoulder girdle muscles in adolescence or young adulthood, with slow progression.
Distal myopathy. Muscle MRI, muscle pathology, and mode of inheritance may be useful in distinguishing between distal myopathies in the differential diagnosis of ANO5 muscle disease (see Table 3).
Table 3.
Genes Associated with Distal Myopathies of Interest in the Differential Diagnosis of ANO5 Muscle Disease
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with ANO5 muscle disease, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with ANO5 Muscle Disease
Treatment of Manifestations
No definitive treatments for the limb-girdle muscular dystrophies exist. Management is tailored to each individual and each specific subtype.
Table 5.
Treatment of Manifestations in Individuals with ANO5 Muscle Disease
Surveillance
Table 6.
Recommended Surveillance for Individuals with ANO5 Muscle Disease
Agents/Circumstances to Avoid
Heavy muscle force training of weak muscles should be avoided as very high levels of CK have been measured after strenuous exercise [Milone et al 2012, Penttilä et al 2012].
The use of statins, which can induce muscle pain and worsen muscle weakness should be avoided. If absolutely necessary for the health of the individual, statin use requires extra monitoring of clinical status especially at the beginning of treatment.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
ANO5 muscle disease is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., carriers of one ANO5 pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing an ANO5 muscle disease.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing an ANO5 muscle disease.
Offspring of a proband. The offspring of an individual with an ANO5 muscle disease are obligate heterozygotes (carriers) for a pathogenic variant in ANO5.
Other family members. Each sib of the proband’s parents is at a 50% risk of being a carrier.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the ANO5 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the ANO5 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic diagnosis are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Genetic and Rare Diseases Information Center (GARD)PO Box 8126Gaithersburg MD 20898-8126Phone: 888-205-2311 (toll-free); 888-205-3223 (toll-free TTY)Fax: 301-251-4911Email: [email protected]
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321Email: [email protected]
- Muscular Dystrophy CanadaCanadaPhone: 800-567-2873Email: [email protected]
- Muscular Dystrophy UKUnited KingdomPhone: 0800 652 6352
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
ANO5 Muscle Disease: Genes and Databases
Table B.
OMIM Entries for ANO5 Muscle Disease (View All in OMIM)
Molecular Pathogenesis
ANO5 (anoctamin 5) belongs to a protein family of Ca2+ activated ion channels and phospholipid scramblases. However, not much is known about its normal function and also the molecular pathogenesis is so far not understood. According to a recent study, it appears that ANO5 deficiency compromises the plasma membrane repair ability of myoblasts [Chandra et al 2019].
Mechanism of disease causation. It seems likely that the main disease mechanism in anoctaminopathy is loss of function. This is based on the observation that in most cases, regardless of the pathogenic variant (truncating or missense), the ANO5 protein is clearly reduced in biopsies of affected individuals.
ANO5-specific laboratory technical considerations. A western blotting method has been developed to detect endogenous ANO5 protein in membrane fractions extracted from human muscle biopsies. This method can be used to quantify the ANO5 protein and evaluate the pathogenicity of novel ANO5 variants of uncertain significance.
Table 7.
Notable ANO5 Pathogenic Variants
References
Literature Cited
- Bolduc V, Marlow G, Boycott KM, Saleki K, Inoue H, Kroon J, Itakura M, Robitaille Y, Parent L, Baas F, Mizuta K, Kamata N, Richard I, Linssen WHJP, Mahjneh I, de Visser M, Bashir R, Brais B. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet. 2010;86:213–21. [PMC free article: PMC2820170] [PubMed: 20096397]
- Chandra G, Defour A, Mamchoui K, Pandey K, Mishra S, Mouly V, Sreetama S, Mahad Ahmad M, Mahjneh I, Morizono H, Pattabiraman N, Menon AK, Jaiswal JK. Dysregulated calcium homeostasis prevents plasma membrane repair in Anoctamin 5/TMEM16E-deficient patient muscle cells. Cell Death Discov. 2019;5:118. [PMC free article: PMC6639303] [PubMed: 31341644]
- Hicks D, Sarkozy A, Muelas N, Koehler K, Huebner A, Hudson G, Chinnery PF, Barresi R, Eagle M, Polvikoski T, Bailey G, Miller J, Radunovic A, Hughes PJ, Roberts R, Krause S, Walter MC, Laval SH, Straub V, Lochmüller H, Bushby K. A founder mutation in anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain. 2011;134:171–82. [PMC free article: PMC4038512] [PubMed: 21186264]
- Jin L, Liu Y, Sun F, Collins MT, Blackwell K, Woo AS, Reichenberger EJ, Hu Y. Three novel ANO5 missense mutations in Caucasian and Chinese families and sporadic cases with gnathodiaphyseal dysplasia. Sci Rep. 2017;7:40935. [PMC free article: PMC5296836] [PubMed: 28176803]
- Magri F, Bo RD, D'Angelo MG, Sciacco M, Gandossini S, Govoni A, Napoli L, Ciscato P, Fortunato F, Brighina E, Bonato S, Bordoni A, Lucchini V, Corti S, Moggio M, Bresolin N, Comi GP. Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb-girdle muscular dystrophy patients. Neuromuscul Disord. 2012;22:934–43. [PMC free article: PMC3500692] [PubMed: 22742934]
- Mahjneh I, Jaiswal J, Lamminen A, Somer M, Marlow G, Kiuru-Enari S, Bashir R. A new distal myopathy with mutation in anoctamin 5. Neuromuscul Disord. 2010;20:791–5. [PMC free article: PMC4206776] [PubMed: 20692837]
- Milone M, Liewluck T, Winder TL, Pianosi PT. Amyloidosis and exercise intolerance in ANO5 muscular dystrophy. Neuromuscul Disord. 2012;22:13–5. [PubMed: 21820307]
- Nallamilli BRR, Chakravorty S, Kesari A, Tanner A, Ankala A, Schneider T, da Silva C, Beadling R, Alexander JJ, Askree SH, Whitt Z, Bean L, Collins C, Khadilkar S, Gaitonde P, Dastur R, Wicklund M, Mozaffar T, Harms M, Rufibach L, Mittal P, Hegde M. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5:1574–87. [PMC free article: PMC6292381] [PubMed: 30564623]
- Papadopoulos C, Laforêt P, Nectoux J, Stojkovic T, Wahbi K, Carlier RY, Carlier PG, Leonard-Louis S, Leturcq F, Romero N, Eymard B, Behin A. Hyperckemia and myalgia are common presentations of anoctamin-5-related myopathy in French patients. Muscle Nerve. 2017;56:1096–100. [PubMed: 28187523]
- Penttilä S, Palmio J, Suominen T, Raheem O, Evilä A, Muelas Gomez N, Tasca G, Waddell LB, Clarke NF, Barboi A, Hackman P, Udd B. Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology. 2012;78:897–903. [PubMed: 22402862]
- Sarkozy A, Hicks D, Hudson J, Laval SH, Barresi R, Hilton-Jones D, Deschauer M, Harris E, Rufibach L, Hwang E, Bashir R, Walter MC, Krause S, van den Bergh P, Illa I, Pénisson-Besnier I, De Waele L, Turnbull D, Guglieri M, Schrank B, Schoser B, Seeger J, Schreiber H, Gläser D, Eagle M, Bailey G, Walters R, Longman C, Norwood F, Winer J, Muntoni F, Hanna M, Roberts M, Bindoff LA, Brierley C, Cooper RG, Cottrell DA, Davies NP, Gibson A, Gorman GS, Hammans S, Jackson AP, Khan A, Lane R, McConville J, McEntagart M, Al-Memar A, Nixon J, Panicker J, Parton M, Petty R, Price CJ, Rakowicz W, Ray P, Schapira AH, Swingler R, Turner C, Wagner KR, Maddison P, Shaw PJ, Straub V, Bushby K, Lochmüller H. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum Mutat. 2013;34:1111–8. [PubMed: 23606453]
- Ten Dam L, Frankhuizen WS, Linssen WHJP, Straathof CS, Niks EH, Faber K, Fock A, Kuks JB, Brusse E, de Coo R, Voermans N, Verrips A, Hoogendijk JE, van der Pol L, Westra D, de Visser M, van der Kooi AJ, Ginjaar I. Autosomal recessive limb-girdle and Miyoshi muscular dystrophies in the Netherlands: The clinical and molecular spectrum of 244 patients. Clin Genet. 2019;96:126–33. [PubMed: 30919934]
- van der Kooi AJ, Ten Dam L, Frankhuizen WS, Straathof CS, van Doorn PA, de Visser M, Ginjaar IB. ANO5 mutations in the Dutch limb girdle muscular dystrophy population. Neuromuscul Disord. 2013;23:456–60. [PubMed: 23607914]
- Vihola A, Luque H, Savarese M, Penttilä S, Lindfors M, Leturq F, Eymard B, Tasca G, Brais B, Conte T, Charton K, Richard I, Udd B. Diagnostic anoctamin 5 protein defect in patients with ANO5 mutated muscular dystrophy. Neuropathol Appl Neurobiol. 2018;44:441–8. [PubMed: 28489263]
- Wahbi K, Béhin A, Bécane HM, Leturcq F, Cossée M, Laforêt P, Stojkovic T, Carlier P, Toussaint M, Gaxotte V, Cluzel P, Eymard B, Duboc D. Dilated cardiomyopathy in patients with mutations in anoctamin 5. Int J Cardiol. 2013;168:76–9. [PubMed: 23041008]
- Witting N, Duno M, Petri H, Krag T, Bundgaard H, Kober L, Vissing J. Anoctamin 5 muscular dystrophy in Denmark: prevalence, genotypes, phenotypes, cardiac findings, and muscle protein expression. J Neurol. 2013;260:2084–93. [PubMed: 23670307]
Chapter Notes
Revision History
- 22 August 2019 (ha) Comprehensive update posted live
- 29 November 2012 (me) Review posted live
- 19 July 2012 (sp) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: November 29, 2012; Last Update: August 22, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Penttilä S, Vihola A, Palmio J, et al. ANO5 Muscle Disease. 2012 Nov 29 [Updated 2019 Aug 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

