Summary
Clinical characteristics.
Beckwith-Wiedemann syndrome (BWS) is a growth disorder variably characterized by macroglossia, hemihyperplasia, omphalocele, neonatal hypoglycemia, macrosomia, embryonal tumors (e.g., Wilms tumor, hepatoblastoma, neuroblastoma, and rhabdomyosarcoma), visceromegaly, adrenocortical cytomegaly, kidney abnormalities (e.g., medullary dysplasia, nephrocalcinosis, and medullary sponge kidney), and ear creases / posterior helical ear pits. BWS is considered a clinical spectrum, in which affected individuals may have many or only one or two of the characteristic clinical features. Although most individuals with BWS show rapid growth in late fetal development and early childhood, growth rate usually slows by age seven to eight years. Adult heights are typically within the normal range. Hemihyperplasia (also known as lateralized overgrowth) is often appreciated at birth and may become more or less evident over time. Hemihyperplasia may affect segmental regions of the body or selected organs and tissues. Hemihyperplasia may be limited to one side of the body (ipsilateral) or involve opposite sides of the body (contralateral). Macroglossia is generally present at birth and can obstruct breathing or interfere with feeding in infants. Neonatal hypoglycemia occurs in approximately 50% of infants with BWS; most episodes are mild and transient. However, in some cases, persistent hypoglycemia due to hyperinsulinism may require consultation with an endocrinologist for therapeutic intervention. With respect to the increased risk for embryonal tumor development, the risk for Wilms tumor appears to be concentrated in the first seven years of life, whereas the risk for developing hepatoblastoma is concentrated in the first three to four years of life. Cognitive and neurobehavioral development is usually normal. After childhood, prognosis is generally favorable, although some adults experience issues requiring medical management (e.g., for renal or skeletal concerns).
Diagnosis/testing.
The clinical diagnosis of BWS can be established in a proband who has two tier 1 characteristic clinical findings OR one tier 1 and one tier 2 clinical finding.
A diagnosis can also be established in a proband with at least one tier 1 or tier 2 clinical finding AND either:
- A constitutional epigenetic or genomic alteration leading to an abnormal methylation pattern at 11p15.5 known to be associated with BWS; OR
- A copy number variant of chromosome 11p15.5 known to be associated with BWS; OR
- A heterozygous BWS-causing pathogenic variant in CDKN1C.
Management.
Treatment of manifestations: Hypoglycemia is treated with oral feeding if it is mild or with glucose supplementation. Hyperinsulinism is treated with standard pharmacotherapy per endocrinologist; partial pancreatectomy may be considered in those with persistent hypoglycemia who are unresponsive to pharmacologic treatment. Feeding and/or respiratory support (sometimes requiring intubation at birth or tracheostomy for severe respiratory insufficiency) may be needed for those with macroglossia and/or upper airway obstruction. Tongue reduction surgery may be considered for this and other indications. Standard treatment is recommended for omphalocele per pediatric surgeon. A shoe lift may be considered in those with a leg length discrepancy. Epiphysiodesis prior to epiphyseal closure in early puberty may be considered in instances with leg length discrepancy >2 cm; alternatively, leg lengthening of the shorter leg may be considered. For those with hemihyperplasia of the face, referral to a craniofacial center for assessment and potential treatments may be considered. If present, standard treatment is recommended for speech delay/impediment, neoplasia, congenital heart defects, and hypercalciuria / kidney anomalies.
Surveillance:
- Tumor surveillance. Perspectives on screening for malignant tumors in childhood differ based on local, national, and international practices. In North America, proactive tumor screening is recommended when the risk of tumor development exceeds 1%. In many European countries, proactive tumor screening protocols are typically undertaken when the risk of tumor development exceeds 5% and are based on molecular mechanism. For most BWS molecular subtypes, tumor screening consists of abdominal ultrasound with views of the liver, adrenal glands, and kidneys every three months until age four years followed by kidney ultrasound only every three months from age four to seven years. Serum alpha-fetoprotein (AFP) levels are performed every three months until age four years. Physical exam by a pediatrician, geneticist, or pediatric oncologist twice a year is also recommended. Proposed screening for neuroblastoma in children with a heterozygous pathogenic variant in CDKN1C includes abdominal ultrasound, urine vanillylmandelic acid and homovanillic acid, and chest radiograph every three months until age six years, then every six months until age ten years.
- Non-tumor surveillance includes measurement of growth parameters, assessment for signs/symptoms of sleep apnea, and monitoring of developmental progress / educational needs at each visit; pre-feed serum glucose level per endocrinologist recommendations in neonates and infants with a history of hypoglycemia/hyperinsulinism or random serum glucose level in neonates and infants with signs/symptoms consistent with hypoglycemia; consideration of blood pressure measurements and measurement of urinary calcium-to-creatinine ratio to screen for occult nephrocalcinosis annually or biannually; consideration of kidney ultrasound to identify findings such as nephrocalcinosis and medullary sponge kidney annually between age eight years and mid-adolescence and periodically in adulthood; assessment of hemihyperplasia and leg length discrepancy at each visit at least until skeletal maturity; dental evaluation with low threshold for orthodontic evaluation as clinically indicated.
Genetic counseling.
BWS is associated with abnormal expression of imprinted genes in the BWS critical region. Reliable recurrence risk assessment requires identification of the genetic mechanism in the proband that underlies the abnormal expression of imprinted genes in the BWS critical region. While the majority of families have a recurrence risk of less than 1%, certain underlying genetic mechanisms (e.g., CDKN1C pathogenic variants and copy number variants involving 11p15) may be associated with a recurrence risk as high as 50% depending on the sex of the transmitting parent and the specific alteration. In families with recurrence of BWS, maternally inherited CDKN1C pathogenic variants account for approximately 40% of genetic alterations and paternally or maternally inherited copy number variants account for approximately 9% of genetic alterations. Of note, some individuals with BWS have methylation alterations in the 11p15 imprinted domain as well as in other imprinted loci. For these individuals review of the maternal history should be undertaken for unexplained spontaneous abortion, hydatidiform moles, or a sib with BWS or another imprinting disorder (e.g., Silver-Russell syndrome); in such cases, a homozygous or heterozygous pathogenic variant in maternal effect gene in the mother's genome may confer a significant recurrence risk.
Prenatal testing and preimplantation genetic testing: If a genomic variant involving chromosome 11p15.5 (i.e., a cytogenetically visible duplication, inversion, or translocation), copy number variant of 11p15.5, or a CDKN1C pathogenic variant has been identified in the proband, prenatal testing via analysis of fetal DNA from samples obtained by chorionic villus sampling (CVS) or amniocentesis is possible. Preimplantation genetic testing is possible for a familial CDKN1C pathogenic variant and may be possible for some familial genomic variants. For evaluation of fetal methylation status, DNA extracted from amniotic fluid is currently felt to provide the most reliable tissue source, although false negative findings have been reported. Tissue obtained via CVS for prenatal testing for methylation status does not yield reliable results. Genetic counseling should emphasize the potential limitations of prenatal testing for epigenetic alterations.
Diagnosis
The phenotypic presentation of Beckwith-Wiedemann syndrome (BWS) is highly variable, and no consensus clinical diagnostic criteria are universally accepted at this time. Clinical diagnostic scoring systems have been proposed and can assist with guiding diagnostic considerations, genetic testing, and management [Weksberg et al 2010, Brioude et al 2018, Duffy et al 2019a]. However, a cautious approach should be adopted especially given the implications for tumor surveillance. That is, children with a "mild" clinical presentation, those whose algorithm scores do not meet the threshold for a BWS clinical diagnosis, and/or those with nondiagnostic genetic testing results may still be at increased risk for tumor development.
Suggestive Findings
Beckwith-Wiedemann syndrome (BWS) should be suspected in a proband who has one or more of the following findings, which are divided into tiers based on their relevance for the diagnosis of BWS.
Tier 1 findings. The features listed below, whether as a single finding or as a combination of findings, are highly suggestive of the diagnosis:
- Macroglossia
- Omphalocele (also sometimes referred to as exomphalos)
- Embryonal tumor, such as Wilms tumor (unilateral or bilateral), hepatoblastoma, or nephroblastomatosis
- Hemihyperplasia (lateralized overgrowth) of one or more body segments
- Macrosomia, defined as pre- and/or postnatal overgrowth, often using a cutoff of >90th or >97th centile, depending on the study
- Hyperinsulinemic hypoglycemia
- Cytomegaly of the adrenal cortex, which is considered pathognomonic for BWS
- Other pathologic findings, including placental mesenchymal dysplasia and pancreatic adenomatosis
- Family history of ≥1 family members with clinical features suggestive of BWS
Tier 2 findings, listed below, are less specific than tier 1 findings:
- Visceromegaly, typically from an imaging study such as ultrasound, involving ≥1 intra-abdominal organs, such as the liver, kidneys, and/or adrenal glands
- Unilateral or bilateral earlobe creases and/or posterior helical ear pits
- Characteristic facies (See Clinical Characteristics, Craniofacial features.)
- Kidney anomalies, such as structural malformations, nephrocalcinosis, or medullary sponge kidney
- Large umbilical hernia that requires surgical correction
- Other embryonal tumors, including rhabdomyoscarcoma, neuroblastoma, or adrenal tumors (pheochromocytoma, adrenocortical carcinoma)
- Transient hypoglycemia requiring medical intervention
Tier 3 findings. These findings are supportive of the diagnosis, particularly if there are tier 1 or tier 2 findings as well:
- Small umbilical hernia or diastasis recti
- Polyhydramnios and/or placentomegaly during pregnancy
- Premature birth
- Nevus simplex, typically on the forehead, glabella, and/or back of the neck AND/OR hemangioma (cutaneous or within organs such as the liver)
- Isolated transient hypoglycemia that does not require medical intervention
- Structural cardiac anomalies or cardiomegaly
- History of assisted reproductive technology (ART) to achieve the proband's pregnancy OR history of subfertility in a parent
- Monozygotic twinning that includes the proband
Establishing the Diagnosis
Clinical diagnosis. The clinical diagnosis of BWS can be established in a proband who has two tier 1 characteristic clinical findings OR one tier 1 and one tier 2 clinical finding.
Molecular diagnosis. A diagnosis of BWS can be established in a proband with at least one tier 1 or tier 2 clinical finding AND either:
- A constitutional epigenetic or genomic alteration leading to an abnormal methylation pattern at 11p15.5 known to be associated with BWS; OR
- A copy number variant of chromosome 11p15.5 known to be associated with BWS; OR
- A heterozygous BWS-causing pathogenic (or likely pathogenic) variant in CDKN1C.
Note: (1) A molecular diagnosis of a constitutional BWS-associated 11p15.5 (epi)genomic alteration in the absence of clinical features associated with BWS does not automatically merit a BWS clinical diagnosis. However, this molecular finding confers an increased risk for tumor development [Scott et al 2008]. Notably, Wilms tumor can be the initial presentation of subtle cases of BWS that were not clinically recognized prior to the tumor diagnosis [MacFarland et al 2018, Fiala et al 2020]. Until further data are available regarding the risk for embryonal tumor development, individuals with constitutional 11p15 (epi)genomic alterations should undergo standard tumor surveillance even in the absence of BWS-associated clinical findings (see Management). (2) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include likely pathogenic variants. (3) Identification of a heterozygous CDKN1C variant of uncertain significance does not establish or rule out the diagnosis.
BWS is associated with abnormal regulation of gene transcription in two imprinted domains on chromosome 11p15.5 (also known as the BWS critical region). Regulation may be disrupted by any one of numerous mechanisms; a simplified description of known etiologic mechanisms is given here to clarify the testing pipelines described in Genetic Testing. See Molecular Pathogenesis for a detailed description of the regulation of gene expression in this genomic region.
The BWS critical region includes two domains: imprinting center 1 (IC1) regulates the expression of IGF2 and H19 in domain 1; imprinting center 2 (IC2) regulates the expression of CDKN1C, KCNQ10T1, and KCNQ1 in domain 2 (see Figure 1). Genomic imprinting is a phenomenon whereby the DNA of the two alleles of a gene is differentially modified so that only one parental allele – parent specific for each gene – is normally expressed. As shown in Figure 1a, differential methylation of IC1 (sometimes also referred to as differentially methylated region 1, DMR1, or H19-DMR) and IC2 (sometimes also referred to as DMR2 or LIT1-DMR) is associated with expression of specific genes on the paternal and maternal alleles in unaffected individuals.

Figure 1.
Map of the BWS locus on 11p15.5 a) A schematic representation of the normal parent of origin-specific imprinted allelic expression. Note: b) and c) show the altered region only. The image is not drawn to scale.
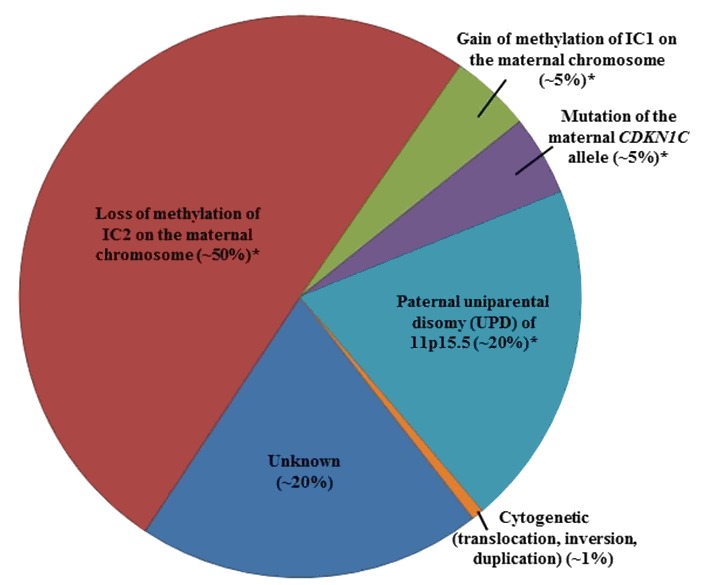
In more than 80% of individuals with BWS, genetic testing can detect one of five alterations [Weksberg et al 2003, Weksberg et al 2005]. It is important to note that some affected individuals do not have a genetic alteration identified through genetic testing due to somatic mosaicism or to limitations in the current genetic testing platforms. For example, small deletions may be missed by routine microarray analysis for which high-density microarrays focused on the BWS critical region may be required [Baker et al 2021, Sobel Naveh et al 2022].
- A schematic of the following four molecular alterations is shown in Figure 1:
- Loss of methylation of IC2 (at the transcriptional start site [TSS] of the KCNQ1OT1 differentially methylated region [DMR]) [on the maternal chromosome] (See Figure 1b.)
- Gain of methylation of IC1 (H19/IGF2:IG [intergenic] DMR) on the maternal chromosome (See Figure 1c.)
- Paternal uniparental disomy (UPD) of 11p15.5 (See Figure 1d.)
- A heterozygous pathogenic variant on the maternal CDKN1C allele (See Figure 1e.)
- Genomic variants involving chromosome 11p15.5 including cytogenetically visible duplications, inversions or translocations, or copy number variants including small duplications or deletions of 11p15.5 are not represented in Figure 1.
Note: Methylation changes may be associated with any of the primary genomic variants above except for pathogenic variants on the maternal CDKN1C allele [Baskin et al 2014, Brioude et al 2018].
Figure 2 summarizes the percentage of individuals with BWS by genetic mechanism.
Genetic Testing
Children who have milder or atypical phenotypes (e.g., ear pits and umbilical hernia) may have mosaic pathogenic BWS-causing alterations but may still be at increased risk (compared to the general population) of developing tumors associated with BWS. This is in part because cells with BWS-associated molecular changes may be present in organs "at risk" for tumor development (e.g., liver or kidneys) but not in tissues that influence external clinical presentation. Therefore, the index of suspicion should be high when evaluating children with minimal clinical features in the BWS phenotypic spectrum, with strong consideration of the use of genetic testing to confirm the diagnosis. Individuals with minimal features of BWS and normal genetic testing may still be at increased risk of developing childhood tumors.
Sample types. Genetic testing frequently is performed on a peripheral blood sample. If genetic testing of a blood sample is normal in the context of clinical findings suggestive of BWS, molecular genetic testing of a different tissue may provide a positive test result. Tissue samples obtained from affected regions (e.g., skin biopsy from larger limb in a person with hemihyperplasia, excised tongue tissue from reduction glossectomy [Alders et al 2014, Duffy et al 2019a]) have a higher likelihood of detecting an alteration; however, samples such as buccal swabs or those obtained opportunistically (e.g., skin sample from incision site) may also provide positive genetic testing results. Consideration should be given to obtaining tissue samples during surgical intervention to support genetic testing.
Genetic testing approaches can include DNA methylation studies, single-gene testing, copy number analysis for (sequences within) 11p15.5, chromosomal microarray, karyotype, and use of multigene panels that include genes in the BWS critical region (see Figure 3).

- DNA methylation studies of IC1 and IC2 should be performed simultaneously.
- Methylation alterations at both IC1 and IC2 suggest uniparental disomy (UPD).
- For recurrence risk purposes, further genetic studies can be undertaken to define the mechanism that leads to the methylation abnormality (see Genetic Counseling).
- Single-gene testing. Sequence analysis followed by gene-targeted deletion/duplication analysis of CDKN1C should be considered in familial cases, in individuals with BWS and midline anomalies (cleft palate, posterior fossa abnormalities, omphalocele, or hypospadias [Gardiner et al 2012, Brioude et al 2015]), or in individuals for whom a strong clinical suspicion for BWS exists but no detectable chromosome 11p15.5 cytogenetic abnormalities, copy number variants, methylation abnormalities, or UPD have been identified.
- Chromosomal microarray (CMA) using oligonucleotide arrays can detect only large deletions or duplications in a proband. CMA may be considered first in a proband with intellectual disability. The ability to size the deletion depends on the type of microarray used and the density of probes in the 11p15.5 region [Keren et al 2013, Baskin et al 2014, Russo et al 2016]. High-density SNP array analysis can be used to detect smaller genetic changes, including duplications, deletions, and segmental paternal UPD as well as genome-wide UPD (see Genetically Related Disorders).
- Karyotype. A karyotype may be considered to test for an inversion or translocation involving 11p15.5. This accounts for fewer than 1% of individuals with BWS.
- A multigene panel that includes CDKN1C and other genes of interest (see Differential Diagnosis and Genetically Related Disorders) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Genetic Testing Used in Beckwith-Wiedemann Syndrome
Clinical Characteristics
Clinical Description
Beckwith-Wiedemann syndrome (BWS) is a growth disorder variably characterized by neonatal hypoglycemia (persistent hypoglycemia or transient hypoglycemia due to hyperinsulinemia), macrosomia, macroglossia, hemihyperplasia, omphalocele, embryonal tumors (e.g., Wilms tumor, hepatoblastoma, neuroblastoma, and rhabdomyosarcoma), visceromegaly, adrenocortical cytomegaly, kidney abnormalities (e.g., medullary dysplasia, nephrocalcinosis, and medullary sponge kidney), and ear creases / posterior helical ear pits. BWS is considered a clinical spectrum, in which affected individuals may have many or only one or two of the characteristic clinical features.
General incidence figures for the clinical findings in Beckwith-Wiedemann syndrome (BWS) are summarized in Table 2; however, specific figures vary widely in published reports, in part due to ascertainment bias and the mosaic nature of the condition in many affected individuals.
Table 2.
Beckwith-Wiedemann Syndrome: Frequency of Select Features
Prenatal and perinatal. The incidence of polyhydramnios, premature birth, and fetal macrosomia may be as high as 50%. Other common features include a long umbilical cord and an enlarged placenta that averages almost twice the normal weight for gestational age. Placental mesenchymal dysplasia has been reported in some babies subsequently found to have features of BWS [Brioude et al 2018].
Infants with BWS are at increased risk for mortality mainly as a result of complications of prematurity, macroglossia, hypoglycemia, and, rarely, cardiomyopathy. However, the previously reported mortality rate of 20% is likely an overestimate given the recent improvements in syndrome recognition and treatment.
Growth parameters. Prenatal and/or postnatal generalized overgrowth is observed in 45%-65% of individuals diagnosed with BWS [Wang et al 2020]. Overgrowth is often defined as a length/height and/or weight that is >90th or >97th centile, or >2 standard deviations above the mean for age and sex, depending on the study. Although most individuals with BWS show rapid growth in early childhood, adult height typically remains at the upper range of normal. Growth rate usually appears to slow around age seven to eight years.
- Growth parameters should be assessed in the context of parental/familial growth parameters (e.g., a child's height at the 85th centile may reflect overgrowth when parental heights plot at ~10th centile).
- Growth parameters obtained in the neonatal period following a premature delivery may not be indicative of subsequent growth patterns.
- Macrocephaly is not a typical feature of BWS.
Hemihyperplasia* (also referred to as lateralized overgrowth, hemihypertrophy*, asymmetric overgrowth, or segmental overgrowth) can often be appreciated at birth; however, it may become more or less evident as the child grows. When asymmetry is present, it is important to distinguish if the asymmetry represents hemihyperplasia or hemihypoplasia (hemiatrophy), which suggests a condition other than BWS. It can be difficult to distinguish mild hemihyperplasia from asymmetry that is considered normal within the general population, such as leg length discrepancies of 1 cm or less. Girth differences can be confounded by placement of the measuring tape, choice of body landmarks, etc., and there are currently no specific recommendations regarding standardized measurements. Diagnostic imaging studies such as CT or MRI may be helpful in defining the tissues involved [Mussa et al 2021].
- Hemihyperplasia may affect segmental regions of the body or selected organs and tissues.
- Hemihyperplasia is typically characterized by overgrowth of muscle tissue leading to differences in bulk but can be associated with bone overgrowth as well.
- When several body segments are involved, hemihyperplasia may be limited to one side of the body (ipsilateral) or involve opposite sides of the body (contralateral).
- Asymmetric growth can remain relatively stable throughout childhood. However, progressive asymmetric growth has also been observed, and this is likely related to mosaicism in the specific tissue (i.e., the percent of cells with the 11p15.5 alteration).
- Referral to an orthopedist for periodic monitoring of leg length discrepancy may include imaging to assess rate of growth and the potential development of scoliosis (see Management).
*Note: Hemihyperplasia refers to an abnormality of cell proliferation leading to asymmetric overgrowth; in BWS, hemihyperplasia, referring to increased cell number, has replaced the term hemihypertrophy, which refers to increased cell size.
Craniofacial features
- Macroglossia (present in ~90%) is generally present at birth, though postnatal development has also been observed [Mussa et al 2016c]. Macroglossia typically consists of an increased size of the tongue in terms of length, width, and/or thickness. When assessing for macroglossia, it is important to ensure that the tongue tissue itself is enlarged. Hypotonia can lead to the appearance of a large tongue, as the tongue sometimes is not retained in the mouth.
- Macroglossia can occasionally obstruct breathing in neonates, who may require respiratory support, tongue reduction, or in some cases tracheostomy.
- Macroglossia may also interfere with feeding in neonates and infants.
- Many affected individuals do not require surgical intervention for macroglossia (see Management, Treatment of Manifestations) depending on the degree of macroglossia and growth velocity. Additionally, growth of the oral cavity may eventually accommodate the enlarged tongue size.
- Indications and timing for surgical correction of macroglossia (reduction glossectomy) vary across different centers. Indications for reduction glossectomy can include airway obstruction leading to sleep apnea, feeding issues, anterior open bite malocclusion, prognathism, and aesthetic concerns [Cielo et al 2018, Cohen et al 2020, Geisler et al 2022].
- Cleft palate is a rare finding in affected individuals (see Phenotype Correlations by Molecular Mechanism).
- Ear findings may be unilateral or bilateral.
- Ear lobe creases are typically on the anterior aspect of the lobe; ear lobe creases that develop in adulthood are not considered to be a feature of BWS.
- Ear pits associated with BWS are located on the posterior aspect of the helix.
- Characteristic facies may include infraorbital creases, midface retrusion, thin vermilion of the upper lip, and prominent jaw (which may become evident in childhood).
Endocrine abnormalities
- Hypoglycemia. Neonatal hypoglycemia is well documented and occurs in approximately 50% of infants with BWS [Mussa et al 2016a]. If hypoglycemia is severe and undetected or untreated, it poses a significant risk for developmental sequelae.
- Most episodes of hypoglycemia are mild and transient and resolve in 72 hours or less.
- In more severe situations, hypoglycemia can persist beyond 72 hours because of hyperinsulinism (defined as increased insulin secretion and/or action at the time of hypoglycemia). Monitoring for hypoglycemia with pre-feed glucose / insulin checks and referral to an endocrinologist for possible fasting studies is recommended if hypoglycemia is persistent (see Management).
- Delayed onset of hypoglycemia (i.e., in the first month of life) is occasionally observed.
- Hypothyroidism has been described but is not common.
Anterior abdominal wall defects including omphalocele, umbilical hernia, and diastasis recti are common. Large umbilical hernias may or may not include the bowel and/or require surgical repair.
Neoplasia. Children with BWS are at increased risk for a variety of tumors, in particular Wilms tumor and hepatoblastoma, but also neuroblastoma, rhabdomyosarcoma, and adrenocortical carcinoma. A wide variety of other tumors, both malignant and benign, may also be seen [Cöktü et al 2020].
- The increased risk for Wilms tumor appears to be concentrated in the first seven years of life [Mussa et al 2019b]. However, Wilms tumor has been reported in children with BWS who are older than age seven years [Gazzin et al 2019].
- The risk for developing hepatoblastoma is concentrated in the first three to four years of life [Mussa et al 2019a].
- The risk of developing a specific tumor in children with BWS is associated with the underlying molecular mechanism and certain phenotypic features (e.g., hemihyperplasia, nephromegaly) [Choufani et al 2013, Maas et al 2016, Mussa et al 2016b, Brzezinksi et al 2021, Duffy et al 2021] (see also Phenotype Correlations by Molecular Mechanism).
Table 3 provides the frequency of select tumors by molecular subtype. Potential limitations of these data include retrospective data collection as well as potential ascertainment bias. In addition, misclassification of BWS subgroups can arise using current clinical testing by methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) [Brzezinski et al 2017]. In the future, prospective studies and improved molecular diagnostics could impact the tumor risks noted below.
Table 3.
Beckwith-Wiedemann Syndrome: Frequency of Select Tumors by Molecular Mechanism
Perspectives on screening for malignant tumors in childhood differ based on local, national, and international practices. In North America, proactive tumor screening is commonly recommended when the risk of tumor development exceeds 1%; however, in many European countries, proactive tumor screening protocols are typically undertaken when the risk of tumor development exceeds 5% [Brzezinski et al 2017, Kalish et al 2017, Brioude et al 2018, Duffy et al 2021, Mussa et al 2021] (see Management, Tables 8 and 9).
Other renal issues
- Kidney abnormalities outside of malignancy can include medullary dysplasia, duplicated collecting system, nephrocalcinosis, medullary sponge kidney, cystic changes, and diverticula [Mussa et al 2012].
- Hypercalciuria can be found in children with BWS even in the absence of renal abnormalities. Of 18 individuals with BWS, 4/18 (22%) had hypercalciuria (as compared to 7%-10% in the general population), and 2/18 (11%) had nephrocalcinosis on imaging (as compared to 7%-10% in the general population) [Goldman et al 2003].
Cognitive and neurobehavioral development is usually normal in children with BWS unless there is a chromosome abnormality, brain malformation, or history of hypoxia or significant untreated hypoglycemia. Neurobehavioral issues such as autism spectrum disorder have been reported with increased frequency in children with BWS ascertained by parental report [Kent et al 2008]. A 2022 study showed that some preschool children with BWS demonstrated psychosocial concerns similar to those reported in children with other chronic health concerns. Additionally, some children with BWS had language comprehension concerns and/or gross motor development [Butti et al 2022], although large body size may transiently impact early gross motor development. Further studies, including longitudinal neurodevelopmental assessments and review of family history for similar issues, are needed to accurately assess whether neurobehavioral and cognitive development is impacted in people with BWS.
Cardiovascular. Much of the information regarding cardiovascular findings in BWS is anecdotal. Cardiomegaly was reported in approximately 20% of affected individuals prior to molecular testing and may be detected in infancy if a chest x-ray is performed, but typically resolves without treatment.
- Cardiomyopathy has been reported but is rare.
- Long QT syndrome has been reported in a child with BWS who had a balanced translocation between chromosomes 11 and 17 that interrupted KCNQ1 [Kaltenbach et al 2013].
Hearing loss is rarely reported in individuals with BWS and is either sensorineural [Kantaputra et al 2013] or conductive due to stapedial fixation [Hopsu et al 2003].
Note: Although parents of children with BWS occasionally raise concerns regarding hearing loss and hypotonia, it is difficult to ascertain whether these and other issues occur with a greater frequency in individuals with BWS compared to the general population.
Brain abnormalities involving the posterior fossa have been rarely reported [Gardiner et al 2012, Brioude et al 2015].
Hematologic. Neonatal polycythemia may occasionally be observed but typically does not require treatment and is self-limited.
Skin. Infantile hemangiomas of the skin and intra-abdominal organs (for example, in the liver) have been noted in some individuals with BWS. However, management is not specific for BWS.
Adulthood. After childhood, prognosis is generally favorable with respect to health and quality of life. However, health concerns (e.g., renal medullary dysplasia, urolithiasis, subfertility in males) may present in older individuals, often stemming from pediatric issues (i.e., azoospermia may result from late surgical correction of cryptorchidism) [Gazzin et al 2019]. In addition, both benign tumors (mammary fibroepithelioma, non-functional adrenal adenoma, hepatic angioma, uterine myoma) and malignant tumors (early T-cell precursor acute lymphoblastic leukemia, intratubular germ cell neoplasia, testicular Sertoli cell tumor) diagnosed outside of early childhood (i.e., after age 7-8 years) were noted in one small study, including one case of hepatoblastoma at age 22 years [Gazzin et al 2019]. As with pediatric presentation, adult health issues in BWS may be associated with specific molecular subtypes, but further study is needed.
Phenotype Correlations by Molecular Mechanism
While general phenotypic correlations by molecular mechanism are provided below, specific clinical outcomes in any individual with BWS cannot be precisely predicted based on the molecular alteration. The remaining variability in individuals with BWS may be due to somatic mosaicism, genetic background, and/or other unidentified factors. For information on tumor risk based on molecular mechanism, see Table 3.
Table 4.
Associated Findings in Individuals with BWS by Molecular Mechanism
Nomenclature
BWS was originally called EMG, based on the three clinical findings of exomphalos, macroglossia, and gigantism.
In terms of syndromic nomenclature, it is well accepted that clinical diagnoses of many genetic disorders caused by germline pathogenic variants can be challenging when there is a wide range of clinical expressivity. This is particularly true for disorders associated with somatic mosaicism of pathogenic variants. Utilization of a dyadic approach to nomenclature as proposed by Sapp et al [2019] and Biesecker et al [2021] will provide increased clarity for diagnostic classification. Examples could include: 11p15.5 IC1-related BWS, 11p15.5 UPD-related BWS, and 11p15.5 IC2-related BWS.
Prevalence
The reported prevalence of ~1:10,000 [Mussa et al 2013] to ~1:13,700 [Thorburn et al 1970] likely represents an underestimate given the existence of undiagnosed individuals with milder phenotypes.
Genetically Related (Allelic) Disorders
Molecular alterations at 11p15 including loss of methylation at imprinting center 2 (IC2), gain of methylation at imprinting center 1 (IC1) [Martin et al 2005], and 11p15 paternal uniparental disomy [Shuman et al 2002] have been reported in individuals who have apparently isolated hemihyperplasia (lateralized overgrowth). Some cases of apparently isolated hemihyperplasia represent one end of the clinical spectrum of BWS; however, other cases of isolated hemihyperplasia are likely caused by somatic mosaicism in genes outside of the 11p15 region.
Isolated Wilms tumor can be associated with constitutional alterations of chromosome 11p15 including hypermethylation at IC1, paternal uniparental disomy (UPD) of 11p15, loss of methylation at IC2 (rare), and genomic abnormalities of 11p15, including deletions and insertions [Scott et al 2008].
Somatic mosaicism for loss of methylation at the paternal IC1 is associated with Silver-Russell syndrome and/or isolated hemihypoplasia [Zeschnigk et al 2008, Eggermann 2009, Eggermann et al 2015].
CDKN1C. A maternally inherited pathogenic variant in CDKN1C leading to increased stability of cyclin-dependent kinase inhibitor 1C was reported in one family with Silver-Russell syndrome [Brioude et al 2013], and maternally inherited gain-of-function heterozygous pathogenic variants in CDKN1C cause IMAGe syndrome [Arboleda et al 2012, Milani et al 2014].
Mosaic genome-wide paternal uniparental isodisomy (UPiD), a sporadic overgrowth disorder, can also be considered in the differential diagnosis of BWS. Like BWS, mosaic genome-wide paternal UPiD is associated with large size for gestational age, placentomegaly, polyhydramnios, macroglossia, hypoglycemia due to hyperinsulinism, umbilical hernia, hepatomegaly, hemangioma, and a high tumor risk (kidney, liver, adrenal gland) [Inbar-Feigenberg et al 2013, Kalish et al 2013, Postema et al 2019]. In contrast to BWS, mosaic genome-wide paternal UPiD can be associated with features of multiple imprinting disorders as well as a higher rate of developmental delay and a more severe presentation. Chimeric genome-wide paternal UPiD has also been reported with similar features to mosaic genome-wide paternal UPiD; in chimeric genome-wide paternal UPiD, there may be mixed cell populations in the internal and external genitalia, including the gonads, which could lead to an increased risk for germ cell tumors [Sheppard et al 2019].
Differential Diagnosis
Overgrowth. Beckwith-Wiedemann syndrome (BWS) is often considered in the differential diagnosis of children presenting with overgrowth (see Table 5). It is important to note the existence of as-yet unclassified overgrowth syndromes that need to be differentiated from BWS.
Of note: In children considered to have BWS and developmental delay who have a normal chromosome study, no history of hypoxia or hypoglycemia, and normal brain imaging, other causes of developmental delay need to be considered.
Table 5.
Overgrowth Disorders to Consider in the Differential Diagnosis of Beckwith-Wiedemann Syndrome
Hemihyperplasia or segmental overgrowth can occur as an isolated finding or may be associated with other syndromes such as Klippel-Trenaunay-Weber syndrome (OMIM 149000) and the disorders summarized in Table 6. Asymmetries, such as of the face, legs, or chest, should be evaluated to exclude plagiocephaly, congenital hip dislocation, and chest wall deformities.
Table 6.
Disorders with Hemihyperplasia or Lateralized Overgrowth to Consider in the Differential Diagnosis of Beckwith-Wiedemann Syndrome
Multilocus imprinting disturbances (MLID). Individuals with MLID may present with single or multiple imprinting disorders (e.g., BWS, Silver-Russell syndrome [SRS]) [Grosvenor et al 2022] or may be phenotypically normal. Although a genetic etiology has not been identified in many children with MLID, 20%-30% of mothers of children with MLID and significant reproductive issues may have trans-acting, compound heterozygous or homozygous pathogenic variants in maternal effect genes encoding subcortical maternal complex (SCMC) proteins [Eggermann et al 2022, Tannorella et al 2022]. SCMC proteins are involved with early embryonic development and maturation of oocytes. Pathogenic variants in the SCMC genes (NLRP2, NLRP5, NLRP7, PADI6, and KHDC3L) can result in reproductive failure, molar pregnancies, and children with imprinting disorders [Eggermann et al 2022]. One individual with MLID and paternal uniparental disomy for chromosome 20 has been reported [Choufani et al 2021].
For individuals with methylation alterations in the 11p15 imprinted domain as well as in other imprinted loci, review of the maternal history should be undertaken for findings such as recurrent pregnancy loss or molar pregnancy. In these situations, consideration should be given to testing for pathogenic variants in maternal effect genes that lead to BWS. If a pathogenic variant in a maternal effect gene is detected in the proband and mother, information regarding the increased risk for reproductive complications such as preeclampsia, recurrent pregnancy loss, and molar pregnancy as well as the significant risk for having children with imprinting disorders should be addressed through genetic counseling [Elbracht et al 2020, Eggermann et al 2022, Tannorella et al 2022].
Note: It is not yet clear if oligogenic or multifactorial causes are relevant in MLID. While it appears that some heterozygous variants in SCMC genes may be disease causing, such cases may also be associated with unidentified pathogenic variants in the second allele or other SCMC genes. In addition, the potential interaction of pathogenic variants with interventions such as assisted reproductive technology will be an important area of future work.
Management
Clinical practice guidelines for Beckwith-Wiedemann syndrome have been published and most frequently focus on the issue of tumor screening protocols (see Kalish et al [2017] and Brioude et al [2018]).
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Beckwith-Wiedemann syndrome, the evaluations summarized in Table 7 (if not performed as part of the evaluation that led to diagnosis) are recommended.
Table 7.
Beckwith-Wiedemann Syndrome: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Table 8.
Beckwith-Wiedemann Syndrome: Treatment of Manifestations
Surveillance
Perspectives on screening for malignant tumors in childhood differ based on local, national, and international practices. In North America proactive tumor screening is often recommended when the risk of tumor development exceeds 1% [Brzezinski et al 2017, Kalish et al 2017, Brioude et al 2018, Duffy et al 2021, Mussa et al 2021]. In many European countries, proactive tumor screening protocols are typically undertaken when the risk of tumor development exceeds 5%, and as such, tumor screening is based on the risk associated with specific molecular mechanism (see Table 3) [Brioude et al 2018]. Table 9 summarizes typical tumor screening protocols in North America. Surveillance has been shown to improve outcomes, including earlier detection of tumors [Mussa et al 2019b].
For general screening guidelines outside of tumor surveillance, see Table 10.
Table 9.
Beckwith-Wiedemann Syndrome: Tumor Surveillance Protocols 1, 2
Table 10.
Beckwith-Wiedemann Syndrome: Recommended General (Non-Tumor) Surveillance
Evaluation of Relatives at Risk
It is appropriate to evaluate the newborn sib of an individual with BWS in order to identify as early as possible those who would benefit from initiation of preventive measures. Evaluations can include:
- Genetic testing if a maternal CDKN1C pathogenic variant or familial duplication, deletion, or cytogenetically visible alteration of 11p15 is known;
- Monitoring of an at-risk newborn sib for hypoglycemia, even in the absence of obvious clinical findings on prenatal investigation;
- Careful evaluation of the apparently unaffected twin of discordant monozygotic twins (MZ), including clinical examination and molecular testing, preferably of multiple tissues, if available. There should be strong consideration of tumor surveillance for the apparently unaffected MZ twin given the possibility of shared fetal circulation and the potential for resulting somatic mosaicism [Weksberg et al 2002, Cohen et al 2019].
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
The following recurrence risk information pertains to individuals who have Beckwith-Wiedemann syndrome (BWS) without other imprinting disorders, such as multilocus imprinting disturbances (MLID). See Differential Diagnosis for more information on MLID.
BWS without MLID is associated with abnormal expression of imprinted genes in the BWS critical region. Abnormal expression of imprinted genes can be caused by an epigenetic or genomic alteration leading to an abnormal methylation pattern at 11p15.5, a copy number variant of chromosome 11p15.5, or a heterozygous maternally inherited CDKN1C pathogenic variant. Reliable recurrence risk assessment requires identification of the genetic mechanism in the proband that underlies the abnormal expression of imprinted genes in the BWS critical region. While the majority of families have a recurrence risk of less than 1%, certain underlying genetic mechanisms involve a recurrence risk as high as 50%. In families with recurrence of BWS, maternally inherited CDKN1C pathogenic variants account for approximately 40% of genetic alterations, and paternally or maternally inherited copy number variants including small duplications or deletions of 11p15.5 account for approximately 9% of genetic alterations [Baskin et al 2014].
Risk to Family Members
Parents of a proband
- Most individuals with BWS do not have an affected parent.
- Recommendations for the clinical evaluation of the parents of a child with BWS and no known family history of BWS include a medical and family history focused on BWS-associated medical issues in early childhood. Infant and childhood photographs of the parents may be useful. Although physical examination may be of limited value in adulthood, ear pits/creases may still be present. Anterior ear lobe creases are not uncommon in the general population; however, posterior ear pits are rarely reported outside of the association with BWS.
- If the proband has BWS associated with MLID, the maternal history should also be reviewed with respect to potential recurrent pregnancy loss or other adverse outcomes that could indicate a maternal effect gene abnormality. Pathogenic variants in maternal effect genes are associated with an increased risk for reproductive complications such as preeclampsia, recurrent pregnancy loss, and molar pregnancy as well as the risk for having children with imprinting disorders [Elbracht et al 2020, Eggermann et al 2022].
- Clarification of the genetic status of the parents of the proband is recommended to allow reliable recurrence risk assessment. Testing recommendations for the parents are based on the genetic alteration identified in the proband. If the genetic alteration identified in the proband is a:
- Cytogenetically visible duplication, inversion, or translocation involving chromosome 11p15.5, then chromosome analysis (karyotyping) should be offered to both parents;
- Copy number variant (e.g., a small 11p15.5 duplication or deletion), then microarray (SNP based) should be offered to both parents;
- Heterozygous CDKN1C pathogenic variant, then targeted testing for the CDKN1C pathogenic variant identified in the proband should be offered to the mother of the proband;
- Paternal uniparental disomy (UPD) of 11p15.5, then parental testing is not indicated/recommended/required, as paternal UPD of 11p15.5 is typically due to postzygotic somatic recombination;
- Loss of methylation of imprinting center 2 (IC2) or gain of methylation of imprinting center 1 (IC1), then maternal history should be reviewed for findings suggestive of a variant in a maternal effect gene.
- If the proband has neither a genomic variant (i.e., a copy number variant at 11p15.5 or a cytogenetically visible chromosome alternation) nor a CDKN1C pathogenic variant and the family history is uninformative (no maternal history of unexplained spontaneous abortion, hydatidiform moles, or a sib with BWS or another imprinting disorder [e.g., Silver-Russell syndrome]), parental testing is typically not indicated.
- Note: If the family history suggests possible MLID, further consideration for genetic testing for pathogenic variants in maternal effect genes should be entertained (see Differential Diagnosis).
Sibs of a proband. See Table 11.
Table 11.
Risk to Sibs of a Proband with Beckwith-Wiedemann Syndrome
Offspring of a proband. See Table 12.
Table 12.
Risk to Offspring of a Proband with Beckwith-Wiedemann Syndrome
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has a BWS-related genetic alteration or chromosome rearrangement, the parent's family members may be at risk.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and genetic counseling regarding prenatal testing is before pregnancy. Decisions about testing to determine the genetic status of at-risk asymptomatic family members are best made before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or who carry genomic alterations that increase the risk of BWS in offspring (e.g., an unaffected female who has inherited a balanced 11p15 translocation from her father).
- If pathogenic variants in maternal effect genes are detected in the proband and mother, information regarding the increased risk for reproductive complications such as preeclampsia, recurrent pregnancy loss, and molar pregnancy as well as the risk for having children with imprinting disorders should be included in genetic counseling [Elbracht et al 2020, Eggermann et al 2022].
Possible imprinting risks associated with assisted reproductive technology (ART). Offspring born following pregnancies conceived naturally following an extended period of subfertility and/or with the use of ART have an increased risk for imprinting disorders [Cortessis et al 2018]. The risk for BWS in live births from ART conception in one study was tenfold higher than from natural conception [Mussa et al 2017].
Monozygotic twinning. Monozygotic twins clinically discordant for BWS (usually females) have been shown to also be discordant for loss of methylation at IC2 in skin fibroblasts but variably concordant in blood cells, probably as a result of shared fetal circulation [Weksberg et al 2002]. Male monozygotic twins are much less frequently observed and demonstrate additional molecular findings including UPD for 11p15 and gain of methylation at IC1. One study demonstrated that in monozygotic twins where the proband with BWS has a higher score on the diagnostic algorithm used, the "unaffected" twin may or may not manifest any BWS-related clinical features [Cohen et al 2019]. This is thought to be due to asymmetric levels of mosaicism, with the majority of embryonic cells with the BWS-related molecular alteration being localized in the embryo with more severe features of BWS. As no recurrences are reported in the sibs of these twins, the recurrence risk is not known.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Positive family history
- Primary genomic variants. If a pathogenic genomic variant involving chromosome 11p15.5 (i.e., a cytogenetically visible duplication, inversion, or translocation), a pathogenic copy number variant of 11p15.5, or a CDKN1C pathogenic variant has been identified in the proband, prenatal testing via analysis of fetal DNA from samples obtained by chorionic villus sampling (CVS) or amniocentesis is possible. Preimplantation genetic testing is possible for a familial CDKN1C pathogenic variant and may be possible for some familial genomic variants.
- Methylation changes. DNA extracted from amniotic fluid is currently felt to provide the most reliable tissue source for evaluating fetal methylation status, although false negative findings have been reported [Eggermann et al 2015]. Cultured amniocytes may demonstrate clonal findings that are not representative of the fetal status, similar to postnatal testing issues related to somatic mosaicism. Similarly, tissue obtained via CVS for prenatal testing for methylation status does not yield reliable results; a false positive test has been reported [Eggermann et al 2015]. This latter issue reflects the timing of methylation establishment in early placental development. Likewise, preimplantation genetic testing may be possible for a pathogenic familial genomic alteration but not for evaluation of methylation status. Genetic counseling regarding the potential limitations of prenatal testing for epigenetic alterations should be undertaken [Eggermann et al 2015].
For all pregnancies at increased risk for BWS, whether or not the genetic mechanism is known:
- Maternal serum alpha-fetoprotein (AFP) concentration may be elevated at 16 weeks' gestation in the presence of omphalocele.
- Ultrasound examination can be performed at 19-20 weeks' gestation and again at 25-32 weeks' gestation to assess growth parameters that may become advanced for gestational age late in the second trimester and to detect abdominal wall defects, organomegaly, kidney anomalies, cleft palate, cardiac abnormalities, and macroglossia.
Note: (1) If ultrasound examination does not show malformations or abnormalities of fetal growth, a residual risk for recurrence of BWS remains, given the variability in clinical presentation. (2) Even in the absence of obvious clinical findings on prenatal investigation, the newborn should be monitored for hypoglycemia.
Negative family history. In pregnancies in which there is no family history of BWS but an abnormality such as apparently isolated omphalocele is detected on ultrasound examination, additional investigations that should be considered [Porter et al 2009, Wilkins-Haug et al 2009] include:
- Molecular genetic testing for methylation alterations in amniocytes, and if no methylation alteration is identified, testing for a CDKN1C pathogenic variant;
- Chromosomal microarray for copy number variants involving chromosome 11p15 and/or cytogenetic testing to evaluate for duplications, inversions, or translocations involving 11p15;
- Serial ultrasound examinations to assess fetal growth and to detect other abnormalities characteristic of BWS.
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements. Molecular genetic testing can be offered if there is a high index of suspicion for BWS.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Associazione Italiana Sindrome di Beckwith-Wiedemann ODV (AIBWS-Onlus)ItalyPhone: 39 345.3121850Email: [email protected]
- Beckwith-Wiedemann Children's Foundation International
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Beckwith-Wiedemann Syndrome: Genes and Databases
Table B.
OMIM Entries for Beckwith-Wiedemann Syndrome (View All in OMIM)
Molecular Pathogenesis
Imprinting is an epigenetic process whereby the DNA of the two alleles of a gene is differentially modified so that only one parental allele (parent specific for each gene) is normally expressed and the other allele is not expressed or is silenced [Barlow 1994]. Imprinted genes cluster to distinct domains in the genome and an imprinting center (IC) controls resetting of a group of closely linked imprinted genes during transmission through the germline [Nicholls 1994].
- Gain of methylation or hypermethylation: increased level of DNA methylation compared to control samples. For imprinted regions this may be associated with methylation of a normally unmethylated allele.
- Loss of methylation or hypomethylation: decreased level of DNA methylation compared to control samples. For imprinted regions, this may be associated with loss of methylation of a normally methylated allele.
An imprinting center (IC) is a region of DNA that can regulate the expression of neighboring imprinted genes on the same allele (in cis) over large distances. ICs are usually characterized by differential DNA methylation and differential histone modifications and may also be referred to as imprinting control regions (ICRs) or differentially methylated regions (DMRs).
Many different molecular alterations in the 11p15 region occur in association with Beckwith-Wiedemann syndrome (BWS). Several imprinted genes, including growth factors and tumor suppressor genes, that are located in the 11p15 region have been implicated in the pathogenesis of this condition:
- IGF2 is an imprinted gene encoding a paternally expressed embryonic growth factor. Disruption of IGF2 imprinting resulting in biallelic expression has been observed in some individuals with BWS as well as in multiple types of tumors, including Wilms tumor. Mice with a pathogenic variant in the paternally derived Igf2 allele are small at birth, whereas the same pathogenic variant on the maternally inherited allele does not affect fetal growth. Overexpression of Igf2 or disruption of the Igf2 receptor has also been shown to cause overgrowth in mice.
- H19 is an imprinted gene encoding a maternally expressed and biologically active noncoding mRNA that may function as a tumor suppressor. Approximately 50% of individuals with BWS exhibit biallelic IGF2 expression in their tissues, demonstrating uncoupled expression of IGF2 and H19; that is, most retain normal maternal monoallelic expression of H19. Less commonly, changes in H19 expression or methylation are reported in individuals with BWS [Joyce et al 1997, Sparago et al 2004].
- CDKN1C encodes the protein p57KIP2, a member of the cyclin-dependent kinase inhibitor family that acts to negatively regulate cell proliferation. This gene is both a putative tumor suppressor gene and a potential negative regulator of fetal growth. Both of these functions and the preferential maternal expression (incomplete repression of transcription of the paternal allele) of this gene support its causal role in BWS. Pathogenic variants in this gene have been reported in approximately 5% of affected individuals. CDKN1C pathogenic variants are found more frequently in individuals with omphalocele, cleft palate, and a positive family history. However, not all instances of vertical transmission of BWS can currently be ascribed to pathogenic variants in CDKN1C [Hatada et al 1997, Lee et al 1997, Cardoso et al 2022].
- KCNQ1. The protein encoded by KCNQ1 forms part of a potassium channel and has also been implicated in at least two cardiac arrhythmia syndromes, Romano-Ward syndrome and Jervell and Lange-Nielsen syndrome. This gene is maternally expressed in most tissues (excluding the heart) and has four alternatively spliced transcripts, two of which are untranslated.
- KCNQ1OT1 is an antisense transcript that originates in intron 10 of KCNQ1. Loss of imprinting occurs in the 5' differentially methylated promoter region (IC2) of KCNQ1OT1 in 50% of individuals with BWS [Bliek et al 2001, Weksberg et al 2001].
Imprinting centers 1 and 2 (IC1 and IC2) within the 11p15.5 region control gene expression across large chromosomal domains:
- IC1 is the telomeric imprinting center on chromosome 11p15.5 that maps upstream of the H19 promoter and regulates H19 and IGF2 expression. IC1 may also be referred to as ICR1, DMR1, or H19DMR. It is normally methylated on the paternally derived allele and unmethylated on the maternally derived allele.
- IC2 is the centromeric imprinting center that regulates expression of several genes, including KCNQ1OT1, KCNQ1, and CDKN1C. It may also be referred to as ICR2, DMR2, or KvDMR1. It is normally methylated on the maternally derived allele and unmethylated on the paternally derived allele.
Overall, there are two imprinted domains in the BWS critical region (see Figure 1a).
- Domain 1 is telomeric and contains the imprinted genes H19 and IGF2. H19 and IGF2 are reciprocally expressed imprinted genes, with H19 expressed from the maternally derived allele and IGF2 expressed from the paternally derived allele. The expression of this domain is regulated by IC1 (see above). IC1 is normally methylated on the paternally derived allele and unmethylated on the maternally derived allele. Regulation of transcription is accomplished by binding of the zinc-finger insulator protein CTCF to its consensus sequence within IC1. CTCF only binds to unmethylated sequence (maternally derived allele) and interferes with downstream enhancers interacting with the IGF2 promoters [Hark et al 2000].
- Domain 2 is centromeric and contains the imprinted genes CDKN1C, KCNQ1, and KCNQ1OT1. Regulation of this domain is controlled by IC2. IC2 is located in intron 10 of KCNQ1. IC2 contains the promoter for KCNQ1OT1, a noncoding RNA with regulatory function [Smilinich et al 1999, Pandey et al 2008]. Although the exact regulation of this region is not clear, it is known that loss of methylation at IC2 on the maternally derived chromosome results in biallelic expression of the normally paternally derived expressed KCNQ1OT1. Additionally, it has been shown that individuals with BWS and loss of methylation at IC2 on the maternally derived chromosome have reduced CDKN1C expression [Diaz-Meyer et al 2003].
Uniparental disomy (UPD). In BWS, somatic mosaicism for 11p15 UPD is found in 20% of affected individuals. The UPD appears to consistently arise from a somatic recombination event resulting in paternal disomy. Most UPD cases demonstrate segmental mosaicism for paternal UPD for 11p15, suggesting that the underlying mechanism is a postzygotic somatic recombination event. Therefore, UPD may not be detected if a low level of mosaicism occurs in the tissue sampled, and testing other tissues (e.g., skin fibroblasts, tumor biopsy) should be considered.
Chapter Notes
Acknowledgments
Sanaa Choufani, Khadine Wiltshire, Evan Hathaway
Author History
J Bruce Beckwith, MD; Loma Linda University (2010-2023)
Jennifer M Kalish, MD, PhD (2023-present)
Cheryl Shuman, MS, CGC (2000-present)
Adam C Smith, PhD; The Hospital for Sick Children (2000-2016)
Rosanna Weksberg, MD, PhD, FRCPC, FCCMG, FACMG (2000-present)
Revision History
- 8 June 2023 (ma) Comprehensive update posted live
- 11 August 2016 (ma) Comprehensive update posted live
- 14 December 2010 (me) Comprehensive update posted live
- 8 September 2005 (me) Comprehensive update posted live
- 10 April 2003 (tk) Comprehensive update posted live
- 3 March 2000 (me) Review posted live
- 28 July 1999 (cs) Original submission
References
Published Guidelines / Consensus Statements
- Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, Boonen SE, Cole T, Baker R, Bertoletti M, Cocchi G, Coze C, De Pellegrin M, Hussain K, Ibrahim A, Kilby MD, Krajewska-Walasek M, Kratz CP, Ladusans EJ, Lapunzina P, Le Bouc Y, Maas SM, Macdonald F, Õunap K, Peruzzi L, Rossignol S, Russo S, Shipster C, Skórka A, Tatton-Brown K, Tenorio J, Tortora C, Grønskov K, Netchine I, Hennekam RC, Prawitt D, Tümer Z, Eggermann T, Mackay DJG, Riccio A, Maher ER. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229-49. [PubMed]
- Eggermann T, Brioude F, Russo S, Lombardi MP, Bliek J, Maher ER, Larizza L, Prawitt D, Netchine I, Gonzales M, Grønskov K, Tümer Z, Monk D, Mannens M, Chrzanowska K, Walasek MK, Begemann M, Soellner L, Eggermann K, Tenorio J, Nevado J, Moore GE, Mackay DJ, Temple K, Gillessen-Kaesbach G, Ogata T, Weksberg R, Algar E, Lapunzina P. Prenatal molecular testing for Beckwith-Wiedemann and Silver-Russell syndromes: a challenge for molecular analysis and genetic counseling. Eur J Hum Genet. 2016;24:784-93. [PubMed]
- Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, Maher ER, Nichols KE, Plon SE, Porter CC, Rednam S, Schultz KAP, States LJ, Tomlinson GE, Zelley K, Druley TE. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. 2017;23:e115-e122. [PubMed]
- Mussa A, Di Candia S, Russo S, Catania S, De Pellegrin M, Di Luzio L, Ferrari M, Tortora C, Meazzini MC, Brusati R, Milani D, Zampino G, Montirosso R, Riccio A, Selicorni A, Cocchi G, Ferrero GB. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur J Med Genet. 2016;59:52-64. [PubMed]
Literature Cited
- Alders M, Maas SM, Kadouch DJ, van der Lip K, Bliek J, van der Horst CM, Mannens MM. Methylation analysis in tongue tissue of BWS patients identifies the (EPI)genetic cause in 3 patients with normal methylation levels in blood. Eur J Med Genet. 2014;57:293-7. [PubMed: 24704790]
- Algar E, Brickell S, Deeble G, Amor D, Smith P. Analysis of CDKN1C in Beckwith Wiedemann syndrome. Hum Mutat. 2000;15:497-508 [PubMed: 10862080]
- Arboleda VA, Lee H, Parnaik R, Fleming A, Banerjee A, Ferraz-de-Souza B, Délot EC, Rodriguez-Fernandez IA, Braslavsky D, Bergadá I, Dell'Angelica EC, Nelson SF, Martinez-Agosto JA, Achermann JC, Vilain E. Mutations in the PCNA-binding domain of CDKN1C cause IMAGe syndrome. Nat Genet. 2012;44:788-92. [PMC free article: PMC3386373] [PubMed: 22634751]
- Baker SW, Duffy KA, Richards-Yutz J, Deardorff MA, Kalish JM, Ganguly A. Improved molecular detection of mosaicism in Beckwith-Wiedemann Syndrome. J Med Genet. 2021;58:178-84. [PMC free article: PMC7959163] [PubMed: 32430359]
- Barlow DP. Imprinting: a gamete's point of view. Trends Genet. 1994;10:194-9 [PubMed: 7864936]
- Baskin B, Choufani S, Chen YA, Shuman C, Parkinson N, Lemyre E, Micheil Innes A, Stavropoulos DJ, Ray PN, Weksberg R. High frequency of copy number variations (CNVs) in the chromosome 11p15 region in patients with Beckwith-Wiedemann syndrome. Hum Genet. 2014;133:321-30. [PubMed: 24154661]
- Biesecker LG, Adam MP, Alkuraya FS, Amemiya AR, Bamshad MJ, Beck AE, Bennett JT, Bird LM, Carey JC, Chung B, Clark RD, Cox TC, Curry C, Dinulos MBP, Dobyns WB, Giampietro PF, Girisha KM, Glass IA, Graham JM Jr, Gripp KW, Haldeman-Englert CR, Hall BD, Innes AM, Kalish JM, Keppler-Noreuil KM, Kosaki K, Kozel BA, Mirzaa GM, Mulvihill JJ, Nowaczyk MJM, Pagon RA, Retterer K, Rope AF, Sanchez-Lara PA, Seaver LH, Shieh JT, Slavotinek AM, Sobering AK, Stevens CA, Stevenson DA, Tan TY, Tan WH, Tsai AC, Weaver DD, Williams MS, Zackai E, Zarate YA. A dyadic approach to the delineation of diagnostic entities in clinical genomics. Am J Hum Genet. 2021;108:8-15. [PMC free article: PMC7820621] [PubMed: 33417889]
- Bliek J, Maas SM, Ruijter JM, Hennekam RC, Alders M, Westerveld A, Mannens MM. Increased tumour risk for BWS patients correlates with aberrant H19 and not KCNQ1OT1 methylation: occurrence of KCNQ1OT1 hypomethylation in familial cases of BWS. Hum Mol Genet. 2001;10:467-76 [PubMed: 11181570]
- Brioude F, Kalish JM, Mussa A, Foster AC, Bliek J, Ferrero GB, Boonen SE, Cole T, Baker R, Bertoletti M, Cocchi G, Coze C, De Pellegrin M, Hussain K, Ibrahim A, Kilby MD, Krajewska-Walasek M, Kratz CP, Ladusans EJ, Lapunzina P, Le Bouc Y, Maas SM, Macdonald F, Õunap K, Peruzzi L, Rossignol S, Russo S, Shipster C, Skórka A, Tatton-Brown K, Tenorio J, Tortora C, Grønskov K, Netchine I, Hennekam RC, Prawitt D, Tümer Z, Eggermann T, Mackay DJG, Riccio A, Maher ER. Expert consensus document: clinical and molecular diagnosis, screening and management of Beckwith-Wiedemann syndrome: an international consensus statement. Nat Rev Endocrinol. 2018;14:229-49. [PMC free article: PMC6022848] [PubMed: 29377879]
- Brioude F, Netchine I, Praz F, Le Jule M, Calmel C, Lacombe D, Edery P, Catala M, Odent S, Isidor B, Lyonnet S, Sigaudy S, Leheup B, Audebert-Bellanger S, Burglen L, Giuliano F, Alessandri JL, Cormier-Daire V, Laffargue F, Blesson S, Coupier I, Lespinasse J, Blanchet P, Boute O, Baumann C, Polak M, Doray B, Verloes A, Viot G, Le Bouc Y, Rossignol S. Mutations of the imprinted CDKN1C gene as a cause of the overgrowth Beckwith-Wiedemann syndrome: clinical spectrum and functional characterization. Hum Mutat. 2015;36:894-902. [PubMed: 26077438]
- Brioude F, Oliver-Petit I, Blaise A, Praz F, Rossignol S, Le Jule M, Thibaud N, Faussat AM, Tauber M, Le Bouc Y, Netchine I. CDKN1C mutation affecting the PCNA-binding domain as a cause of familial Russell Silver syndrome. J Med Genet. 2013;50:823-30. [PubMed: 24065356]
- Brzezinski J, Shuman C, Choufani S, Ray P, Stavropoulos DJ, Basran R, Steele L, Parkinson N, Grant R, Thorner P, Lorenzo A, Weksberg R. Wilms tumour in Beckwith-Wiedemann Syndrome and loss of methylation at imprinting centre 2: revisiting tumour surveillance guidelines. Eur J Hum Genet. 2017;25:1031-9. [PMC free article: PMC5558170] [PubMed: 28699632]
- Brzezinksi J, Shuman C, Weksberg R. Hereditary overgrowth syndromes. In: Malkin D, ed. The Hereditary Basis of Childhood Cancer. Switzerland: Springer Cham; 2021:163-88.
- Butti N, Castagna A, Montirosso R. Psychosocial difficulties in preschool-age children with Beckwith-Wiedemann syndrome: an exploratory study. Children (Basel). 2022;9:551. [PMC free article: PMC9024744] [PubMed: 35455595]
- Cardoso LCA, Parra A, Gil CR, Arias P, Gallego N, Romanelli V, Kantaputra PN, Lima L, Llerena Júnior JC, Arberas C, Guillén-Navarro E, Nevado J, Tenorio-Castano J, Lapunzina P, et al. Clinical spectrum and tumour risk analysis in patients with Beckwith-Wiedemann syndrome due to CDKN1C pathogenic variants. Cancers (Basel). 2022;14:3807. [PMC free article: PMC9367242] [PubMed: 35954470]
- Carli D, Operti M, Russo S, Cocchi G, Milani D, Leoni C, Prada E, Melis D, Falco M, Spina J, Uliana V, Sara O, Sirchia F, Tarani L, Macchiaiolo M, Cerrato F, Sparago A, Pignata L, Tannorella P, Cardaropoli S, Bartuli A, Riccio A, Ferrero GB, Mussa A. Clinical and molecular characterization of patients affected by Beckwith-Wiedemann spectrum conceived through assisted reproduction techniques. Clin Genet. 2022;102:314-23. [PMC free article: PMC9545072] [PubMed: 35842840]
- Choufani S, Ko JM, Lou Y, Shuman C, Fishman L, Weksberg R. Paternal uniparental disomy of the entire chromosome 20 in a child with Beckwith-Wiedemann syndrome. Genes (Basel). 2021;12:172. [PMC free article: PMC7911624] [PubMed: 33513760]
- Choufani S, Shuman C, Weksberg R. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2010;154C:343-54. [PubMed: 20803657]
- Choufani S, Shuman C, Weksberg R. Molecular findings in Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2013;163C:131-40. [PubMed: 23592339]
- Cielo CM, Duffy KA, Vyas A, Taylor JA, Kalish JM. Obstructive sleep apnoea and the role of tongue reduction surgery in children with Beckwith-Wiedemann syndrome. Paediatr Respir Rev. 2018;25:58-63. [PMC free article: PMC5890299] [PubMed: 28366681]
- Cohen JL, Cielo CM, Kupa J, Duffy KA, Hathaway ER, Kalish JM, Taylor JA. The utility of early tongue reduction surgery for macroglossia in Beckwith-Wiedemann syndrome. Plast Reconstr Surg. 2020;145:803e-813e. [PMC free article: PMC8038320] [PubMed: 32221229]
- Cohen JL, Duffy KA, Sajorda BJ, Hathaway ER, Gonzalez-Gandolfi CX, Richards-Yutz J, Gunter AT, Ganguly A, Kaplan J, Deardorff MA, Kalish JM. Diagnosis and management of the phenotypic spectrum of twins with Beckwith-Wiedemann syndrome. Am J Med Genet A. 2019;179:1139-47. [PMC free article: PMC7959854] [PubMed: 31067005]
- Cöktü S, Spix C, Kaiser M, Beygo J, Kleinle S, Bachmann N, Kohlschmidt N, Prawitt D, Beckmann A, Klaes R, Nevinny-Stickel-Hinzpeter C, Döhnert S, Kraus C, Kadgien G, Vater I, Biskup S, Kutsche M, Kohlhase J, Eggermann T, Zenker M, Kratz CP. Cancer incidence and spectrum among children with genetically confirmed Beckwith-Wiedemann spectrum in Germany: a retrospective cohort study. Br J Cancer. 2020;123:619-23. [PMC free article: PMC7434760] [PubMed: 32451468]
- Cortessis VK, Azadian M, Buxbaum J, Sanogo F, Song AY, Sriprasert I, Wei PC, Yu J, Chung K, Siegmund KD. Comprehensive meta-analysis reveals association between multiple imprinting disorders and conception by assisted reproductive technology. J Assist Reprod Genet. 2018;35:943-52. [PMC free article: PMC6030010] [PubMed: 29696471]
- De Pellegrin M, Brogioni L, Laskow G, Barera G, Pajno R, Osimani S, Russo S, Marcucci L. Guided growth in leg length discrepancy in Beckwith-Wiedemann syndrome: a consecutive case series. Children (Basel). 2021;8:1152. [PMC free article: PMC8700625] [PubMed: 34943348]
- Diaz-Meyer N, Day CD, Khatod K, Maher ER, Cooper W, Reik W, Junien C, Graham G, Algar E, Der Kaloustian VM, Higgins MJ. Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J Med Genet. 2003;40:797-801 [PMC free article: PMC1735305] [PubMed: 14627666]
- Duffy KA, Cielo CM, Cohen JL, Gonzalez-Gandolfi CX, Griff JR, Hathaway ER, Kupa J, Taylor JA, Wang KH, Ganguly A, Deardorff MA, Kalish JM. Characterization of the Beckwith-Wiedemann spectrum: diagnosis and management. Am J Med Genet C Semin Med Genet. 2019a;181:693-708. [PMC free article: PMC7959855] [PubMed: 31469230]
- Duffy KA, Cohen JL, Elci OU, Kalish JM. Development of the serum α-fetoprotein reference range in patients with Beckwith-Wiedemann spectrum. J Pediatr. 2019b;212:195-200.e2. [PMC free article: PMC6707865] [PubMed: 31235384]
- Duffy KA, Deardorff MA, Kalish JM. The utility of alpha-fetoprotein screening in Beckwith-Wiedemann syndrome. Am J Med Genet A. 2017;173:581-4. [PMC free article: PMC5928504] [PubMed: 28160403]
- Duffy KA, Getz KD, Hathaway ER, Byrne ME, MacFarland SP, Kalish JM. Characteristics associated with tumor development in individuals diagnosed with Beckwith-Wiedemann spectrum: novel tumor-(epi)genotype-phenotype associations in the BWSp population. Genes (Basel). 2021;12:1839. [PMC free article: PMC8621885] [PubMed: 34828445]
- Eggermann T. Silver-Russell and Beckwith-Wiedemann syndromes: opposite (epi)mutations in 11p15 result in opposite clinical pictures. Horm Res. 71 Suppl. 2009;2:30-5. [PubMed: 19407494]
- Eggermann T, Soellner L, Buiting K, Kotzot D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol Med. 2015;21:77-87. [PubMed: 25547535]
- Eggermann T, Yapici E, Bliek J, Pereda A, Begemann M, Russo S, Tannorella P, Calzari L, de Nanclares GP, Lombardi P, Temple IK, Mackay D, Riccio A, Kagami M, Ogata T, Lapunzina P, Monk D, Maher ER, Tümer Z. Trans-acting genetic variants causing multilocus imprinting disturbance (MLID): common mechanisms and consequences. Clin Epigenetics. 2022;14:41. [PMC free article: PMC8928698] [PubMed: 35296332]
- Elbracht M, Mackay D, Begemann M, Kagan KO, Eggermann T. Disturbed genomic imprinting and its relevance for human reproduction: causes and clinical consequences. Hum Reprod Update. 2020;26:197-213. [PubMed: 32068234]
- Fiala EM, Ortiz MV, Kennedy JA, Glodzik D, Fleischut MH, Duffy KA, Hathaway ER, Heaton T, Gerstle JT, Steinherz P, Shukla N, McNeer N, Tkachuk K, Bouvier N, Cadoo K, Carlo MI, Latham A, Dubard Gault M, Joseph V, Kemel Y, Kentsis A, Stadler Z, La Quaglia M, Papaemmanuil E, Friedman D, Ganguly A, Kung A, Offit K, Kalish JM, Walsh MF. 11p15.5 epimutations in children with Wilms tumor and hepatoblastoma detected in peripheral blood. Cancer. 2020;126:3114-21. [PMC free article: PMC7383476] [PubMed: 32320050]
- Fontana L, Bedeschi MF, Maitz S, Cereda A, Faré C, Motta S, Seresini A, D'Ursi P, Orro A, Pecile V, Calvello M, Selicorni A, Lalatta F, Milani D, Sirchia SM, Miozzo M, Tabano S. Characterization of multi-locus imprinting disturbances and underlying genetic defects in patients with chromosome 11p15.5 related imprinting disorders. Epigenetics. 2018;13:897-909. [PMC free article: PMC6284780] [PubMed: 30221575]
- Gardiner K, Chitayat D, Choufani S, Shuman C, Blaser S, Terespolsky D, Farrell S, Reiss R, Wodak S, Pu S, Ray PN, Baskin B, Weksberg R. Brain abnormalities in patients with Beckwith-Wiedemann syndrome. Am J Med Genet A. 2012;158A:1388-94. [PubMed: 22585446]
- Gazzin A, Carli D, Sirchia F, Molinatto C, Cardaropoli S, Palumbo G, Zampino G, Ferrero GB, Mussa A. Phenotype evolution and health issues of adults with Beckwith-Wiedemann syndrome. Am J Med Genet A. 2019;179:1691-702. [PubMed: 31339634]
- Geisler EL, Jeffers J, Salhi S, Perlyn CA. Reduction glossectomy for macroglossia in Beckwith-Wiedemann syndrome: is post-op intubation necessary? Cleft Palate Craniofac J. 2022;59:126-31. [PubMed: 33550827]
- Goldman M, Shuman C, Weksberg R, Rosenblum ND. Hypercalciuria in Beckwith-Wiedemann syndrome. J Pediatr. 2003;142:206-8 [PubMed: 12584548]
- Grosvenor SE, Davies JH, Lever M, Sillibourne J, Mackay DJG, Temple IK. A patient with multilocus imprinting disturbance involving hypomethylation at 11p15 and 14q32, and phenotypic features of Beckwith-Wiedemann and Temple syndromes. Am J Med Genet A. 2022;188:1896-903. [PMC free article: PMC9310769] [PubMed: 35266280]
- Guemes M, Shah P, Rozenkova K, Gilbert C, Morgan K, Hussain K. Severe hyperinsulinaemic hypoglycaemia in Beckwith-Widemann syndrome due to paternal uniparental disomy of 11p15.5 managed with sirolimus therapy. Horm Res Paediar. 2016;85:353-7. [PubMed: 26863215]
- Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature. 2000;405:486-9 [PubMed: 10839547]
- Hatada I, Nabetani A, Morisaki H, Xin Z, Ohishi S, Tonoki H, Niikawa N, Inoue M, Komoto Y, Okada A, Steichen E, Ohashi H, Fukushima Y, Nakayama M, Mukai T. New p57KIP2 mutations in Beckwith-Wiedemann syndrome. Hum Genet. 1997;100:681-3 [PubMed: 9341892]
- Hopsu E, Aarnisalo A, Pitkaranta A. Progressive stapedial fixation in Beckwith-Wiedemann syndrome. Arch Otolaryngol Head Neck Surg. 2003;129:1131-4. [PubMed: 14568801]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389-97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Inbar-Feigenberg M, Choufani S, Cytrynbaum C, Chen YA, Steele L, Shuman C, Ray PN, Weksberg R. Mosaicism for genome-wide paternal uniparental disomy with features of multiple imprinting disorders: diagnostic and management issues. Am J Med Genet A. 2013;161A:13-20. [PubMed: 23239666]
- Joyce JA, Lam WK, Catchpoole DJ, Jenks P, Reik W, Maher ER, Schofield PN. Imprinting of IGF2 and H19: lack of reciprocity in sporadic Beckwith-Wiedemann syndrome. Hum Mol Genet. 1997;6:1543-8 [PubMed: 9285792]
- Kalish JM, Conlin LK, Bhatti TR, Dubbs HA, Harris MC, Izumi K, Mostoufi-Moab S, Mulchandani S, Saitta S, States LJ, Swarr DT, Wilkens AB, Zackai EH, Zelley K, Bartolomei MS, Nichols KE, Palladino AA, Spinner NB, Deardorff MA. Clinical features of three girls with mosaic genome-wide paternal uniparental isodisomy. Am J Med Genet A. 2013;161A:1929-39. [PMC free article: PMC4082120] [PubMed: 23804593]
- Kalish JM, Doros L, Helman LJ, Hennekam RC, Kuiper RP, Maas SM, Maher ER, Nichols KE, Plon SE, Porter CC, Rednam S, Schultz KAP, States LJ, Tomlinson GE, Zelley K, Druley TE. Surveillance recommendations for children with overgrowth syndromes and predisposition to Wilms tumors and hepatoblastoma. Clin Cancer Res. 2017;23:e115-e122. [PMC free article: PMC5538793] [PubMed: 28674120]
- Kaltenbach S, Capri Y, Rossignol S, Denjoy I, Soudée S, Aboura A, Baumann C, Verloes A. Beckwith-Wiedemann syndrome and long QT syndrome due to familial-balanced translocation t(11;17)(p15.5;q21.3) involving the KCNQ1 gene. Clin Genet. 2013;84:78-81. [PubMed: 23061425]
- Kantaputra PN, Sittiwangkul R, Sonsuwan N, Romanelli V, Tenorio J, Lapunzina P. A novel mutation in CDKN1C in sibs with Beckwith-Wiedemann syndrome and cleft palate, sensorineural hearing loss, and supernumerary flexion creases. Am J Med Genet A. 2013;161A:192-7. [PubMed: 23197429]
- Kent L, Bowdin S, Kirby GA, Cooper WN, Maher ER. Beckwith Weidemann syndrome: a behavioral phenotype-genotype study. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1295-7. [PubMed: 18314872]
- Keren B, Chantot-Bastaraud S, Brioude F, Mach C, Fonteneau E, Azzi S, Depienne C, Brice A, Netchine I, Le Bouc Y, Siffroi JP, Rossignol S. SNP arrays in Beckwith-Wiedemann syndrome: an improved diagnostic strategy. Eur J Med Genet. 2013;56:546-50. [PubMed: 23892181]
- Lam WW, Hatada I, Ohishi S, Mukai T, Joyce JA, Cole TR, Donnai D, Reik W, Schofield PN, Maher ER. Analysis of germline CDKN1C (p57KIP2) mutations in familial and sporadic Beckwith-Wiedemann syndrome (BWS) provides a novel genotype- phenotype correlation. J Med Genet. 1999;36:518-23 [PMC free article: PMC1734395] [PubMed: 10424811]
- Lee MP, DeBaun M, Randhawa G, Reichard BA, Elledge SJ, Feinberg AP. Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am J Hum Genet. 1997;61:304-9 [PMC free article: PMC1715913] [PubMed: 9311734]
- Li M, Squire J, Shuman C, Fei YL, Atkin J, Pauli R, Smith A, Nishikawa J, Chitayat D, Weksberg R. Imprinting status of 11p15 genes in Beckwith-Wiedemann syndrome patients with CDKN1C mutations. Genomics 2001;74:370-6 [PubMed: 11414765]
- Li M, Squire JA, Weksberg R. Molecular genetics of Wiedemann-Beckwith syndrome. Am J Med Genet 1998;79:253-9 [PubMed: 9781904]
- Maas SM, Vansenne F, Kadouch DJ, Ibrahim A, Bliek J, Hopman S, Mannens MM, Merks JH, Maher ER, Hennekam RC. Phenotype, cancer risk, and surveillance in Beckwith-Wiedemann syndrome depending on molecular genetic subgroups. Am J Med Genet A. 2016;170:2248-60. [PubMed: 27419809]
- MacFarland SP, Duffy KA, Bhatti TR, Bagatell R, Balamuth NJ, Brodeur GM, Ganguly A, Mattei PA, Surrey LF, Balis FM, Kalish JM. Diagnosis of Beckwith-Wiedemann syndrome in children presenting with Wilms tumor. Pediatr Blood Cancer. 2018; 65:e27296. [PMC free article: PMC6107414] [PubMed: 29932284]
- Martin RA, Grange DK, Zehnbauer B, Debaun MR. LIT1 and H19 methylation defects in isolated hemihyperplasia. Am J Med Genet A. 2005;134A:129-31. [PubMed: 15651076]
- Milani D, Pezzani L, Tabano S, Miozzo M. Beckwith-Wiedemann and IMAGe syndromes: two very different diseases caused by mutations on the same gene. Appl Clin Genet. 2014;7:169-75. [PMC free article: PMC4173641] [PubMed: 25258553]
- Mussa A, Carli D, Cardaropoli S, Ferrero GB, Resta N. Lateralized and segmental overgrowth in children. Cancers (Basel). 2021;13:6166. [PMC free article: PMC8699773] [PubMed: 34944785]
- Mussa A, Di Candia S, Russo S, Catania S, De Pellegrin M, Di Luzio L, Ferrari M, Tortora C, Meazzini MC, Brusati R, Milani D, Zampino G, Montirosso R, Riccio A, Selicorni A, Cocchi G, Ferrero GB. Recommendations of the Scientific Committee of the Italian Beckwith-Wiedemann Syndrome Association on the diagnosis, management and follow-up of the syndrome. Eur J Med Genet. 2016a;59:52-64. [PubMed: 26592461]
- Mussa A, Duffy KA, Carli D, Ferrero GB, Kalish JM. Defining an optimal time window to screen for hepatoblastoma in children with Beckwith-Wiedemann syndrome. Pediatr Blood Cancer. 2019a;66:e27492. [PMC free article: PMC7955797] [PubMed: 30270492]
- Mussa A, Duffy KA, Carli D, Griff JR, Fagiano R, Kupa J, Brodeur GM, Ferrero GB, Kalish JM. The effectiveness of Wilms tumor screening in Beckwith-Wiedemann spectrum. J Cancer Res Clin Oncol. 2019b;145:3115-23. [PMC free article: PMC6876630] [PubMed: 31583434]
- Mussa A, Molinatto C, Baldassarre G, Riberi E, Russo S, Larizza L, Riccio A, Ferrero GB. Cancer risk in Beckwith-Wiedemann syndrome: a systematic review and meta-analysis outlining a novel (epi)genotype specific histotype targeted screening protocol. J Pediatr. 2016b Sep;176:142-149.e1. [PubMed: 27372391]
- Mussa A, Molinatto C, Cerrato F, Palumbo O, Carella M, Baldassarre G, Carli D, Peris C, Riccio A, Ferrero GB. Assisted reproductive techniques and risk of Beckwith-Wiedemann syndrome. Pediatrics. 2017;140:e20164311. [PubMed: 28634246]
- Mussa A, Peruzzi L, Chiesa N, De Crescenzo A, Russo S, Melis D, Tarani L, Baldassarre G, Larizza L, Riccio A, Silengo M, Ferrero GB. Nephrological findings and genotype-phenotype correlation in Beckwith-Wiedemann syndrome. Pediatr Nephrol. 2012;27:397-406. [PubMed: 22015620]
- Mussa A, Russo S, De Crescenzo A, Chiesa N, Molinatto C, Selicorni A, Richiardi L, Larizza L, Silengo MC, Riccio A, Ferrero GB. Prevalence of Beckwith-Wiedemann syndrome in north west of Italy. Am J Med Genet A. 2013;161A:2481-6. [PubMed: 23918458]
- Mussa A, Russo S, De Crescenzo A, Freschi A, Calzari L, Maitz S, Macchiaiolo M, Molinatto C, Baldassarre G, Mariani M, Tarani L, Bedeschi MF, Milani D, Melis D, Bartuli A, Cubellis MV, Selicorni A, Cirillo Silengo M, Larizza L, Riccio A, Ferrero GB. (Epi)genotype-phenotype correlations in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2016c;24:183-90. [PMC free article: PMC4717210] [PubMed: 25898929]
- Naujokat H, Möller B, Terheyden H, Birkenfeld F, Caliebe D, Krause MF, Fischer-Brandies H, Wiltfang J. Tongue reduction in Beckwith-Wiedemann syndrome: outcome and treatment algorithm. Int J Oral Maxillofac Surg. 2019;48:9-16. [PubMed: 30057238]
- Nicholls RD. New insights reveal complex mechanisms involved in genomic imprinting [editorial; comment]. Am J Hum Genet. 1994;54:733-40 [PMC free article: PMC1918270] [PubMed: 8178814]
- Niemitz EL, DeBaun MR, Fallon J, Murakami K, Kugoh H, Oshimura M, Feinberg AP. Microdeletion of LIT1 in familial Beckwith-Wiedemann syndrome. Am J Hum Genet. 2004;75:844-9 [PMC free article: PMC1182113] [PubMed: 15372379]
- O'Keefe D, Dao D, Zhao L, Sanderson R, Warburton D, Weiss L, Anyane-Yeboa K, Tycko B. Coding mutations in p57KIP2 are present in some cases of Beckwith-Wiedemann syndrome but are rare or absent in Wilms tumors. Am J Hum Genet. 1997;61:295-303 [PMC free article: PMC1715902] [PubMed: 9311733]
- Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232-46. [PubMed: 18951091]
- Pignata L, Cecere F, Verma A, Hay Mele B, Monticelli M, Acurzio B, Giaccari C, Sparago A, Hernandez Mora JR, Monteagudo-Sánchez A, Esteller M, Pereda A, Tenorio-Castano J, Palumbo O, Carella M, Prontera P, Piscopo C, Accadia M, Lapunzina P, Cubellis MV, de Nanclares GP, Monk D, Riccio A, Cerrato F. Novel genetic variants of KHDC3L and other members of the subcortical maternal complex associated with Beckwith-Wiedemann syndrome or Pseudohypoparathyroidism 1B and multi-locus imprinting disturbances. Clin Epigenetics. 2022;14:71. [PMC free article: PMC9148495] [PubMed: 35643636]
- Porter A, Benson CB, Hawley P, Wilkins-Haug L. Outcome of fetuses with a prenatal ultrasound diagnosis of isolated omphalocele. Prenat Diagn. 2009;29:668-73 [PubMed: 19367563]
- Postema FAM, Bliek J, van Noesel CJM, van Zutven LJCM, Oosterwijk JC, Hopman SMJ, Merks JHM, Hennekam RC. Multiple tumors due to mosaic genome-wide paternal uniparental disomy. Pediatr Blood Cancer. 2019;66:e27715. [PubMed: 30882989]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Russo S, Calzari L, Mussa A, Mainini E, Cassina M, Di Candia S, Clementi M, Guzzetti S, Tabano S, Miozzo M, Sirchia S, Finelli P, Prontera P, Maitz S, Sorge G, Calcagno A, Maghnie M, Divizia MT, Melis D, Manfredini E, Ferrero GB, Pecile V, Larizza L. A multi-method approach to the molecular diagnosis of overt and borderline 11p15.5 defects underlying Silver-Russell and Beckwith-Wiedemann syndromes. Clin Epigenetics. 2016;8:23. [PMC free article: PMC4772365] [PubMed: 26933465]
- Sanchez-Delgado M, Riccio A, Eggermann T, Maher ER, Lapunzina P, Mackay D, Monk D. Causes and consequences of multi-locus imprinting disturbances in humans. Trends Genet. 2016;32:444-55. [PubMed: 27235113]
- Sapp JC, Buser A, Burton-Akright J, Keppler-Noreuil KM, Biesecker LG. A dyadic genotype-phenotype approach to diagnostic criteria for Proteus syndrome. Am J Med Genet C Semin Med Genet. 2019;181:565-70. [PMC free article: PMC7520016] [PubMed: 31692258]
- Sasaki K, Soejima H, Higashimoto K, Yatsuki H, Ohashi H, Yakabe S, Joh K, Niikawa N, Mukai T. Japanese and North American/European patients with Beckwith-Wiedemann syndrome have different frequencies of some epigenetic and genetic alterations. Eur J Hum Genet. 2007;15:1205-10. [PubMed: 17700627]
- Scott RH, Douglas J, Baskcomb L, Huxter N, Barker K, Hanks S, Craft A, Gerrard M, Kohler JA, Levitt GA, Picton S, Pizer B, Ronghe MD, Williams D, Cook JA, Pujol P, Maher ER, Birch JM, Stiller CA, Pritchard-Jones K, Rahman N, et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat Genet. 2008;40:1329-34. [PubMed: 18836444]
- Sheppard SE, Lalonde E, Adzick NS, Beck AE, Bhatti T, De Leon DD, Duffy KA, Ganguly A, Hathaway E, Ji J, Linn R, Lord K, Randolph LM, Sajorda B, States L, Conlin LK, Kalish JM. Androgenetic chimerism as an etiology for Beckwith-Wiedemann syndrome: diagnosis and management. Genet Med. 2019;21:2644-9. [PMC free article: PMC7848850] [PubMed: 31147633]
- Shuman C, Steele L, Fei YL, Ray PN, Zackai E, Parisi M, Squire J, Weksberg R. Paternal uniparental disomy of 11p15 is associated with isolated hemihyperplasia and expands Beckwith-Wiedemann syndrome spectrum. Am J Hum Genet 2002;71:A1800, 477
- Slavotinek A, Gaunt L, Donnai D. Paternally inherited duplications of 11p15.5 and Beckwith-Wiedemann syndrome. J Med Genet. 1997;34:819-26 [PMC free article: PMC1051088] [PubMed: 9350814]
- Smilinich NJ, Day CD, Fitzpatrick GV, Caldwell GM, Lossie AC, Cooper PR, Smallwood AC, Joyce JA, Schofield PN, Reik W, Nicholls RD, Weksberg R, Driscoll DJ, Maher ER, Shows TB, Higgins MJ. A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A. 1999;96:8064-9 [PMC free article: PMC22188] [PubMed: 10393948]
- Sparago A, Cerrato F, Vernucci M, Ferrero GB, Silengo MC, Riccio A. Microdeletions in the human H19 DMR result in loss of IGF2 imprinting and Beckwith-Wiedemann syndrome. Nat Genet. 2004;36:958-60 [PubMed: 15314640]
- Tannorella P, Calzari L, Daolio C, Mainini E, Vimercati A, Gentilini D, Soli F, Pedrolli A, Bonati MT, Larizza L, Russo S. Germline variants in genes of the subcortical maternal complex and multilocus imprinting disturbance are associated with miscarriage/infertility or Beckwith-Wiedemann progeny. Clin Epigenetics. 2022;14:43. [PMC free article: PMC8941822] [PubMed: 35317853]
- Thorburn MJ, Wright ES, Miller CG, Smith-Read EH. Exomphalos-macroglossia-gigantism syndrome in Jamaican infants. Am J Dis Child. 1970;119:316-21. [PubMed: 5434588]
- Trobaugh-Lotrario AD, Venkatramani R, Feusner JH. Hepatoblastoma in children with Beckwith-Wiedemann syndrome: does it warrant different treatment? J Pediatr Hematol Oncol. 2014;36:369-73. [PubMed: 24608075]
- Wang KH, Kupa J, Duffy KA, Kalish JM. Diagnosis and management of Beckwith-Wiedemann syndrome. Front Pediatr. 2020;7:562. [PMC free article: PMC6990127] [PubMed: 32039119]
- Weksberg R, Nishikawa J, Caluseriu O, Fei YL, Shuman C, Wei C, Steele L, Cameron J, Smith A, Ambus I, Li M, Ray PN, Sadowski P, Squire J. Tumor development in the Beckwith-Wiedemann syndrome is associated with a variety of constitutional molecular 11p15 alterations including imprinting defects of KCNQ1OT1. Hum Mol Genet. 2001;10:2989-3000 [PubMed: 11751681]
- Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2010 Jan;18(1):8-14. [PMC free article: PMC2987155] [PubMed: 19550435]
- Weksberg R, Shuman C, Caluseriu O, Smith AC, Fei YL, Nishikawa J, Stockley TL, Best L, Chitayat D, Olney A, Ives E, Schneider A, Bestor TH, Li M, Sadowski P, Squire J. Discordant KCNQ1OT1 imprinting in sets of monozygotic twins discordant for Beckwith-Wiedemann syndrome. Hum Mol Genet. 2002;11:1317-25 [PubMed: 12019213]
- Weksberg R, Shuman C, Smith AC. Beckwith-Wiedemann syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:12-23. [PubMed: 16010676]
- Weksberg R, Smith AC, Squire J, Sadowski P. Beckwith-Wiedemann syndrome demonstrates a role for epigenetic control of normal development. Hum Mol Genet. 2003;12 Spec No 1:R61-8 [PubMed: 12668598]
- Wilkins-Haug L, Porter A, Hawley P, Benson CB. Isolated fetal omphalocele, Beckwith-Wiedemann syndrome, and assisted reproductive technologies. Birth Defects Res A Clin Mol Teratol. 2009;85:58-62. [PubMed: 19107956]
- Zeschnigk M, Albrecht B, Buiting K, Kanber D, Eggermann T, Binder G, Gromoll J, Prott EC, Seland S, Horsthemke B. IGF2/H19 hypomethylation in Silver-Russell syndrome and isolated hemihypoplasia. Eur J Hum Genet. 2008;16:328-34 [PubMed: 18159214]
Publication Details
Author Information and Affiliations
Research Scientist
Children's Hospital of Philadelphia;
Assistant Professor of Pediatrics and Genetics
Perelman School of Medicine at the University of Pennsylvania
Philadelphia, Pennsylvania
Senior Associate Scientist, Research Institute
The Hospital for Sick Children;
Professor, Department of Pediatrics
University of Toronto
Toronto, Ontario
Publication History
Initial Posting: March 3, 2000; Last Revision: September 21, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Shuman C, Kalish JM, Weksberg R. Beckwith-Wiedemann Syndrome. 2000 Mar 3 [Updated 2023 Sep 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.