Summary
Clinical characteristics.
Caffey disease is characterized by massive subperiosteal new bone formation (usually involving the diaphyses of the long bones as well as the ribs, mandible, scapulae, and clavicles) typically associated with fever, soft-tissue swelling, and pain, with onset between birth and five months and spontaneous resolution by age two years. Recurrence of bone hyperostosis, fever, soft-tissue swelling, and pain can occur later in life. Adults with a history of Caffey disease in childhood may have joint laxity, skin hyperextensibility, hernias, short stature, and an increased risk for bone fractures and/or deformities.
Diagnosis/testing.
The diagnosis of Caffey disease is established in a proband with typical clinical and radiographic findings; identification of a heterozygous COL1A1 pathogenic variant associated with Caffey disease on molecular genetic testing can confirm the diagnosis.
Management.
Treatment of manifestations: Anti-inflammatory agents, antipyretics, and analgesics can be used in the short term to decrease swelling and fever and to relieve pain; standard treatments for joint hypermobility, skin hyperextensibility, and hernias.
Surveillance: Annual evaluation of stature, fracture history, joint extensibility, and hernias throughout childhood. Consider assessment of bone mineral density in adults with a history of recurrent fractures.
Genetic counseling.
Caffey disease is inherited in an autosomal dominant manner. Some individuals diagnosed with Caffey disease have a parent who had Caffey disease in childhood; others have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with Caffey disease caused by a de novo pathogenic variant is unknown. Each child of an individual who had Caffey disease in childhood has a 50% chance of inheriting the pathogenic variant. Once a molecular diagnosis has been established in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
No consensus clinical diagnostic criteria for Caffey disease have been published.
Suggestive Findings
Caffey disease should be suspected in probands with the following clinical, radiographic, laboratory, and family history findings. Clinical and radiographic findings typically appear between birth and age five months and resolve spontaneously by age two years, although recurrence in adolescence is possible.
Clinical findings
- Irritability, fever, and/or pallor
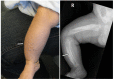
- Soft-tissue swelling and pain adjacent to involved bones (See Figure 2.)
Radiographic findings
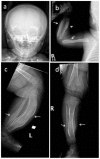
- Subperiosteal cortical hyperostosis of the diaphyses of the long bones (with sparing of the epiphyses)
Laboratory findings
- Serum biochemical markers of inflammation (white blood cell count, erythrocyte sedimentation rate, C-reactive protein) have been elevated a few affected individuals [Gensure et al 2005].
- Alkaline phosphatase may be elevated.
Family history is consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of Caffey disease is established in a proband with typical clinical and radiographic findings. Identification of a heterozygous COL1A1 pathogenic variant associated with Caffey disease (p.Arg1014Cys, p.Arg918Cys) on molecular genetic testing can confirm the diagnosis in those with atypical clinical and radiographic features (see Table 1).
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (see Option 1), whereas comprehensive genomic testing does not (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of Caffey disease, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of COL1A1 to detect the only reported pathogenic variants associated with Caffey disease to date, p.Arg1014Cys or p.Arg918CysNote: To date, no large multiexon COL1A1 deletions or duplications have been identified in individuals with Caffey disease.
- A multigene panel that includes COL1A1 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of Caffey disease is not considered because an individual has atypical phenotypic features, comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. To date, all COL1A1 pathogenic variants associated with Caffey disease are within the coding region and are likely to be identified on exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Caffey Disease
Clinical Characteristics
Clinical Description
Caffey disease is characterized by massive subperiosteal new bone formation (hyperostosis) usually involving the diaphyses of the long bones, as well as the ribs, mandible, scapulae, and clavicles [Caffey & Silverman 1945, Caffey 1957].
Onset. The clinical findings most often appear at age two months (typically between birth and age five months). Rarely, hyperostosis can be detected by ultrasound examination late in the third trimester of pregnancy [Schweiger et al 2003]. One report describes prenatal periosteal inflammation in a fetus with heterozygous COL1A1 pathogenic variant p.Arg1014Cys [Kamoun-Goldrat et al 2008].
Skeletal manifestations. Typically the skeletal manifestations of Caffey disease first appear with soft-tissue swelling and pain over the affected bones between birth and age five months. Massive subperiosteal new bone formation usually involving the diaphyses of the long bones can be seen on imaging. Hyperostosis of the long bones is typically asymmetric, although symmetric hyperostosis has been reported [Tilva et al 2023].
Hyperostosis can also involve the ribs, mandible, scapulae, and clavicles. The hyperostosis resolves before age two years [Kamoun-Goldrat & le Merrer 2008, Cerruti-Mainardi et al 2011, Ranganath et al 2011].
Constitutional manifestations. Skeletal manifestations are accompanied by fever and elevated serum biochemical markers of inflammation (e.g., white blood cell count, erythrocyte sedimentation rate, C-reactive protein) [Gensure et al 2005].
Recurrence of hyperostosis, joint swelling, pain, and fever have been reported multiple times, until late adolescence in individuals with the typical infantile presentation [Borochowitz et al 1991; Navarre et al 2013; ALBagshi & ALZoayed 2015; A Guerin, unpublished data]. Etiology and precipitating factors for recurrence remain unclear [Navarre et al 2013].
Additional findings reported. In one reported family, an individual with COL1A1 pathogenic variant p.Arg1014Cys had a history of Caffey disease as a child and developed joint laxity, skin hyperextensibility, hernias, and multiple fractures in adulthood [Gensure et al 2005]. Additional individuals in the family with the COL1A1 pathogenic variant p.Arg1014Cys had varying degrees of joint laxity and skin hyperextensibility. Skin biopsy of affected individuals showed collagen fibrils that were larger, more variable in shape, and less densely packed than age- and sex-matched controls. Granulofilamentous material was also visible in the matrix along the collagen fibrils. Cultured fibroblasts showed a mix of normal type I collagen and abnormal disulfide crosslinking, either within or between abnormal collagen fibrils. These findings have not been identified in other individuals/families with the same COL1A1 pathogenic variant [Cho et al 2008, Cerruti-Mainardi et al 2011, Ranganath et al 2011].
Other
- Tumoral calcinosis (1 individual); thought to be due to constant remodeling after repeated inflammatory events [Issa El Khoury et al 2012]
- Anemia (1 individual) [Restrepo et al 2004]
- Thrombocytosis (1 individual) [Krishnamurthy & Srinivasan 2012].
Prognosis. In many individuals, the manifestations of Caffey disease resolve spontaneously by age two years and do not predispose to long-term bone abnormalities. Affected individuals from one family had short stature in adulthood and residual bone deformities [Suphapeetiporn et al 2007]. Fractures have been reported in some individuals [Gensure et al 2005, Suphapeetiporn et al 2007].
Histopathology. Bone and muscle biopsy of affected sites in a few individuals have demonstrated an inflammatory reaction [Katz et al 1981].
Genotype-Phenotype Correlations
There are no known genotype-phenotype correlations.
Penetrance
Reduced penetrance based on family history or molecular genetic testing has been reported [Cho et al 2008, Kutty et al 2010, Prior et al 2012, Kitaoka et al 2014, Dhooge et al 2021].
Nomenclature
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], Caffey disease known to be caused by a heterozygous COL1A1 pathogenic variant is referred to as COL1A1-related Caffey disease and included in the osteosclerotic disorders group.
"Prenatal lethal forms of hyperostosis," also referred to as "prenatal Caffey disease" or "Caffey dysplasia" [Nemec et al 2012], are distinct from Caffey disease (also known as infantile cortical hyperostosis) (see Differential Diagnosis).
Prevalence
The number of clinical reports of Caffey disease described to date is no more than a few hundred; however, given the spontaneous resolution of this condition in early childhood, it is likely underdiagnosed.
Genetically Related (Allelic) Disorders
Other phenotypes known to be associated with germline pathogenic variants in COL1A1 are summarized in Table 2.
Table 2.
COL1A1 Allelic Disorders
Differential Diagnosis
Other genetic and acquired conditions may manifest as joint swelling and hyperostosis and thus need to be distinguished from Caffey disease.
Table 3.
Genes of Interest in the Differential Diagnosis of Caffey Disease
Lethal prenatal Caffey disease (prenatal Caffey disease / Caffey dysplasia). This condition typically presents before 35 weeks' gestation and is characterized by cortical hyperostosis as well as bowing or angulation of the long bones and the presence of polyhydramnios and fetal lung disease [Langer & Kaufmann 1986, Lécolier et al 1992, Drinkwater et al 1997, Dahlstrom et al 2001, Savarirayan et al 2002, Hochwald & Osiovich 2011, Nemec et al 2012]. Autosomal recessive inheritance involving genes other than COL1A1 has been proposed [de Jong & Muller 1995, Drinkwater et al 1997, Schweiger et al 2003, Gensure et al 2005].
Acquired conditions
- Hypervitaminosis A can result in bone pain and swelling similar to that seen in Caffey disease. In addition, hyperostosis has been documented in adults with hypervitaminosis A [Wendling et al 2009].
- Prostaglandin E1 (PGE1) exposure. Reversible hyperostosis and long bone swelling has been noted in neonates on PGE1 therapy for several weeks for maintenance of ductus arteriosus patency in the context of congenital heart disease [de Almeida et al 2007].
- Bone malignancies can present similarly to Caffey disease but can be distinguished on bone biopsy.
- Osteomyelitis may be mistakenly diagnosed as joint swelling. Febrile episodes can be common to both conditions; however, the finding of hyperostosis on radiographs helps distinguish between these two entities.
Non-accidental childhood injury (child physical abuse / non-accidental trauma). The prevalence of physical abuse is much greater than the prevalence of Caffey disease. Often the clinical history and presence of fractures, which are not usually a presenting feature of Caffey disease, aid in distinguishing the two [Lee et al 2021].
Management
No clinical practice guidelines for Caffey disease have been published. In the absence of published guidelines, the following recommendations are based on the authors' personal experience managing individuals with this disorder.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Caffey disease, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Caffey Disease: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
Caffey Disease: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 6 are recommended.
Table 6.
Caffey Disease: Recommended Surveillance
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Caffey disease is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Some individuals diagnosed with Caffey disease have a parent who had Caffey disease in childhood.
- An individual diagnosed with Caffey disease may have the disorder as the result of a de novo pathogenic variant. The proportion of individuals with Caffey disease caused by a de novo pathogenic variant is unknown.
- If the proband appears to be the only family member with Caffey disease (i.e., a simplex case), recommendations for the evaluation of the parents of the proband include molecular genetic testing (if a molecular diagnosis has been established in the proband) and a detailed medical history focusing on features of hyperostosis in infancy and current bone health.
- If a molecular diagnosis has been established in the proband, the pathogenic variant identified in the proband is not identified in either parent, and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ (gonadal) cells only.
- The family history of some individuals diagnosed with Caffey disease may appear to be negative because of failure to recognize or remember the occurrence of the disorder in family members or because of reduced penetrance in a parent. Therefore, an apparently negative family history cannot be confirmed unless a molecular diagnosis has been established in the proband and molecular genetic testing has established that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband had Caffey disease in childhood and/or is known to have a Caffey disease-related pathogenic variant, the risk to the sibs is 50%.
- If the proband has a known Caffey disease-related pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the possibility of parental germline mosaicism [Rahbari et al 2016].
- If the genetic status of the parents has not been established but neither parent is known to have had Caffey disease in childhood, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for Caffey disease because of the possibility of reduced penetrance in a parent or parental germline mosaicism.
Offspring of a proband. Each child of an individual who had Caffey disease in childhood has a 50% chance of inheriting a Caffey disease-related pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent had Caffey disease in childhood, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who were affected as children.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once a molecular diagnosis has been established in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Caffey Disease: Genes and Databases
Table B.
OMIM Entries for Caffey Disease (View All in OMIM)
Molecular Pathogenesis
COL1A1 encodes collagen type I, a heterotrimer consisting of two alpha-1 chains and one alpha-2 chain (encoded by COL1A2), which is a fibril-forming collagen found in most connective tissues and is abundant in bone, cornea, dermis, and tendon.
Both COL1A1 pathogenic variants associated with Caffey disease impact an Arg-to-Cys substitution in the Xaa position of the Gly-Xaa-Yaa triplet repeat of the pro-α1(I) chain and reside in regions hypothesized to interact with interleukin-2 and α1β1 integrin [Makar et al 1975, Sweeney et al 2008, Dhooge et al 2021]. Transmission electron microscopy analysis of affected probands' skin biopsies suggests that the introduction of an Arg-Cys substitution interferes with collagen fibril organization [Dhooge et al 2021].
Mechanism of disease causation. The mechanism of disease is unclear. The two COL1A1 pathogenic variants associated with Caffey disease could impact cell signaling. The location of the variants near domains hypothesized to interact with interleukin-2 and α1β1 integrin may play a role in temporary inflammation. Prostaglandin E (PGE) and transforming growth factor beta (TGF-β) may also play a role in pathogenesis, as both can promote cortical hyperostosis [Dhooge et al 2021].
Table 7.
COL1A1 Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Acknowledgments
The authors would like to acknowledge the patients and families who allowed the use of photographs in this review.
Author History
Revision History
- 23 May 2024 (sw) Comprehensive update posted live
- 13 June 2019 (ha) Comprehensive update posted live
- 2 August 2012 (me) Review posted live
- 17 February 2012 (ag) Original submission
References
Literature Cited
- ALBagshi MH, ALZoayed HI. Infantile cortical hyperostosis - a report of Saudi family. Sudan J Paediatr. 2015;15:61-4. [PMC free article: PMC4949861] [PubMed: 27493423]
- Borochowitz Z, Gozal D, Misselevitch I, Aunallah J, Boss JH. Familial Caffey's disease and late recurrence in a child. Clin Genet. 1991;40:329-35. [PubMed: 1756606]
- Caffey J. Infantile cortical hyperostosis; a review of the clinical and radiographic features. Proc R Soc Med. 1957;50:347–54. [PMC free article: PMC1889299] [PubMed: 13431894]
- Caffey J, Silverman WA. Infantile cortical hyperostosis. Preliminary report on a new syndrome. Am J Roentgenol Radium Ther Nucl Med. 1945;54:1.
- Cerruti-Mainardi P, Venturi G, Spunton M, Favaron E, Zignani M, Porvera S, Dallapiccola B. Infantile cortical hyperostosis and COL1A1 mutation in four generations. Eur J Pediatr. 2011;170:1385–90. [PMC free article: PMC3197908] [PubMed: 21567126]
- Cho TJ, Moon HJ, Cho DY, Park MS, Lee DY, Yoo WJ, Chung CY, Choi IH. The c.3040C>T mutation in COL1A1 is recurrent in Korean patients with infantile cortical hyperostosis (Caffey disease). J Hum Genet. 2008;53:947–9. [PubMed: 18704262]
- Dahlstrom JE, Arbuckle SM, Kozlowski K, Peek MJ, Thomson M, Reynolds GJ, Sillence DO. Lethal prenatal onset infantile cortical hyperostosis (Caffey disease). Pathology. 2001;33:521–5. [PubMed: 11827425]
- de Almeida JF, Kimura H, Hercowitz LH, Korkes H, Troster EJ. Cortical hyperostosis secondary to prolonged use of prostaglandin E1. Clinics (Sao Paulo). 2007;62:363–6. [PubMed: 17589681]
- de Jong G, Muller LM. Perinatal death in two sibs with infantile cortical hyperostosis (Caffey disease). Am J Med Genet. 1995;59:134–8. [PubMed: 8588573]
- Dhooge T, Syx D, Hermanns-Lê T, Hausser I, Mortier G, Zonana J, Symoens S, Byers PH, Malfait F. Caffey disease is associated with distinct arginine to cysteine substitutions in the proα1(I) chain of type I procollagen. Genet Med. 2021;23:2378-85. [PubMed: 34272483]
- Drinkwater BM, Crino JP, Garcia J, Ogburn J, Hecht JT. Recurrent severe infantile cortical hyperostosis (Caffey disease) in siblings. Prenat Diagn. 1997;17:773–6. [PubMed: 9267903]
- Gensure RC, Mäkitie O, Barclay C, Chan C, Depalma SR, Bastepe M, Abuzahra H, Couper R, Mundlos S, Sillence D, Ala Kokko L, Seidman JG, Cole WG, Jüppner H. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest. 2005;115:1250–7. [PMC free article: PMC1087158] [PubMed: 15864348]
- Hochwald O, Osiovich H. Prenatal Caffey disease. Isr Med Assoc J. 2011;13:113–4. [PubMed: 21443040]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Issa El Khoury F, Kreichati G, Kharrat K, Ghanem I. Tumoral calcinosis of the cervical spine and its association with Caffey disease in a 4-month-old boy: case report and review of the literature. J Pediatr Orthop B. 2012;21:286–91. [PubMed: 22080299]
- Kamoun-Goldrat A, le Merrer M. Infantile cortical hyperostosis (Caffey disease): a review. J Oral Maxillofac Surg. 2008;66:2145–50. [PubMed: 18848116]
- Kamoun-Goldrat A, Martinovic J, Saada J, Sonigo-Cohen P, Razavi F, Munnich A, Le Merrer M. Prenatal cortical hyperostosis with COL1A1 gene mutation. Am J Med Genet A. 2008;146A:1820–4. [PubMed: 18553566]
- Katz JM, Kirkpatrick JA, Papanicolaou N, Desai P. Case report 139. Infantile cortical hyperostosis (Caffey disease). Skeletal Radiol. 1981;6:77–80. [PubMed: 7008205]
- Kitaoka T, Miyoshi Y, Namba N, Miura K, Kubota T, Ohata Y, Fujiwara M, Takagi M, Hasegawa T, Jüppner H, Ozono K. Two Japanese familial cases of Caffey disease with and without the common COL1A1 mutation and normal bone density, and review of the literature. Eur J Pediatr. 2014;173:799–804. [PubMed: 24390061]
- Krishnamurthy S, Srinivasan S. Severe thrombocytosis as initial manifestation of Caffey disease in a 4 month old infant. Pediatr Blood Cancer. 2012;59:345–6. [PubMed: 22213629]
- Kutty N, Thomas D, George L, John TB. Caffey disease or infantile cortical hyperostosis: a case report. Oman Med J. 2010;25:134–6. [PMC free article: PMC3215490] [PubMed: 22125716]
- Langer R, Kaufmann HJ. Case report 363: infantile cortical hyperostosis (Caffey disease ICH) iliac bones, femora, tibiae and left fibula. Skeletal Radiol. 1986;15:377–82. [PubMed: 3526563]
- Lécolier B, Bercau G, Gonzalès M, Afriat R, Rambaud D, Mulliez N, de Kermadec S. Radiographic, haematological, and biochemical findings in a fetus with Caffey disease. Prenat Diagn. 1992;12:637–41. [PubMed: 1359527]
- Lee DY, Kim WJ, Kim B, Nho JH, Hong CH, Lee SM, Yoo ID, Lee C, Jung KJ. Differential diagnosis between child abuse and infantile cortical hyperostosis: a case report and literature review. Int J Environ Res Public Health. 2021;18:12269. [PMC free article: PMC8623057] [PubMed: 34832024]
- Malfait F, Symoens S, De Backer J, Hermanns-Lê T, Sakalihasan N, Lapière CM, Coucke P, De Paepe A. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28:387–95. [PubMed: 17211858]
- Makar AB, McMartin KE, Palese M, Tephly TR. Formate assay in body fluids: application in methanol poisoning. Biochem Med. 1975;13:117-26. [PubMed: 1]
- Merdler-Rabinowicz R, Grinberg A, Jacobson JM, Somekh I, Klein C, Lev A, Ihsan S, Habib A, Somech R, Simon AJ. Fetuin-A deficiency is associated with infantile cortical hyperostosis (Caffey disease). Pediatr Res. 2019;86:603-7. [PMC free article: PMC7086575] [PubMed: 31288248]
- Nemec SF, Rimoin DL, Lachman RS. Radiological aspects of prenatal-onset cortical hyperostosis. Eur J Radiol. 2012;81:e565–72. [PubMed: 21726971]
- Prior AR, Moldovan O, Azevedo A, Moniz C. Caffey disease in neonatal period: the importance of the family! BMJ Case Rep. 2012;2012:bcr2012006996. [PMC free article: PMC4543740] [PubMed: 23047998]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Ranganath P, Laine CM, Gupta D, Mäkitie O, Phadke SR. COL1A1 mutation in an Indian child with Caffey disease. Indian J Pediatr. 2011;78:877–9. [PubMed: 21249479]
- Restrepo S, Sánchez AM, Palacios E. Infantile cortical hyperostosis of the mandible. Ear Nose Throat J. 2004;83:454–5. [PubMed: 15372912]
- Savarirayan R, Cormier-Daire V, Amor DJ, Wilcox WR, Lachman RS, Rimoin DL. Prenatal cortical hyperostosis (Caffey disease). Pediatr Radiol. 2002;32:694. [PubMed: 12422848]
- Schweiger S, Chaoui R, Tennstedt C, Lehmann K, Mundlos S, Tinschert S. Antenatal onset of cortical hyperostosis (Caffey disease): case report and review. Am J Med Genet A. 2003;120A:547–52. [PubMed: 12884437]
- Suphapeetiporn K, Tongkobpetch S, Mahayosnond A, Shotelersuk V. Expanding the phenotypic spectrum of Caffey disease. Clin Genet. 2007;71:280–4. [PubMed: 17309652]
- Sweeney SM, Orgel JP, Fertala A, McAuliffe JD, Turner KR, Di Lullo GA, Chen S, Antipova O, Perumal S, Ala-Kokko L, Forlino A, Cabral WA, Barnes AM, Marini JC, San Antonio JD. Candidate cell and matrix interaction domains on the collagen fibril, the predominant protein of vertebrates. J Biol Chem. 2008;283:21187-97. [PMC free article: PMC2475701] [PubMed: 18487200]
- Tilva H, Kanjiya A, Umate R, Wanjari MB, Tivaskar S. A rare case of infantile cortical hyperostosis (ICH) of the bilateral tibia or Caffey disease. Cureus. 2023;15:e33655. [PMC free article: PMC9913865] [PubMed: 36788874]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191:1164-209. [PMC free article: PMC10081954] [PubMed: 36779427]
- Wendling D, Hafsaoui C, Laurain JM, Runge M, Magy-Bertrand N, Prati C. Dysphagia and hypervitaminosis A: cervical hyperostosis. Joint Bone Spine. 2009;76:409–11. [PubMed: 19289294]
Publication Details
Author Information and Affiliations
Department of Pediatrics
Queen's University
Kingston, Canada
Department of Pædiatrics
The Hospital for Sick Children and University of Toronto
Toronto, Canada
Department of Pædiatrics
The Hospital for Sick Children and University of Toronto
Toronto, Canada
Publication History
Initial Posting: August 2, 2012; Last Update: May 23, 2024.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Guerin A, Dupuis L, Mendoza-Londono R. Caffey Disease. 2012 Aug 2 [Updated 2024 May 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

