Molecular Pathogenesis
OTOF encodes otoferlin, a member of the FER-1 family of transmembrane proteins characterized by the presence of C2 domains [Yasunaga et al 1999]. Otoferlin contains six C2 domains (C2A-F), a transmembrane domain, and two Fer domains [Roux et al 2006, Lek et al 2010]. In the ear, otoferlin is expressed in cochlear inner hair cells. Otoferlin is a Ca2+ sensor that plays a significant role in the synaptic transmission in inner hair cells by tethering glutamatergic synaptic vesicles to the plasma membrane and triggering their fusion and pool replenishment at ribbon synapses [Strenzke et al 2016, Michalski et al 2017].
Normal hearing relies on a temporally precise and sustained glutamate release at these ribbon synapses. Defects in otoferlin affect this process by compromising exocytosis of synaptic vesicles and their reformation leading to impaired signal transmission to the auditory nerve. Consequently, individuals with biallelic pathogenic variants in OTOF have congenital severe-to-profound hearing loss characterized by normal otoacoustic emission (OAE), indicating normal cochlear function, and abnormal auditory brain stem responses (ABRs), indicating altered transmission of auditory signal from the synapse to the brain and impaired speech discrimination.
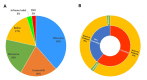
To date, more than 220 pathogenic variants have been reported in OTOF [Azaiez et al 2018]. They are distributed all over the gene and affect all domains except C2A domain. Of these variants, 38% were missense, 20% nonsense, 20% frameshift, 17% splice, 3% inframe indel, and 2% copy number variants (CNV) (see ). Truncating variants (nonsense, frameshift, splice, and CNV) in OTOF lead to absent or shortened nonfunctional protein. Missense variants impair protein folding, stability, or function. Both types of variants typically result in congenital or prelingual severe-to-profound hearing loss (see ).
Certain pathogenic variants in OTOF cause temperature-sensitive auditory neuropathy spectrum disorder (see Table 6). Missense variants cause normal to mild hearing loss when homozygous or compound heterozygous with another missense variant, but when in compound heterozygosity with a truncating variant, the hearing loss is moderate. At normal body temperature, hearing ranges from normal to a moderate loss, while an elevation in body temperature severely worsens hearing loss and speech perception. When temperature decreases, hearing returns to baseline. Studies in mouse models showed that otoferlin is sensitive to heat and this instability is exacerbated by certain pathogenic variants such as Ile515Thr. At increased temperatures, mutated otoferlin undergoes faster degradation and loss from the plasma membrane [Strenzke et al 2016].
Mechanism of disease causation.
OTOF-related deafness is caused by loss-of-function pathogenic variants.
OTOF-specific laboratory technical considerations. Variants in OTOF should be annotated on transcript NM_001287489.2, the cochlea-specific transcript.
Table 6.
Notable OTOF Pathogenic Variants
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
TS-ANSD = temperature-sensitive auditory neuropathy spectrum disorder