Clinical Description
The following information is based on case reports reviewed in Patel et al [2007], Pober et al [2009], Shaheen et al [2010], Roane et al [2012], Storm et al [2013], Dachy et al [2015], Anglani et al [2018], and Khan & Ghazi [2018].
Craniofacial features. Widely spaced eyes, depressed nasal bridge, short nose with a broad and occasionally indented tip, broad forehead, and prominent parietal frontal bossing are characteristic findings and are consistently present in individuals with DBS and confirmed biallelic LRP2 pathogenic variants. Downslanted palpebral fissures and low-set posteriorly angulated ears are very frequent findings. While some of these features may change over time, the facial gestalt remains characteristic in the few reported adults.
Ophthalmologic abnormalities. Progressive visual loss resulting in legal blindness is common and seemingly the result of severe myopia and an attendant risk for retinal detachment, although prompt treatment can improve the chances of useful vision. Retinal detachment can be unilateral or bilateral, complete or partial, and can occur in young children. Prophylactic treatment with peripheral barrier photocoagulation to prevent retinal detachment has been successful in several individuals. Progressive retinal dystrophy has been observed in a few individuals; the exact nature and rate of progression of the retinal dystrophy remain to be determined. Optic nerve hypoplasia has been reported in at least one individual. The combination of a small and recessed optic nerve head, surrounded by multiple rings of abnormal pigmentation masking the neuroretinal rim, and the presence of multiple thin vessels emanating from the optic disk was proposed as a characteristic finding on fundoscopy. Iris coloboma, frequently bilateral or affecting the right eye, is common and often present alongside hypoplastic iris stroma. Glaucoma and cataracts have only rarely been reported.
Sensorineural hearing loss. Profound bilateral sensorineural hearing loss is frequently acquired and diagnosed in early childhood. Despite a relative paucity of data, it appears that hearing can deteriorate over time, but not every individual progresses to complete loss. Some individuals have useful hearing using hearing aids and seven reported individuals have successfully received cochlear implants (see Deafness and Hereditary Hearing Loss Overview for definitions).
Renal disease. The majority of individuals with DBS present with asymptomatic low-molecular weight proteinuria (LMWP). In the adult kidney, megalin is highly expressed in proximal tubules where it plays a key role in the reuptake of several proteins by receptor-mediated endocytosis. The characteristic urinary protein profile can be detected by Coomassie staining of a random urine sample. Increased excretion of vitamin D-binding protein (VDBP) and retinol-binding protein (RBP) are consistent with the presence of DBS and may lead to hypovitaminoses in some individuals. Urinary loss of VDBP and RBP, however, are not specific to DBS and may be present in individuals with tubular dysfunction. Urinary cubilin and its ligands are detectable in urine samples of individuals with DBS. Hypercalciuria, nephrocalcinosis, nephrolithiasis, and focal segmental glomerulosclerosis have been reported. Progression to nephropathy and end-stage renal disease is a rare but life-threatening complication, especially in adults, and urinary function should be monitored in individuals with DBS. One of the first reported individuals died of renal failure at age 30 years. No renal phenotype has been convincingly documented in individuals with a heterozygous LRP2 pathogenic variant.
Developmental delay, intellectual disability, and behavioral features. Motor milestones are only slightly delayed and most children become continent. No systematic studies of intellectual functioning exist, but available data suggest that all individuals with DBS have intellectual disabilities of varying degrees ranging from mild to moderate. Formal assessment is difficult because of the severe vision and hearing deficits. Roane at al [2012] described a child age five years engaging in self-injurious behavior that was responsive to behavioral therapy; however, self-injurious behavior is not typically observed in individuals with DBS. Teenagers and young adults attend school with special provisions for hearing and vision problems. Some communicate using sign language, and can perform routine tasks and retain a variable degree of independence. An adult individual reportedly suffered from psychosis since childhood. Mood swings and depressive symptoms in this individual were compatible with her diagnosis of schizoaffective disorder. Overall, interactions with her were described as pleasant. She did not exhibit deterioration of cognitive or functional abilities beyond what was caused by her co-occurring psychiatric disorder but she did not live independently.
Diaphragmatic hernia (or eventration) and/or omphalocele (or umbilical hernia) are each reported in approximately 40% of affected individuals. The two defects infrequently occur together [Gripp et al 1997, Kantarci et al 2007, Patel et al 2007]. The absence of either or both does not exclude the diagnosis.
Seizures. A few individuals have developed tonic-clonic seizures in childhood or adolescence. In one individual, the first episode of seizures developed into status epilepticus leading to her demise. No single antiepileptic drug has been demonstrated to be the treatment of choice specifically for this disorder, and therapy should follow standard guidelines.
Growth. Relatively high birth weight (~4 kg) has been recorded in several infants. Macrocephaly is present in many individuals with DBS and it is often apparent at birth. Height and weight eventually appear to be within the normal range but a few of the older children are tall, with heights on the 90th centile.
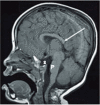
Neuroimaging. Agenesis of corpus callosum, either complete or partial, is reported in most individuals with DBS (see ). The presence of additional brain anomalies, such as periventricular nodular heterotopia and abnormalities of gyral patterns, is increasingly appreciated.
MRI in a female age three years demonstrates numerous abnormalities including hypogenesis of the corpus callosum (most notably involving the rostrum and splenium) (long white arrow), partially empty sella turcica (S), and small pons (P).
Other. Involvement of other organ systems (e.g., ventriculoseptal defect, bicornuate uterus) has been reported but is rare. Pubertal development occurs at the appropriate times.
Prognosis. Information on long-term follow up and natural history of DBS is limited to a few individuals because many affected pregnancies are interrupted or result in perinatal death secondary to congenital malformations. Major risks are due to complications of retinal detachment leading to visual loss in individuals already compromised by hearing defects, and progression of renal dysfunction. Only two adults older than age 50 years with molecularly confirmed LRP2 pathogenic variants have been reported [Stora et al 2009, Anglani et al 2018]. The most notable health concern is deteriorating renal function, resulting in renal transplantation in one individual and renal rickets in the other.