Clinical Description
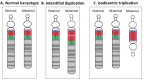
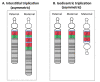
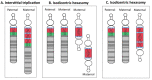
Maternal 15q duplication syndrome (maternal dup15q) is characterized by hypotonia and motor delays, intellectual disability, autism spectrum disorder (ASD), and epilepsy including infantile spasms. These clinical findings differ significantly between people with a maternal interstitial duplication and those with a maternal isodicentric supernumerary chromosome, or idic(15) (Table 2). Those with a maternal idic(15) are typically more severely affected than those with an interstitial duplication. However, severity varies even among individuals who have increased dosage by the same genetic mechanism. Some phenotypic features, such as ASD, are more consistently observed in individuals with a maternal idic(15) or large (>5-Mb) interstitial duplications that extend beyond the PWACR [Hogart et al 2010].
Table 2.
Maternal 15q Interstitial Duplication and Idic(15): Comparison of Clinical Features
View in own window
| Feature | Maternal Interstitial Duplication | Maternal Isodicentric Supernumerary Chromosome |
|---|
|
Hypotonia
| Mild to moderate | Severe |
Developmental delay /
intellectual disability
| Moderate | Severe |
|
Autism spectrum disorder
| ≥50% 1, 2 | ≥80% 1, 3 |
|
Epilepsy
| ~25% 4 | ~65% 4 |
Hypotonia and motor skills. Hypotonia in newborns and infants with maternal dup15q is associated with feeding difficulties and gross motor delays [Depienne et al 2009, Hogart et al 2010, Urraca et al 2013]. Hypotonia also contributes to gastrointestinal issues in maternal dup15q, such as constipation.
Although childhood hypotonia impairs motor development, most children achieve independent walking after age two to three years (younger in children with an interstitial duplication) [Hogart et al 2010, Piard et al 2010, Al Ageeli et al 2014].
A wide-based or ataxic gait is common [Bundey et al 1994]. Delays and persistent impairment in both fine and gross motor skills affect adaptive living skills and distinguish children with maternal dup15q from children with nonsyndromic autism spectrum disorder [DiStefano et al 2016].
Developmental delay and intellectual disability. Developmental delay in early childhood is nearly universal. This can be more specifically diagnosed as intellectual disability after age five years.
In addition to motor delays, speech and language development is particularly affected, with universal delays ranging from moderate to severe [Hogart et al 2010]. Some individuals exhibit echolalia, pronoun reversal, and stereotyped utterances, while others may lack functional speech [Battaglia et al 1997, Battaglia 2008].
Most children and adults with maternal dup15q function in the moderate-to-severe range of intellectual disability; however, there is some variability, with a higher range of cognitive abilities seen in those with an interstitial duplication [DiStefano et al 2020].
Individuals with maternal dup15q who have a diagnosis of epilepsy have lower verbal, daily living, socialization, fine motor, and gross motor skills compared to individuals with maternal dup15q who do not have epilepsy [DiStefano et al 2016, DiStefano et al 2020].
Autism spectrum disorder (ASD). Most children and adults with maternal dup15q meet criteria for ASD. In one study, 25/27 individuals with a maternal idic(15) met criteria for autism on the ADOS, while the remaining 2/27 met criteria for ASD [DiStefano et al 2020]. Among individuals with a maternal interstitial dup15q duplication, 10/12 met criteria for autism, one individual met criteria for ASD, and one individual did not meet criteria for either. Compared to other CNVs known to cause ASD, maternal dup15q confers the greatest risk (odds ratio ≥2.6) [Malhotra & Sebat 2012, Moreno-De-Luca et al 2013]. Manifestations of ASD, particularly difficulties with social interaction, may increase from early to late childhood [Simon et al 2010].
Compared to children with nonsyndromic ASD, children with maternal dup15q-ASD demonstrate a distinctive behavioral profile, including preserved responsive social smile and directed facial expressions towards others – features that may inform behavioral interventions [DiStefano et al 2016].
Epilepsy. More than half of individuals with maternal dup15q have epilepsy, usually involving multiple seizure types including infantile spasms and myoclonic, tonic-clonic, absence, and/or focal seizures [Conant et al 2014]. Seizures most often begin between ages six months and nine years [Battaglia 2008]. Incidence of epilepsy is higher in individuals with maternal dup15q resulting from an idic(15) compared to those with maternal dup15q resulting from interstitial duplication (57% and 6% respectively) [DiStefano et al 2020].
Maternal dup15q is one of the most common known causes of infantile spasms [Conant et al 2014]. Infantile spasms in individuals with maternal dup15q often progress to Lennox Gastaut syndrome and other complex seizure patterns that may be difficult to control. As many as 40% of individuals with seizures present initially with infantile spasms; of this group, approximately 90% subsequently develop other seizure types.
Intractable epilepsy in individuals with maternal dup15q may result in disabling secondary effects, including falls or developmental regression. This occurs in more than half of individuals with frequent, uncontrolled seizures or nonconvulsive status epilepticus [Battaglia et al 1997].
Children with epilepsy have been found to have lower cognitive and adaptive function than those without epilepsy [DiStefano et al 2020].
Dysmorphic features. Minor dysmorphic features often reported in individuals with maternal dup15q include flat occiput, downslanting palpebral fissures, depressed nasal bridge, short nose with an upturned nasal tip, low-set ears, long philtrum, high-arched palate, thick vermilion of the upper and lower lips, and micrognathia [Battaglia et al 1997, Borgatti et al 2001, Hogart et al 2010, Urraca et al 2013]. These features are typically subtle and may be missed in infancy.
Psychosis. Although maternal idic(15) has been reported in schizophrenia cohorts [Rees et al 2014], psychosis is not a commonly ascertained comorbidity in maternal dup15q – a finding that may reflect the difficulty of recognizing and diagnosing psychosis in individuals with low cognitive functioning and limited verbal skills. For instance, psychosis is a common comorbidity in Prader-Willi syndrome caused by uniparental disomy, which similarly involves a duplication of the maternally contributed 15q11.2-13.1 [Bassett 2011]. These individuals tend to have higher cognitive and verbal abilities than individuals with maternal dup15q. Conversely, with a high rate of ASD in individuals with maternal dup15q, psychosis related to mood disorder may be misdiagnosed as schizophrenia.
Sudden unexpected death in epilepsy (SUDEP) occurs in a small but significant minority of individuals with maternal dup15q [Friedman et al 2016, Devinsky 2011, Wegiel et al 2012]. These deaths almost always occur during sleep and most (though not all) have occurred in teenagers and young adults with epilepsy. Nonambulatory status and poor seizure control appear to be risk factors for SUDEP in individuals with maternal dup15q [Friedman et al 2016].
The mechanism underlying SUDEP is not well understood; however, available evidence suggests that in most instances a tonic-clonic seizure is followed by a shutdown of brain function and cardio-respiratory arrest. SUDEP occurs in 9% of individuals with epilepsy; the incidence of SUDEP in individuals with maternal dup15q is unknown.