Clinical Description
To date, at least 85 individuals with a clinical and/or molecular diagnosis of encephalocraniocutaneous lipomatosis (ECCL) have been reported [Moog 2009, Bennett et al 2016, McDonell et al 2018, Valera et al 2018]. Of these, 14 individuals have been identified with a pathogenic variant in FGFR1 [Bennett et al 2016, Bavle et al 2018, Valera et al 2018, Chacon-Camacho et al 2019, Córdoba et al 2019, Kordacka et al 2019] and a few with a pathogenic variant in KRAS [Boppudi et al 2016, McDonell et al 2018, Chacon-Camacho et al 2019, Nagatsuma et al 2019]. The following description of the phenotypic features in individuals with a clinical and/or molecular diagnosis associated with ECCL is based on these reports.
Note: There is evidence that oculoectodermal syndrome (OES) may constitute a clinical spectrum with ECCL, with OES on the mild end and ECCL on the more severe end of the spectrum. Individuals who do not fulfill the clinical diagnostic criteria for ECCL but display some features have sometimes been reported as having OES (see Nomenclature).
ECCL comprises a spectrum of predominantly congenital anomalies. In its typical form, ECCL is characterized by congenital skin, eye, and brain anomalies, in particular intracranial and spinal lipomas. To a much lesser degree, the bones and the heart can be affected. About 40% of affected individuals have bilateral abnormalities of the skin or the eyes. Although variable in its extent and severity, the pattern of skin and eye findings is often consistent and recognizable.
Since 2016, ECCL has also been recognized as a tumor predisposition syndrome [Bennett et al 2016].
Table 2.
Encephalocraniocutaneous Lipomatosis: Frequency of Select Features
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
|
Skin
| Nevus psiloliparus | 75% | |
| Nodular skin tags | 70%-75% | Periocular or between outer canthus & tragus |
| Alopecia | Common | A specific percentage is difficult to define. |
| Subcutaneous fatty lipomas | 40% | Frontotemporal or zygomatic |
| Focal scalp aplasia/hypoplasia | 25% | |
| Pigmentary abnormalities | Rare | Either as spots or following lines of Blaschko |
|
Eye
| Choristomas | 80%-85% | |
|
CNS
| Intracranial lipomas | 65% | Often in cerebello-pontine angle |
| Other findings | Common | See Table 3. |
|
Neurologic
| Developmental delay /
intellectual disability | 65% | |
| Seizures | 50% | |
|
Tumors
| Brain tumors | Unknown | Mostly low-grade glioma |
| Jaw tumors | Unknown | |
CNS = central nervous system
Skin. Non-scarring alopecia (with or without underlying fatty tissue) and subcutaneous fatty masses are the most typical skin anomalies.
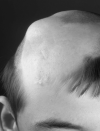
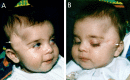
A smooth, hairless fatty tissue nevus of the scalp, the so-called nevus psiloliparus (NP) (), is the dermatologic hallmark and found in about 75% of affected individuals.
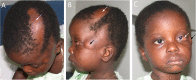
In about 40%, fatty subcutaneous masses are seen in the frontotemporal or zygomatic region (, , and ).
Rarely, subcutaneous lipomas are seen outside the craniofacial region.
Alopecia without a fatty nevus can be linear or patchy, and may follow the lines of Blaschko (, ). Some affected individuals have scarring alopecia resulting from focal skin aplasia.
Areas of focal skin aplasia are usually not extensive (up to a few cm in size) and there have been no reports of affected individuals requiring surgical treatment, such as skin graft, for focal skin aplasia.
Small nodular skin tags (which histologically are fibromas, lipomas, fibrolipomas, or choristomas) are found in about 70%-75% of affected individuals, most often on the eyelids or following a line from the outer canthus to the tragus.
Some individuals have several café au lait spots. Linear hyperpigmentation following the lines of Blaschko and (rarely) asymmetric growth may be seen [
Moog et al 2007b].
Eyes. Choristomas, with or without other eye anomalies, are seen in the majority of affected individuals (). Several affected individuals have had irregular or disrupted eyebrows.
Central nervous system (CNS) findings in 52 individuals who underwent imaging are summarized in Table 3. Note: The presence and extent of intracranial abnormalities does not correlate well with the severity of the developmental, skin, and eye findings.
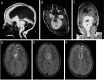
Lipomas. Intracranial lipomas () are the most common and specific CNS feature, found on neuroimaging in about 65% of affected individuals.
Almost 60% of these are located in the cerebello-pontine angle.
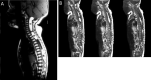
Spinal lipomas, which can extend over the whole length of the spinal cord (), are also commonly seen on spinal cord imaging.
Subcutaneous fatty masses may overlie the spinal lipomas or be found adjacent to the spinal cord ().
Vascular abnormalities. Abnormal intracranial blood vessels, such as leptomeningeal angiomatosis, thrombosed vascular malformations, and abnormal vessels, were found in nine individuals with ECCL in whom vascular imaging was performed.
While this finding could represent ascertainment bias, it is suspected that these vascular abnormalities are common, as many of the other brain abnormalities observed could result from prenatal (or, less likely, postnatal) vascular perfusion defects or hemorrhage, including asymmetric brain atrophy, porencephaly, ventriculomegaly, and calcifications [
Moog et al 2007a].
Table 3.
Encephalocraniocutaneous Lipomatosis: Frequency of Central Nervous System Anomalies in 52 Affected Individuals Who Underwent Imaging
View in own window
| Intracranial Area / Feature | Type of Anomaly | Frequency 1 | Comment |
|---|
|
Cerebral hemispheres
| Any anomaly | 43 (83%) | |
| Ventriculomegaly | 30 (57.7%) | Hydrocephalus in 9/30 (30%) |
| Asymmetric atrophy | 26 (50%) | |
| Calcifications | 24 (46.1%) | |
| Arachnoid cysts | 19 (36.5%) | |
| Porencephaly | 17 (32.7%) | |
| Cortical dysplasia | 15 (28.8%) | |
| Callosal abnormalities | 10 (19.2%) | Agenesis (partial or total) in 3/10 (30%); thin corpus callosum in 7/10 (70%) |
|
Posterior fossa
| Cerebellar hypoplasia | 6 (11.5%) | Of those w/posterior fossa abnormalities, 6/6 (100%) had cerebellar hypoplasia; of the 6, 3 also had megacisterna magna. |
|
Lipomas
| Any lipoma | 33 (63.5%) | Intracranial &/or spinal |
| Intracranial lipomas | 31 (59.6%) | In 18/31 (58%), lipomas were located in cerebello-pontine angle. |
| Spinal lipomas | 12/13 (92.3%) 2 | Not all persons underwent spinal MRI imaging. |
|
Intracranial vessels
| Abnormal or excessive vessels | 5/9 (55%) 2 | |
| Meningeal angiomatosis | 6/9 (66%) 2 | |
- 1.
Unless otherwise specified, the denominator used for calculating the percentages is 52.
- 2.
The numbers shown include only those individuals for whom information about the respective abnormality was available. Many others had no information available; thus, actual numbers and percentages are very likely higher.
Neurology. The spectrum of neurologic dysfunction is broad, ranging from normal cognitive development and no seizures to a very disabling disorder with severe intellectual disability (ID), intractable seizures, and other physical disabilities.
Among reported individuals with ECCL, about one third have normal cognitive development, one third have mild developmental delay (DD) or ID, and the final one third have severe or unspecified DD/ID.
About half of affected individuals had a history of typically infant- or childhood-onset epileptic seizures of different types that may be refractory or difficult to treat.
Musculoskeletal
Jaw tumors identified as osteomas, odontomas, or ossifying fibromas have been described in affected individuals [
Zielińska-Kaźmierska et al 2005,
Moog 2009] (see
Tumors below). These tumors are typically not malignant but can grow into the surrounding tissues, thus displacing teeth and other facial tissues.
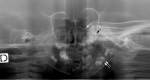
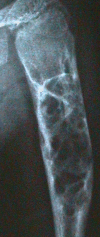
In severely affected individuals, possibly progressive, multiple lytic bone lesions affecting the long bones have been described () [
Moog et al 2007b].
Tumors (cancers). Apart from lipomas and jaw tumors, ECCL is associated with increased risk for:
Pilocytic astrocytomas have been reported in six of 14 (43%) individuals with
FGFR1-related ECCL; they have not been reported in those with
KRAS-related ECCL (see
Phenotype Correlations by Gene).
Possibly Wilms tumor
The first reported child with ECCL and Wilms tumor also had severe growth restriction, which is unusual in individuals with ECCL, such that Wilms tumor in this situation could represent a rare co-occurrence with ECCL [
Damar et al 2017].
Currently, insufficient evidence has accumulated to support a general recommendation for Wilms tumor screening in individuals with ECCL. Such screening may, however, be considered in individuals with KRAS-related ECCL in whom a pathogenic variant involving codon 12 is identified.
Note: Wilms tumor and nephroblastomatosis have been reported in two individuals with postzygotic
KRAS variants c.35G>A (p.Gly12Asp) and c.35G>T (p.Gly12Val), respectively, and phenotypes different from ECCL and oculoectodermal syndrome [
Chang et al 2021] (see
Genetically Related Disorders).
Other findings include:
Macrocephaly (in ~20% of affected individuals);
Congenital heart malformations, in particular aortic coarctation.
Oculoectodermal syndrome (OES; OMIM 600268) shares many features with ECCL, including focal alopecia or aplasia of the scalp, NP (possibly), periorbital skin tags, linear hyperpigmentation, ocular choristomas, macrocephaly, benign bone tumors, and coarctation of the aorta [Ardinger et al 2007]. Cranial MRI has been performed in about 60% of individuals with OES and may show arachnoid cysts, asymmetry of hemispheres, or mild dilatation of ventricles. Unlike ECCL, no lipomas or brain tumors (including pilocytic astrocytomas) have been reported in individuals with OES to date.
In at least seven individuals with a clinical diagnosis of OES, postzygotic activating variants in KRAS have been identified, many of which involve codon p.Ala146 [Boppudi et al 2016, McDonell et al 2018, Chacon-Camacho et al 2019]; pathogenic gain-of-function variants involving the p.Ala146 codon have also been identified in individuals who meet the clinical diagnostic criteria for ECCL. As noted above, recent data suggests that ECCL and OES are manifestations of a single spectrum (see Phenotype Correlations by Gene), as was first suggested in 2007 [Ardinger et al 2007, Moog et al 2007a]. Further research is needed to determine the phenotypes related to specific postzygotic activating pathogenic variants in KRAS.
Phenotype Correlations by Gene
Comparing FGFR1-related ECCL to KRAS-related ECCL, a gene-phenotype correlation emerges, especially when including individuals reported as having the OES phenotype who were found to have a KRAS pathogenic variant (see Nomenclature).
Compared to individuals with FGFR1-related ECCL, individuals with KRAS-related ECCL are:
Although these are preliminary considerations based on a small number of known affected individuals, the difference between FGFR1-related ECCL and KRAS-related ECCL (including those with an OES phenotype) needs to be reviewed in future, as different surveillance and management needs are likely ‒ underscoring the need for molecular confirmation of the diagnosis.