Summary
Clinical characteristics.
GLB1-related disorders comprise two phenotypically distinct lysosomal storage disorders: GM1 gangliosidosis and mucopolysaccharidosis type IVB (MPS IVB).
The phenotype of GM1 gangliosidosis constitutes a spectrum ranging from severe (infantile) to intermediate (late-infantile and juvenile) to mild (chronic/adult).
- Type I (infantile) GM1 gangliosidosis begins before age 12 months. Prenatal manifestations may include nonimmune hydrops fetalis, intrauterine growth restriction, and placental vacuolization; congenital dermal melanocytosis (Mongolian spots) may be observed. Macular cherry-red spot is detected on eye exam. Progressive central nervous system dysfunction leads to spasticity and rapid regression; blindness, deafness, decerebrate rigidity, seizures, feeding difficulties, and oral secretions are observed. Life expectancy is two to three years.
- Type II can be subdivided into the late-infantile (onset age 1-3 years) and juvenile (onset age 3-10 years) phenotypes. Central nervous system dysfunction manifests as progressive cognitive, motor, and speech decline as measured by psychometric testing. There may be mild corneal clouding, hepatosplenomegaly, and/or cardiomyopathy; the typical course is characterized by progressive neurologic decline, progressive skeletal disease in some individuals (including kyphosis and avascular necrosis of the femoral heads), and progressive feeding difficulties leading to aspiration risk.
- Type III begins in late childhood to the third decade with generalized dystonia leading to unsteady gait and speech disturbance followed by extrapyramidal signs including akinetic-rigid parkinsonism. Cardiomyopathy develops in some and skeletal involvement occurs in most. Intellectual impairment is common late in the disease with prognosis directly related to the degree of neurologic impairment.
MPS IVB is characterized by skeletal dysplasia with specific findings of axial and appendicular dysostosis multiplex, short stature (below 15th centile in adults), kyphoscoliosis, coxa/genu valga, joint laxity, platyspondyly, and odontoid hypoplasia. First signs and symptoms may be apparent at birth. Bony involvement is progressive, with more than 84% of adults requiring ambulation aids; life span does not appear to be limited. Corneal clouding is detected in some individuals and cardiac valvular disease may develop.
Diagnosis/testing.
The diagnosis of a GLB1-related disorder is established in a proband with suggestive clinical findings by identification of biallelic pathogenic (or likely pathogenic) variants in GLB1 by molecular genetic testing, or of significantly reduced activity of the beta-galactosidase enzyme activity in peripheral blood leukocytes or fibroblasts.
Management.
Treatment of manifestations: Best provided by specialists in biochemical genetics, cardiology, orthopedics, and neurology and therapists knowledgeable about GLB1-related disorders; surgery is best performed in centers with surgeons and anesthesiologists experienced in the care of individuals with lysosomal storage disorders; early and ongoing interventions to optimize comfort, mobility, educational and social outcomes. Anesthetic expertise and precautions are necessary to anticipate and manage complications relating to skeletal involvement and airway compromise.
Surveillance:
- GM1 gangliosidosis. Assess yearly: quality of life by neurologist, physiotherapist and nutritionist; seizure risk by a neurologist; cervical spine stability; and hip dislocation risk. Perform every one to three years: electrocardiogram, echocardiogram, and eye examination.
- MPS IVB. Yearly: ocular exam; assess lower extremities for malalignment, hips for dysplasia/subluxation, thoracolumbar spine for kyphosis, and cervical spine for instability; assessment by physiotherapist to optimize ambulation; perform endurance tests to evaluate functional status of the cardiovascular, pulmonary, musculoskeletal, and nervous systems. Perform electrocardiogram and echocardiogram every one to three years.
Agents/circumstances to avoid: Unplanned anesthesia management due to increased risk for complications; psychotropic medications because of the risk of worsening neurologic disease; positioning that increases aspiration risk during feedings, seizure medication dosages that result in excessive sedation, and circumstances that exacerbate fall risk for those with GM1 gangliosidosis; excessive weight gain in those with MPS IVB.
Genetic counseling.
GLB1-related disorders are inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a GLB1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the GLB1 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal and preimplantation genetic testing are possible.
GeneReview Scope
Table
GM1 gangliosidosis Type I (infantile)
Diagnosis
GLB1-related disorders comprise two phenotypically unique disorders, GM1 gangliosidosis and mucopolysaccharidosis type IVB (MPS IVB). Based on age of presentation, three types of GM1 gangliosidosis have been described: type I (infantile), type II (late infantile and juvenile) and type III (chronic/adult).
Suggestive Findings
A GLB1-related disorder is suspected in individuals with the following clinical, radiographic, neuroimaging, and laboratory findings and family history.
Clinical Findings
Type I (infantile; onset age <1 year) GM1 gangliosidosis should be suspected in infants with the following clinical findings:
- Macular cherry-red spots
- Developmental delay
- Developmental regression generally observed by age six months
- Hepatosplenomegaly
- Hypertrophic or dilated cardiomyopathy
- Coarse facial features
- Generalized skeletal dysplasia of varying severity
- Congenital dermal melanocytosis (Mongolian spots)
- Elevated serum concentrations of AST with normal ALT, and increased serum chitotriosidase activity
- Vacuolated lymphocytes and abnormally granulated eosinophils in peripheral blood smear
Type II GM1 gangliosidosis should be suspected in individuals with the following clinical findings:
- Late infantile (onset age 1-3 years)
- Developmental arrest followed by regression
- Corneal clouding (Figure 1)
- Motor abnormalities
- Progressive and diffuse atrophy on brain imaging
- Possible hepatosplenomegaly, cardiomyopathy, and/or skeletal abnormalities
- Elevated serum concentrations of AST with normal ALT, and increased serum chitotriosidase activity
- Juvenile (onset age 3-10 years)
- Ataxia and progressive impairment of gross and fine motor skills
- Progressive dysarthria
- Brain MRI findings of progressive atrophy
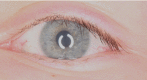
Figure 1.
Image obtained with a slit lamp demonstrating mild-to-moderate corneal clouding in an adolescent with the juvenile form of GM1 gangliosidosis Picture courtesy of Dr Wahdi Zein, National Eye Institute, National Institutes of Health, Bethesda, MD
Type III (chronic/adult) GM1 gangliosidosis should be suspected in individuals with the following clinical findings:
- Dystonia leading to gait and speech/swallowing difficulty
- Cognitive/intellectual impairment, behavioral/psychiatric disorders, short stature, and below-normal weight [Giugliani et al 2019]
- Auditory startle (rare clinical finding; may or may not be present) [Pillai et al 2018]
Mucopolysaccharidosis type IVB (MPS IVB) should be suspected in individuals with the following clinical findings:
- Corneal clouding (rare)
- Cardiac valvular disease
- Severe skeletal abnormalities
- Short stature
- Normal developmental milestones, cognitive function, and neurologic function
- Abnormal pulmonary function, including obstructive or restrictive lung disease
Radiographic Findings
GM1 gangliosidosis (Figure 2)
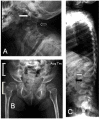
Figure 2.
Radiographs of the late-infantile form of GM1 gangliosidosis A. The odontoid process is under-ossified (white arrow). The vertebral bodies are flattened (black arrow).
- Type I. Findings observed in many (but not all) persons include vertebral beaking, kyphosis, scoliosis, hip dislocation, and osteoporosis [Jarnes Utz et al 2017].
- Type II. Odontoid hypoplasia is only observed in the late-infantile subtype, while irregularity and central indentation of endplates of vertebral bodies is only seen in the juvenile subtype. The finding of pear-shaped vertebral bodies favors a diagnosis of late-infantile disease, while squared and flat vertebral bodies is suggestive of juvenile disease. Other findings include scoliosis, anterior lumbar vertebral hypoplasia, lower ileum and fossa acetabulae hypoplasia, femoral head subluxation, coxa valga, and short femoral necks. Significantly decreased bone mineral density is typical, while bone age and tubular bones may remain unaffected [Ferreira et al 2020].
- Type III. Beaked and/or flattened vertebrae, hip dysplasia, osteopenia, J-shaped sella turcica, scoliosis, kyphosis, pectus carinatum, and odontoid hypoplasia may be observed in some affected persons [Giugliani et al 2019].
MPS IVB. See Note. Findings on skeletal survey that suggest the diagnosis of MPS IVB include the following:
- Odontoid hypoplasia with subsequent risk for cervical instability
- Kyphosis (curving of the spine that causes a bowing or rounding of the back, which leads to a hunchback or slouching posture)
- Gibbus (structural kyphosis due to wedging of one or more adjacent vertebrae)
- Scoliosis
- Pectus carinatum or excavatum
Note: (1) Based on wide variations and subtleties of the radiographic findings in MPS IV, multiple body regions should be evaluated. (2) While the radiographic findings in MPS IVA (caused by biallelic GALNS pathogenic variants) and MPS IVB are extensive and can be diagnostic, they cannot distinguish MPS IVA from MPS IVB. (See Mucopolysaccharidosis Type IVA for a detailed discussion of the radiographic findings.)
Neuroimaging Findings
GM1 gangliosidosis. Brain MRI can show the following:
- Type I. Increase in ventricular and total brain tissue volume, atrophy of cerebellar white matter, and atrophy of the caudate, putamen, corpus callosum, and basal ganglia [Nestrasil et al 2018]
- Type II. Decrease in total brain tissue volume with increase in ventricular volume, diffuse hypomyelination and white matter abnormalities, atrophy of caudate, putamen, corpus callosum, and basal ganglia, and T2-weighted hypointensity in the basal ganglia/globus pallidus that is not specific (Figure 3) [Deodato et al 2017, Feng et al 2018, Lee et al 2018, Nestrasil et al 2018, King et al 2020, Uchino et al 2020]
- Type III. General brain atrophy, subcortical white matter changes, ventriculomegaly, atrophy of the basal ganglia, hypomyelination, hyperintensity in the putamen, mild cerebral atrophy, and/or a "wish bone" pattern of iron accumulation [Pillai et al 2018, Giugliani et al 2019]
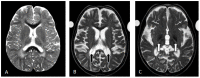
Figure 3.
Brain MRI findings in a child with the late-infantile form of GM1 gangliosidosis A. At age 3 years 6 months: T2-weighted axial view showing minimal atrophy
Laboratory Findings
GM1 gangliosidosis. A specific glycosaminoglycan pattern in urine noted in persons with GM1 gangliosidosis raises suspicion – but is not diagnostic – for this disorder. A characteristic urinary oligosaccharide profile can also be detected in urine and dried urine spots via UHPLC-MS/MS analysis [Semeraro et al 2021].
Elevated glycan biomarkers may be detectable via MS/MS in dried blood spots of newborns with GM1 gangliosidosis [Su et al 2021].
MPS IVB. Excretion of keratan sulfate in the urine can be diagnostic of MPS IV; however, the presence of keratan sulfate in the urine does not distinguish MPS IVA from MPS IVB; thus, additional studies are warranted (see Establishing the Diagnosis).
Note: A glycosaminoglycan screen can be falsely negative; thus, negative results should not deter the clinician from additional testing in an individual with a suspected diagnosis of a GLB1-related disorder on the basis of clinical and imaging findings.
Family History
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of a GLB1-related disorder is established in a proband with suggestive clinical findings by identification of (a) biallelic pathogenic (or likely pathogenic) variants in GLB1 by molecular genetic testing (see Table 1) or (b) significantly reduced activity of the beta-galactosidase enzyme activity in peripheral blood leukocytes or fibroblasts. Although only one method is required to establish the diagnosis, the other may be used in tandem to confirm the diagnosis or in some cases, to identify a genotype-phenotype correlation.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include any likely pathogenic variants. (2) Identification of biallelic GLB1 variants of uncertain significance (or of one known GLB1 pathogenic variant and one GLB1 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other lysosomal storage disorders are more likely to be diagnosed using comprehensive genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of GLB1 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
A lysosomal storage disorder multigene panel that includes GLB1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in GLB1-Related Disorders
Beta-Galactosidase Enzyme Analysis
The definitive diagnosis of a GLB1-related disorder can also be made by measuring beta-galactosidase enzyme activity in peripheral blood leukocytes or fibroblasts.
The diagnosis of MPS IVB can be confirmed by the combination of keratan sulfate in the urine and decreased enzyme activity for beta-galactosidase enzyme activity in peripheral blood leukocytes or fibroblasts in the absence of intellectual disability.
Table 2.
Beta-Galactosidase Enzyme Activity in GLB1-Related Disorders by Phenotype
Clinical Characteristics
Clinical Description
GLB1-related disorders comprise two phenotypically distinct disorders: GM1 gangliosidosis and mucopolysaccharidosis type IVB (MPS IVB).
To date, more than 200 individuals have been identified with GM1 gangliosidosis [Suzuki et al 2014, Regier et al 2016, Jarnes Utz et al 2017] and approximately 60 individuals have been described with MPS IVB [Bleier et al 2018, Abumansour et al 2019]. The following description of the phenotypic features associated with each GLB1-related disorder is based on these reports.
Table 3.
Clinical, Skeletal, Neuroimaging, and Biochemical Findings in GLB1-Related Disorders
GM1 Gangliosidosis
The phenotype of GM1 gangliosidosis constitutes a spectrum ranging from severe (infantile) to intermediate (late infantile and juvenile) to mild (chronic/adult). While classification into these types is arbitrary, it is helpful in understanding the variation observed in the timing of disease onset, symptoms, rate of progression, and longevity.
Type I (infantile) GM1 gangliosidosis
- Onset of symptoms is before age 12 months. In some infants, prenatal manifestations include nonimmune hydrops fetalis (6%), intrauterine growth restriction (1%), and placental vacuolization [Brunetti-Pierri & Scaglia 2008, Jarnes Utz et al 2017, Lang et al 2020]. Congenital dermal melanocytosis (Mongolian spots) may be observed [Mishra et al 2021].
- Laboratory findings can include elevated serum concentrations of AST with normal ALT, and increased serum chitotriosidase activity [Arash-Kaps et al 2019] and vacuolated lymphocytes and abnormally granulated eosinophils in the peripheral blood smear [Lynch & Czuchlewski 2016].
- Eye findings. Macular cherry-red spot is detected.
- Central nervous system dysfunction manifests as:
- Early developmental delay with hypotonia;
- Exaggerated startle response, followed by spasticity and rapid regression;
- Severe central nervous system dysfunction by the end of the first year leading to blindness and deafness; decerebrate rigidity; seizures; and poor feeding, difficulties in swallowing, excess secretions, and risk of aspiration.
- Hepatosplenomegaly occurs in high prevalence (85%) and is typically observed within the first 7.5-10 months of life, but may not always be present at the time of diagnosis [Lang et al 2020].
- Cardiac involvement manifests as hypertrophic or dilated cardiomyopathy, cardiomegaly, and/or hypertension [Jarnes Utz et al 2017, Lang et al 2020].
- Coarsened facial appearance may include frontal bossing, depressed nasal bridge with a broad nasal tip, long philtrum, large low-set ears, gingival hypertrophy with macroglossia, coarse thickened skin, and hirsutism.
- Skeletal dysplasia can be seen at the time of diagnosis and may result in scoliosis, kyphosis, and restrictive lung disease.
- Prognosis. Progression is rapid with death by age two to three years, frequently secondary to aspiration pneumonia.
Type II (late-infantile and juvenile) GM1 gangliosidosis
- Onset of the late-infantile form is typically between ages one and three years with life expectancy until ages five to ten years; onset of the juvenile form is typically between ages three and ten years with life expectancy well into the second decade. Laboratory findings can include elevated serum concentrations of AST with normal ALT, and increased serum chitotriosidase activity [Lee at al 2018, Arash-Kaps et al 2019].
- Eye findings. Mild corneal clouding
- Central nervous system dysfunction manifests as progressive cognitive, motor, and speech decline, as measured by the Vineland Adaptive Behavior Scale and other age-appropriate psychometric testing.
- Hepatosplenomegaly and/or cardiac involvement (manifesting as dilated or hypertrophic cardiomyopathy) is variably present [Arash-Kaps et al 2019].
- Coarsened facial appearance is not observed in this subtype of GM1 gangliosidosis.
- Skeletal dysplasia may or may not be present.
- Prognosis. The typical course is characterized by progressive neurologic decline, progressive skeletal disease in some individuals (including kyphosis and avascular necrosis of the femoral heads), and progressive feeding difficulties leading to aspiration risk. Aspiration pneumonia and resulting respiratory complications may necessitate artificial respiratory support [Uchino et al 2020]. Life expectancy of the late-infantile subtype is up to age 9-10 years, while individuals with the juvenile form may survive until early adulthood.
Type III (chronic/adult) GM1 gangliosidosis
- Onset of symptoms is between age ten and 30 years.
- Eye findings. Cloudy corneas may or may not be present.
- Central nervous system dysfunction
- Typically presents with generalized dystonia leading to unsteady gait and speech disturbance
- Shortly thereafter, most will develop extrapyramidal signs including akinetic-rigid parkinsonism.
- Intellectual impairment is common in late stages of the disease.
- Behavioral/psychiatric disorders may occur.
- Cardiac involvement in the form of cardiomyopathy develops in some individuals.
- Hepatosplenomegaly and coarse facial features are not typically observed in this subtype of GM1 gangliosidosis [Giugliani et al 2019].
- Skeletal abnormalities, found in 95% of individuals, are most commonly short stature, kyphosis, hip dysplasia, and scoliosis of varying severity.
- Prognosis is directly related to the degree of neurologic impairment. Most affected individuals have a shorter life span than their unaffected relatives [Suzuki et al 2014].
Mucopolysaccharidosis Type IVB
Onset of first signs and symptoms occurs as early as birth.
Eye findings. Corneal clouding is detected in some individuals.
Central nervous system dysfunction, hepatosplenomegaly and coarsened facial appearance are not typically present in this disorder.
Cardiac involvement manifests as valvular disease. Although previously considered a frequent finding, a study of 60 individuals with MPS IVB showed infrequency of cardiac valve pathology and no reports of cardiomyopathy [Abumansour et al 2019].
Skeletal dysplasia is the main feature of this disorder with specific findings of axial and appendicular dysostosis multiplex [Abumansour et al 2019], short stature (below 15th centile in adults) [Bleier et al 2018], kyphoscoliosis, coxa/genu valga, joint laxity, platyspondyly, and odontoid hypoplasia. Structural abnormalities of the chest may cause restrictive lung disease. Spinal cord compression is a common complication of spinal dysostosis.
Dental findings including small and widely spaced teeth, caries, and tooth enamel abnormalities occur in a minority of individuals.
Auditory findings. Although not well documented in MPS IVB, sensorineural and/or conductive hearing loss is common in MPS IVA and may occur in MPS IVB through a similar mechanism.
Prognosis. Neurologic symptoms such as pain, bladder incontinence, and spasticity may occur secondary to spinal cord compression in some individuals, with a reported frequency of approximately 10%. Bony involvement is progressive, with more than 84% of adults requiring ambulation aids [Bleier et al 2018]. Based on current data, life span is not limited by this condition. More than two thirds of participants in a survey of the natural history of MPS IVB were between ages 30 and 64 years [Bleier et al 2018].
Genotype-Phenotype Correlations
Due to extensive molecular heterogeneity, no clear genotype-phenotype correlations have been identified in GM1 gangliosidosis. Generally, pathogenic variants that affect the surface of the beta-galactosidase enzyme tend to result in a less severe phenotype and are associated with type III (adult/chronic) GM1 gangliosidosis, while pathogenic variants that affect the protein core or enzyme active site are associated with the more severe type I (infantile) form of the disease [Ohto et al 2012, Feng et al 2018, Arash-Kaps et al 2019].
MPS IVB is associated with pathogenic variants of GLB1 that impair the catalytic degradation of keratan sulfate bound oligosaccharides while GM1 gangliosidosis is associated with pathogenic variants that impair the degradation of gangliosides.
However, some pathogenic variants are associated with both MPS IVB and GM1 gangliosidosis. In the homozygous state, a pathogenic variant typically associated with MPS IVB, when present in compound heterozygosity with another GLB1 pathogenic variant, may manifest as type II or type III GM1 gangliosidosis or as a blended MPSIVB/GM1 gangliosidosis phenotype, including dysostosis as well as neurodegenerative manifestations.
Nomenclature
In the past GM1 gangliosidosis was referred to as beta-galactosidase-1 deficiency or beta-galactosidosis; mucopolysaccharidosis type IVB was referred to as Morquio syndrome type B. These terms should be used when searching for older literature on GM1 gangliosidosis.
Prevalence
GM1 gangliosidosis of all types is estimated to occur in one in 100,000 to 300,000 [Suzuki et al 2014]. The most common is the infantile form. The prevalence in Brazil (1:17,000), in persons of Roma ancestry (1:10,000), and in the Maltese Islands (1:3,700) is much higher than in other areas and likely represents founder effects (reviewed in Brunetti-Pierri & Scaglia [2008]).
The prevalence of chronic/adult GM1 gangliosidosis is higher in the Japanese population, likely due both to a founder effect and possibly a greater awareness of the disorder among Japanese healthcare providers [Higaki et al 2011].
MPS IVB. Before 1980, MPS IVA and IVB were indistinguishable. The overall prevalence of MPS IV was reported as 1:75,000 to 1:640,000 (reviewed in Ohto et al [2012]). Subsequently the prevalence of MPS IVB has been reported as 1:250,000-1:1,000,000 (reviewed in Bleier et al [2018]).
Genetically Related (Allelic) Disorders
No phenotypes other than those described in this GeneReview are known to be associated with germline pathogenic variants in GLB1.
Differential Diagnosis
GM1 Gangliosidosis
Type I (infantile) GM1 gangliosidosis. See Table 4.
Table 4.
Genetic Disorders of Interest in the Differential Diagnosis of Type I (Infantile) GM1 Gangliosidosis
Type II (late-infantile and juvenile) GM1 gangliosidosis. See Table 5.
Table 5.
Genetic Disorders of Interest in the Differential Diagnosis of Type II (Late-Infantile and Juvenile) GM1 Gangliosidosis
Some spinocerebellar ataxia syndromes (e.g., ataxia caused by mutation of FGF14, MTCL1, or TXN2 or SCA7 with extreme anticipation) may be associated with early onset and can be considered in the differential diagnosis of type II (late-infantile and juvenile) GM1 gangliosidosis (see Hereditary Ataxia Overview).
Type III (chronic/adult) GM1 gangliosidosis. See Table 6.
Table 6.
Genetic Disorders of Interest in the Differential Diagnosis of Type III (Chronic/Adult) GM1 Gangliosidosis
Note: A positive test for anti-GM1 ganglioside antibodies is not indicative of a diagnosis of GM1 gangliosidosis. These test results are associated with multifocal motor neuropathy or Guillain-Barré syndrome [Hasan et al 2021].
Mucopolysaccharidosis Type IVB (MPS IVB)
Mucopolysaccharidosis IVA (MPS IVA) and MPS IVB are clinically indistinguishable. Of individuals with the MPS IV phenotype, MPS IVA accounts for more than 95% of affected individuals and MPS IVB accounts for fewer than 5% of affected individuals. The diagnosis of MPS IVA is confirmed by detection either of deficient N-acetylgalactosamine 6-sulfatase (GALNS) enzyme activity or of biallelic GALNS pathogenic variants.
Management
No clinical practice guidelines for GLB1-related disorders (GM1 gangliosidosis and MPS IVB) have been published.
It is important to note that all GLB1-related disorders may be associated with anesthetic risks.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with a GLB1-related disorder, the evaluations summarized in Tables 7, 8, 9, and 10 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 7.
Recommended Evaluations Following Initial Diagnosis in Individuals with Type I (Infantile) GM1 Gangliosidosis
Table 8.
Recommended Evaluations Following Initial Diagnosis in Individuals with Type II (Late-Infantile and Juvenile) GM1 Gangliosidosis
Table 9.
Recommended Evaluations Following Initial Diagnosis in Individuals with Type III (Chronic/Adult) GM1 Gangliosidosis
Table 10.
Recommended Evaluations Following Initial Diagnosis in Individuals with MPS IVB
Treatment of Manifestations
Treatment and quality of life can be optimized when care is provided by specialists in biochemical genetics, cardiology, orthopedics, and neurology and therapists knowledgeable about GLB1-related disorders.
- Surgery is best performed in centers with surgeons and anesthesiologists experienced in the care of individuals with lysosomal storage disorders.
- Early and ongoing interventions to optimize comfort, mobility, educational and social outcomes are recommended.
Table 11.
Treatment of Manifestations in Individuals with Type I (Infantile) GM1 Gangliosidosis
Table 12.
Treatment of Manifestations in Individuals with Type II (Late-Infantile and Juvenile) GM1 Gangliosidosis
Table 13.
Treatment of Manifestations in Individuals with Type III (Adult/Chronic) GM1 Gangliosidosis
MPS IVB. Since MPS IVA and IVB are clinically indistinguishable; details of interventions are based on those recommended for MPS in general or specifically for MPS IVA.
Table 14.
Treatment of Manifestations in Individuals with MPS IVB
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
- An estate plan should be carefully drafted with the support of a special needs estate attorney, to ensure that the individual with special needs remains qualified for government and state benefits. The plan must include a special needs trust; non-profit organizations may be helpful by serving as the trustee and/or managing the trust [Carney 2021].
- Eligibility and plans for guardianship will be established at age 18. Special needs attorneys and social workers may be helpful in navigating guardianship decisions. Depending on the individual's cognitive status and decision-making abilities, the family may elect guardianship, a durable power of attorney for medical purposes, and/or supported decision making. If the affected individual has sibs, a simple will should be drafted at this time to protect the child against disqualification for government benefits [Carney 2021].
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Surveillance
Table 15.
Recommended Surveillance for Individuals with GM1 Gangliosidosis
Table 16.
Recommended Surveillance for Individuals with MPS IVB
Agents/Circumstances to Avoid
The following should be avoided:
- Unplanned anesthesia management. Because children with MPS IVB and those with GM1 gangliosidosis with skeletal involvement (spine anomalies, short neck, large head, and risk for atlantoaxial instability due to odontoid hypoplasia) are at increased risk for complications of anesthesia [Walker et al 2013] anesthesia is best done with advanced planning by experienced specialists whenever possible.
- Psychotropic medications. Because psychotropic medications have been associated with worsening neurologic disease in adults with Tay-Sachs disease (which is caused by deficiency of the second enzyme in the beta-galactosidase pathway) [Shapiro et al 2006], use of these medications in individuals with a GLB1-related disorder should be avoided whenever possible.
- For individuals with type I (infantile) GM1 gangliosidosis
- Positioning that increases aspiration risk during feedings
- Seizure medication dosages that result in excessive sedation
- For individuals with types II and III (juvenile and adult) GM1 gangliosidosis. Circumstances that exacerbate fall risk
- For persons with MPS IVB. Excessive weight gain, which causes undue stress on the axial skeleton and may decrease the ability to ambulate independently. It is important that nutrition optimize growth while maintaining a lean habitus.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
There are currently several therapies under investigation that offer some promise in altering the outcome for individuals with GM1 gangliosidosis:
- Intracisternal injection of AAVrh.10 carrying GLB1 for the treatment of type I (infantile) GM1 gangliosidosis (NCT04273269)
- Intra-cisterna magna injection of AAVhu68 carrying GLB1 for the treatment of types I and II (infantile and late-infantile) GM1 gangliosidosis (NCT04713475)
- Intravenous administration of AAV9 carrying GLB1 for the treatment of type I and type II GM1 gangliosidosis (NCT03952637)
- "Syner-G" regimen (combination of miglustat and the ketogenic diet) for improved outcomes in individuals with types I and II (infantile and juvenile) GM1 gangliosidosis (NCT02030015)
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
GLB1-related disorders (GM1 gangliosidosis and mucopolysaccharidosis type IVB) are inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one GLB1 pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a GLB1 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a GLB1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Sibs who inherit biallelic GLB1 pathogenic variants may exhibit variability in both the severity of disease manifestations and age of onset.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband
- Individuals with a severe GLB1-related disorder do not reproduce.
- The offspring of an individual with a mild GLB1-related disorder are obligate heterozygotes (carriers) for a pathogenic variant in GLB1.Note: No studies addressing fertility in individuals with a mild GLB1-related disorder have been published.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a GLB1 pathogenic variant.
Carrier Detection
Molecular genetic testing. Carrier testing for at-risk relatives requires prior identification of the GLB1 pathogenic variants in the family.
Biochemical testing. Enzyme activity may not be predictive of carrier status in family members of individuals with a GLB1-related disorder.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the GLB1 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Cure GM1 FoundationPO Box 6890Albany 94706Phone: 510-560-6164Email: [email protected]
- National Tay-Sachs and Allied Diseases Association, Inc. (NTSAD)Phone: 617-277-4463Email: [email protected]
- Canadian Society for Mucopolysaccharide and Related DiseasesCanadaPhone: 800-667-1846Email: [email protected]
- MedlinePlus
- MedlinePlus
- Metabolic Support UKUnited KingdomPhone: 0845 241 2173
- MPS SocietyUnited KingdomPhone: 0345 389 9901Email: [email protected]
- National MPS SocietyPhone: 877-MPS-1001
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
GLB1-Related Disorders: Genes and Databases
Table B.
OMIM Entries for GLB1-Related Disorders (View All in OMIM)
Molecular Pathogenesis
GLB1-related disorders comprise two phenotypically distinct disorders: GM1 gangliosidosis with varying degrees of neurologic involvement and mucopolysaccharidosis type IVB (MPS IVB), a progressive skeletal dysplasia.
Both disorders are caused by pathogenic variants in GLB1 leading to decreased activity of beta-galactosidase, a lysosomal enzyme involved in the metabolism of the sphingolipid, GM1 ganglioside, and the glycosaminoglycan, keratan sulfate.
GM1 gangliosidosis is caused by pathogenic variants that lead to accumulation of sphingolipid intermediates in the lysosome and, thus, interfere with appropriate functioning of the organelle. A hallmark of GM1 gangliosidosis is degeneration of the CNS, where ganglioside synthesis is the highest. An inverse ratio of enzyme activity and substrate storage has been observed, with the lowest amounts of enzyme activity and highest amounts of storage material noted in neural tissue from individuals with the most severe form: infantile GM1 gangliosidosis [Brunetti-Pierri & Scaglia 2008].
MPS IVB is caused by pathogenic variants that impair the catabolism of keratan sulfate and have little effect on GM1 ganglioside accumulation. Keratan sulfate accumulation is thought to be the cause of severe skeletal abnormalities.
The mechanism by which GM1 ganglioside or keratan sulfate primarily accumulates has been explored but not yet established. It is hypothesized that the protein structure coded by GLB1 has specific binding sites for the degradation of keratan sulfate and GM1 ganglioside. For example, genetic changes that affect the binding site for keratan sulfate (e.g., Trp273Leu) lead to the MPS IVB presentation [Yuskiv et al 2020].
Mechanism of disease causation. GLB1-related disorders occur via a loss-of-function mechanism.
Table 17.
Notable GLB1 Pathogenic Variants
Chapter Notes
Author Notes
The Medical Genetics Branch of the National Human Genome Research Institute continues to study the natural history of patients with GM1. Careful phenotyping and advanced imaging facilitate the characterization of disease progression necessary to evaluate the efficacy of therapeutic interventions. The laboratory is also engaged in studying biomarkers of disease progression in clinical samples, particularly CSF. Careful, repeated observations especially in later-onset patients have identified significantly decreased bone density and an increased incidence of odontoid hypoplasia, particularly in juvenile patients [Author, unpublished observations]. These findings may impact surgical decision making and the activities of daily living. A Phase I/II clinical trial investigating the safety and efficacy of intravenous administration of AAV9-GLB1 vector into individuals with type I and type II GM1 gangliosidosis, sponsored by the National Human Genome Research Institute, is currently underway.
Acknowledgments
Precilla D'Souza, PNP, DNP and Jean Johnston, BA, MS are gratefully acknowledged for providing excellent care for patients and their families.
Revision History
- 22 April 2021 (ha) Comprehensive update posted live
- 29 August 2019 (aa) Revision: Clinical Characteristics [Regier et al 2016] and Management (miglustat) [Deodato et al 2017, Jarnes Utz et al 2017]
- 17 October 2013 (me) Review posted live
- 24 January 2013 (ct) Original submission
References
Literature Cited
- Abumansour IS, Yuskiv N, Paschke E, Stockler-Ipsiroglu S. Morquio-B disease: Clinical and genetic characteristics of a distinct GLB1-related dysostosis multiplex. JIMD Rep. 2019;51:30-44. [PMC free article: PMC7012745] [PubMed: 32071837]
- Akyol MU, Alden TD, Amartino H, Ashworth J, Belani K, Berger KI, Borgo A, Braunlin E, Eto Y, Gold JI, Jester A, Jones SA, Karsli C, Mackenzie W, Marinho DR, McFadyen A, McGill J, Mitchell JJ, Muenzer J, Okuyama T, Orchard PJ, Stevens B, Thomas S, Walker R, Wynn R, Giugliani R, Harmatz P, Hendriksz C, Scarpa M, et al. Recommendations for the management of MPS IVA: systematic evidence- and consensus-based guidance. Orphanet J Rare Dis. 2019;14:137. [PMC free article: PMC6567385] [PubMed: 31196221]
- Arash-Kaps L, Komlosi K, Seegräber M, Diederich S, Paschke E, Amraoui Y, Beblo S, Dieckmann A, Smitka M, Hennermann JB. The clinical and molecular spectrum of GM1 gangliosidosis. J Pediatr. 2019;215:152-157.e3. [PubMed: 31761138]
- Baiotto C, Sperb F, Matte U, da Silva CD, Sano R, Coelho JC, Giugliani R. Population analysis of the GLB1 gene in South Brazil. Genet Mol Biol. 2011;34:45-8. [PMC free article: PMC3085372] [PubMed: 21637542]
- Bleier M, Yuskiv N, Priest T, Popurs MAM, Stockler-Ipsiroglu S. Morquio B patient/caregiver survey: First insight into the natural course of a rare GLB1 related condition. Mol Genet Metab Rep. 2018;16:57-63. [PMC free article: PMC6072644] [PubMed: 30094186]
- Brunetti-Pierri N, Scaglia F. GM1 gangliosidosis: review of clinical, molecular, and therapeutic aspects. Mol Genet Metab. 2008;94:391-6. [PubMed: 18524657]
- Carney MT. Planning for the future of a pediatric kidney transplant recipient with special needs: A parent's perspective. Pediatr Transplant. 2021;25:e13906. [PubMed: 33152150]
- Deodato F, Procopio E, Rampazzo A, Taurisano R, Donati MA, Dionisi-Vici C, Caciotti A, Morrone A, Scarpa M. The treatment of juvenile/adult GM1-gangliosidosis with Miglustat may reverse disease progression. Metab Brain Dis. 2017;32:1529-36. [PubMed: 28577204]
- Feng Y, Huang Y, Zhao X, Sheng H, Feng Y, Zhang W, Liu L. Clinical and molecular characteristics of 11 Chinese probands with GM1 gangliosidosis. Metab Brain Dis. 2018;33:2051-7. [PubMed: 30267299]
- Ferreira CR, Regier DS, Yoon R, Pan KS, Johnston JM, Yang S, Spranger JW, Tifft CJ. The skeletal phenotype of intermediate GM1 gangliosidosis: clinical, radiographic and densitometric features, and implications for clinical monitoring and intervention. Bone. 2020;131:115142. [PMC free article: PMC6937522] [PubMed: 31704340]
- Giugliani L, Steiner CE, Kim CA Lourenço CM, Santos MLSF, de Souza CFM, Brusius-Facchin AC, Baldo G, Riegel M, Giugliani R. Clinical findings in Brazilian patients with adult GM1 gangliosidosis. JIMD Rep. 2019;49:96-106. [PMC free article: PMC6718113] [PubMed: 31497487]
- Hasan I, Papri N, Hayat S, Jahan I, Ara G, Islam B, Islam Z. Clinical and serological prognostic factors in childhood Guillain‐Barré syndrome: a prospective cohort study in Bangladesh. J Peripher Nerv Syst. 2021:26:83-9. [PubMed: 33555098]
- Higaki K, Li L, Bahrudin U, Okuzawa S, Takamuram A, Yamamoto K, Adachi K, Paraguison RC, Takai T, Ikehata H, Tominaga L, Hisatome I, Iida M, Ogawa S, Matsuda J, Ninomiya H, Sakakibara Y, Ohno K, Suzuki Y, Nanba E. Chemical chaperone therapy: chaperone effect on mutant enzyme and cellular pathophysiology in β-galactosidase deficiency. Hum Mutat. 2011;32:843–52. [PubMed: 21520340]
- Jarnes Utz JR, Kim JR, Kim S, King K, Ziegler R, Schema L, Redtree ES, Whitley CB. Infantile gangliosidoses: Mapping a timeline. Mol Genet Metab. 2017;121:170-9. [PMC free article: PMC5727905] [PubMed: 28476546]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549: 519-22. [PubMed: 28959963]
- King KE, Kim S, Whitley CB, Jarnes-Utz JR. The juvenile gangliosidoses: a timeline of clinical change. Mol Genet Metab Rep. 2020;25:100676. [PMC free article: PMC7674119] [PubMed: 33240792]
- Lang FM, Korner P, Harnett M, Karunakara A, Tifft CJ. The natural history of Type 1 infantile GM1 gangliosidosis: a literature-based meta-analysis. Mol Genet Metab. 2020;129:228-35. [PMC free article: PMC7093236] [PubMed: 31937438]
- Lee JS, Choi J-M, Lee M, Kim SY, Lee S, Lim BC, Cheon JE, Kim IO, Kim KJ, Choi M, Seong MW, Chae JH. Diagnostic challenge for the rare lysosomal storage disease: late infantile GM1 gangliosidosis. Brain Dev. 2018;40:383-90. [PubMed: 29439846]
- Lynch DT, Czuchlewski DR. Peripheral blood findings in GM1 gangliosidosis. Blood. 2016;127:2161. [PubMed: 28092877]
- Mishra S, Pai P, Uttarilli A, Girisha KM. Mongolian spots in GM1 gangliosidosis: a pictorial report. Clin Dysmorphol. 2021;30:6-9. [PubMed: 33038107]
- Nestrasil I, Ahmed A, Utz JM, Rudser K, Whitley CB, Jarnes-Utz JR. Distinct progression patterns of brain disease in infantile and juvenile gangliosidoses: volumetric quantitative MRI study. Mol Genet Metab. 2018;123:97-104. [PMC free article: PMC5832355] [PubMed: 29352662]
- Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T. Crystal structure of human β-galactosidase: structural basis of GM1 gangliosidosis and morquio B diseases. J Biol Chem. 2012;287:1801-12. [PMC free article: PMC3265862] [PubMed: 22128166]
- Pillai SH, Sundaram S, Zafer SM, Rajan R. Expanding the phenotypic spectrum of type III GM1 gangliosidosis: progressive dystonia with auditory startle. Neurol India. 2018;66:S149-S150. [PubMed: 29503341]
- Regier DS, Kwon HJ, Johnston J, Golas G, Yang S, Wiggs E, Latour Y, Thomas S, Portner C, Adams D, Vezina G, Baker EH, Tifft CJG. MRI/MRS as surrogage marker for clinical progression in GM1 gangliosidosis. Am J Med Genet A. 2016;170:634–44. [PubMed: 26646981]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Semeraro M, Sacchetti E, Deodato F, Coşkun T, Lay I, Catesini G, Olivieri G, Rizzo C, Boenzi S, Dionisi-Vici C. A new UHPLC-MS/MS method for the screening of urinary oligosaccharides expands the detection of storage disorders. Orphanet J Rare Dis. 2021;16:24. [PMC free article: PMC7796585] [PubMed: 33422100]
- Shapiro BE, Hatters-Friedman S, Fernandes-Filho JA, Anthony K, Natowicz MR. Late-onset Tay-Sachs disease: adverse effects of medications and implications for treatment. Neurology. 2006;67:875-7. [PubMed: 16966555]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Su P, Khaledi H, Waggoner C, Gelb MH. Detection of GM1-gangliosidosis in newborn dried blood spots by enzyme activity and biomarker assays using tandem mass spectrometry. J Inherit Metab Dis. 2021;44:264-71. [PubMed: 32506457]
- Sun YM, Lu C, Wu ZY. Spinocerebellar ataxia: relationship between phenotype and genotype - a review. Clin Genet. 2016;90:305–14. [PubMed: 27220866]
- Suzuki Y, Nanba E, Matsuda J, Higaki K, Oshima A. β-galactosidase deficiency (β-galactosidosis): GM1 gangliosidosis and Morquio B disease. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G, eds. The Online Metabolic and Molecular Bases of Inherited Disease (OMMBID). Chap 151. New York, NY: McGraw-Hill. 2014.
- Uchino A, Nagai M, Kanazawa N, Ichinoe M, Yanagisawa N, Adachi K, Nanba E, Ishiura H, Mitsui J, Tsuji S, Suzuki K, Murayama S, Nishiyama K. An autopsy case of GM1 gangliosidosis type II in a patient who survived a long duration with artificial respiratory support. Neuropathology. 2020;40:379-88. [PubMed: 32219895]
- Walker R, Belani KG, Braunlin EA, Bruce IA, Hack H, Harmatz PR, Jones S, Rowe R, Solanki GA, Valdemarsson B. Anaesthesia and airway management in mucopolysaccharidosis. J Inherit Metab Dis. 2013;36:211-9. [PMC free article: PMC3590422] [PubMed: 23197104]
- Yuskiv N, Higaki K, Stockler-Ipsiroglu S. Morquio B disease. Disease characteristics and treatment options of a distinct GLB1-related dysostosis multiplex. Int J Mol Sci. 2020;21:9121. [PMC free article: PMC7729736] [PubMed: 33266180]
Publication Details
Author Information and Affiliations
Children's National Hospital
Washington, DC
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
Publication History
Initial Posting: October 17, 2013; Last Update: April 22, 2021.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Regier DS, Tifft CJ, Rothermel CE. GLB1-Related Disorders. 2013 Oct 17 [Updated 2021 Apr 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


