Summary
Clinical characteristics.
Hypokalemic periodic paralysis (hypoPP) is a condition in which affected individuals may experience paralytic episodes with concomitant hypokalemia (serum potassium <3.5 mmol/L). The paralytic attacks are characterized by decreased muscle tone (flaccidity) more marked proximally than distally with normal to decreased deep tendon reflexes. The episodes develop over minutes to hours and last several minutes to several days with spontaneous recovery.
Some individuals have only one episode in a lifetime; more commonly, crises occur repeatedly: daily, weekly, monthly, or less often. The major triggering factors are cessation of effort following strenuous exercise and carbohydrate-rich evening meals. Additional triggers can include cold, stress/excitement/fear, salt intake, prolonged immobility, use of glucosteroids or alcohol, and anesthetic procedures. The age of onset of the first attack ranges from two to 30 years; the duration of paralytic episodes ranges from one to 72 hours with an average of nearly 24 hours. Long-lasting interictal muscle weakness may occur in some affected individuals and in some stages of the disease and in myopathic muscle changes. A myopathy may occur independent of paralytic symptoms and may be the sole manifestation of hypoPP.
Diagnosis/testing.
The diagnosis of hypoPP is established in a proband who meets the consensus diagnostic criteria based on a history of attacks of muscle weakness associated with documented serum potassium <3.5 mmol/L during attacks and/or the identification of a heterozygous pathogenic variant in CACNA1S or SCN4A. Of all individuals meeting diagnostic criteria for hypoPP, approximately 30% do not have a pathogenic variant identified in either of these known genes.
In the case of long-lasting interictal flaccid muscle weakness, imaging techniques can inform on the pathogenesis, potential therapy, and prognosis. Muscle ultrasound and muscle 1H-MRI are reliable image techniques with high accuracy for the disease. The weakness can be caused by edemas, fatty muscle degeneration, and muscle atrophy or a combination of these pathologies.
Management.
Treatment of manifestations. Treatment varies depending on the intensity and duration of the paralytic attack. Minor attacks may resolve spontaneously. Moderate attacks may be self-treated in a non-medical setting by ingestion of oral potassium salts. Severe attacks typically require more intensive medical management with intravenous potassium infusion, serial measurement of serum potassium concentration, clinical evaluation of possible respiratory involvement, and continuous electrocardiogram monitoring. There is no known curative treatment for hypoPP-related myopathy; physiotherapy may help to maintain strength and motor skills.
Prevention of primary manifestations. The goal of preventive treatment is to reduce the frequency and intensity of paralytic attacks. This may be achieved by avoidance of triggering factors, adherence to a diet low in sodium and carbohydrate and rich in potassium, and with the use of oral potassium supplementation. If dietary intervention and oral potassium supplementation are not effective in preventing attacks, treatment with a carbonic anhydrase inhibitor (acetazolamide or dichlorphenamide) may be necessary. If carbonic anhydrase inhibitors are not tolerated or not effective after prolonged use, alternatives include potassium-sparing diuretics such as triamterene, spironolactone, or eplerenone.
Prevention of secondary complications. Creating a safe environment, getting help in case of paralytic attack, and preventing falls and accidents are critical; an affected person experiencing a paralytic attack must have access to potassium as well as physical assistance and companions must be informed of the risk in order to enable rapid treatment. Anesthetic complications should be prevented by strict control of serum potassium concentration, avoidance of large glucose and salt load, maintenance of body temperature and acid-base balance, and careful use of neuromuscular blocking agents with continuous monitoring of neuromuscular function. It is unknown whether prevention of paralytic attacks also prevents the development of myopathy. Individuals with known pathogenic variants in one of the genes associated with hypoPP who developed myopathy without having experienced episodes of weakness have been reported.
Surveillance. The frequency of consultations is adapted to the individual's signs/symptoms and response to preventive treatment. Periodic neurologic examination with attention to muscle strength in the legs should be performed to detect long-lasting weakness associated with myopathy. For those taking acetazolamide, the following are indicated every three months: complete blood count; electrolytes; and glucose, uric acid, and liver enzyme levels. Renal ultrasound should be performed annually.
Agents/circumstances to avoid. Factors that trigger paralytic attacks (e.g., unusually strenuous effort, carbohydrate-rich meals or sweets, cold, stress/excitement/fear, high salt intake, prolonged immobility, oral or intravenous glucosteroids, certain anesthetic procedures, alcohol) should be avoided when possible.
Evaluation of relatives at risk. When the family-specific pathogenic variant is known, molecular genetic testing of at-risk asymptomatic family members can identify those at risk for unexpected acute paralysis and/or possible anesthetic complications.
Genetic counseling.
HypoPP is inherited in an autosomal dominant manner. Most individuals diagnosed with hypoPP have an affected parent. The proportion of cases caused by a de novo pathogenic variant is unknown. Offspring of a proband are at a 50% risk of inheriting the pathogenic variant. Penetrance is about 90% in males and reduced in females. Once the pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Diagnosis
Hypokalemic periodic paralysis (hypoPP) can be a primary condition or a symptom of an overarching syndrome or disease (see Differential Diagnosis). This GeneReview focuses on primary hypoPP resulting from a genetic ion channel abnormality.
Suggestive Findings
HypoPP should be suspected in individuals who describe episodic paralytic attacks with the following symptoms and signs (Note: Strictly speaking, "periodic" is a misnomer, as the attacks do not occur at regular intervals; "episodic" is a better descriptor.):
Decreased muscle tone (flaccidity)
Bilateral, symmetric, ascending (lower limbs affected before upper limbs) paralysis that is more marked in proximal than in distal muscles with sparing of the cranial muscles
Deep tendon reflexes that are normal or decreased and plantar reflexes that are normal (downward movement of toes)
Concomitant hypokalemia that is usually pronounced (0.9-3.5 mmol/L)
The typical evolution of symptoms is as follows:
Symptoms tend to occur under the following circumstances:
At rest after strong physical exertion
With fever
At times of mental stress
On awakening after a carbohydrate-rich meal the previous evening
After prolonged immobility (e.g., with long-distance travel)
Suspicion for hypoPP is also raised in individuals who have
A familial history of paralytic attack in earlier generations (father or mother, grandfather or grandmother) and in sibs;
A personal history of previous spontaneously regressive episodes of paralysis or acute muscle weakness with the above-mentioned characteristics;
Long-lasting flaccid weakness, especially if the weakness is only moderate and is prevalent in the family.
Establishing the Diagnosis
The diagnosis of hypoPP is established in a proband who meets the consensus diagnostic criteria for primary hypokalemic periodic paralysis as published in a Cochrane review [Sansone et al 2008] and/or with the identification of a heterozygous pathogenic variant in one of the genes listed in Table 1.
Consensus diagnostic criteria
Two or more attacks of muscle weakness with documented serum potassium <3.5 mEq/L
OR
One attack of muscle weakness in the proband and one attack of weakness in one relative with documented serum potassium <3.5 mEq/L
OR
Three or more of the following six clinical/laboratory features:
Onset in the first or second decade
Duration of attack (muscle weakness involving ≥1 limbs) longer than two hours
The presence of triggers (previous carbohydrate rich meal, symptom onset during rest after exercise, stress)
Improvement in symptoms with potassium intake
A family history of the condition or genetically confirmed skeletal calcium or sodium channel mutation
AND
Exclusion of other causes of hypokalemia (renal, adrenal, thyroid dysfunction; renal tubular acidosis; diuretic and laxative abuse)
In individuals who have had one or more paralytic episodes, several tests can be used to differentiate between primary hypoPP and the other possible causes.
See Clinical Testing Available (pdf) if the diagnosis is less apparent.
Atypical clinical presentation. In some individuals, hypoPP may present solely as long-lasting weakness due to a progressive limb-girdle myopathy with no history of paralytic episodes. The diagnosis may be considered if paralytic episodes are present in family members, whereas in simplex cases (i.e., a single occurrence in a family) the diagnosis is often revealed only after muscle biopsy (see Clinical Description).
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive
genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of hypoPP is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of hypoPP has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of hypoPP, the recommended approach is the use of a multigene panel.
A multigene panel that includes CACNA1S, SCN4A, and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition at the most reasonable cost while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. Given the dominant-negative mechanism of disease and lack of reported large deletions or duplications in the genes associated with hypoPP, a multigene panel that also includes deletion/duplication analysis is not typically recommended.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the diagnosis of hypoPP is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is the most commonly used genomic testing method; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Hypokalemic Periodic Paralysis
View in own window
| Gene 1, 2 | Proportion of HypoPP Attributed to Pathogenic Variants in Gene | Proportion of Pathogenic Variants 3 Detectable by Method |
|---|
| Sequence analysis 4 | Gene-targeted deletion/duplication analysis 5 |
|---|
|
CACNA1S
| ~40%-60% 6 | ~100% | Unknown 7 |
|
SCN4A
| 7%-14% 6 | ~100% | Unknown 7 |
| Unknown | ~30% 8 | NA |
- 1.
Genes are listed in alphabetic order.
- 2.
- 3.
- 4.
Sequence analysis detects variants that are benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. Variants may include small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. For issues to consider in interpretation of sequence analysis results, click here.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
- 7.
No data on detection rate of gene-targeted deletion/duplication analysis are available.
- 8.
Author, personal observation
Clinical Characteristics
Clinical Description
Large-scale studies of the natural history of primary hypokalemic periodic paralysis (hypoPP) have not been performed. Thus, knowledge of the natural history relies largely on personal observations and on individual cases that have been published with a retrospective description of the individual disease history.
Pattern of attacks. The natural history and expressivity vary greatly over time. Frequency ranges from a single occurrence of a paralytic attack that may be triggered by exceptional circumstances (extreme physical effort, specific medical intervention) to spontaneous recurrent attacks of variable frequency (ranging from multiple attacks daily to less frequent attacks). The pattern of recurrent attacks may be linked more or less to identifiable temporal and behavioral circumstances, and may evolve over years.
Triggers of paralytic attacks. There may be identifiable triggers for paralytic attacks in hypoPP. In cohort studies by Miller et al [2004] and Cavel-Greant et al [2012], affected individuals were given a list of triggers and asked which trigger applied to their own case:
Cessation of effort following strenuous exercise (e.g., football match) may trigger a paralytic attack. Approximately 67% of affected individuals identified exercise as a trigger.
Carbohydrate-rich evening meals followed by nocturnal rest may result in an immobilizing paralytic attack in the morning. Approximately 45% of affected individuals identified heavy meals or sweets as a trigger.
Cold. In ~24%. Re-warming usually recovers muscle strength.
Stress/excitement/fear/epinephrine. In ~12%. Stress, excitement, or fear results in the body producing epinephrine, which makes episodes of paralysis more likely in some individuals. This appears to be due to the effect of epinephrine in reducing blood potassium.
Salt intake. In ~11%. One of the most potent triggers of hypokalemic periodic paralysis is consumption of sodium chloride. The salt effect is far less known than the carbohydrate trigger. For many people it is easier to reduce salt than it is to reduce carbohydrates. Many foods contain huge amounts of salt, particularly snacks and tomato sauce. Soda drinks that contain both sodium and sugar are a particular problem.
Prolonged immobility is a trigger for most affected individuals. "To keep moving" is the general clinical recommendation.
Use of glucosteroids, especially parenterally, can trigger an attack.
Anesthesia/ICU. During anesthesia there are many changes that can contribute to paralysis, including cooling, glucose, mannitol, sodium, and certain anesthetics such as succinylcholine, the risk for which requires preventive measures and careful anesthetic follow up (see
Management). It is not clear that people with hypoPP are at any increased risk for
malignant hyperthermia.
Alcohol. It is unclear why alcohol sometimes triggers periodic paralysis. It could be from electrolyte imbalance, dehydration, or increased exercise or dietary indiscretion that often accompanies the inebriated state.
Age of onset of paralytic attacks. Three cohort studies have addressed this issue [Sternberg et al 2001, Miller et al 2004, Cavel-Greant et al 2012].
Frequency of paralytic episodes
In a cohort study by
Miller et al [2004], the mean frequency of paralytic attacks was seven per month (2/week), with a range of one attack per day to one attack every four months.
It was formerly assumed that the frequency of attacks peaks and then decreases with age, but in a recent survey only 21% of the patients reported decreased frequency with age [
Cavel-Greant et al 2012].
Duration of paralytic episodes
Potassium levels during paralytic episodes
The mean level of serum potassium during attacks was reported as 1.8 mmol/L in a cohort study by
Sternberg et al [2001] and as 2.3 mmol/L in a cohort study by
Miller et al [2004]. The lowest reported value was 1.2 mmol/L.
Occasionally, normal potassium values are noted. The ictal potassium level depends on the pathogenic variant (see
Molecular Pathogenesis).
Respiratory involvement and fatal outcome. The involvement of respiratory muscles during paralytic attacks is a rare but life-threatening complication of primary hypoPP. There are three possible life-threatening elements of paralytic attacks:
Hypokalemia leading to possible cardiac dysrhythmia
Weakness or paralysis of respiratory muscles leading to acute respiratory insufficiency
Inability to move that can lead to death if it occurs in a hostile environment (i.e., drowning if the paralytic attack occurs in a swimming pool)
Long-lasting interictal flaccid weakness. Most individuals have normal muscle strength and physical activity during the interictal period (i.e., between paralytic attacks), but in some affected individuals and in some stages of the disease, there may be long-lasting weakness manifesting as abortive paralytic attacks occurring in rapid succession over a long period (weeks, months). Such attacks may respond to treatment with carbonic anhydrase inhibitors or aldosterone inhibitors (see Treatment of Manifestations).
Myopathy. The development of more permanent fixed weakness due to myopathic muscle changes (fibrosis and fatty replacement) appears to vary widely from one individual to another. The myopathy may occur independent of paralytic attacks. Rarely, early signs of myopathy (e.g., Achilles' tendon shortening or scoliosis) may be present in childhood – possibly the result of more severe pathogenic variants.
In the case of long-lasting interictal flaccid muscle weakness, imaging techniques can inform on the pathogenesis, potential therapy, and prognosis. Muscle ultrasound and muscle 1H-MRI are reliable image techniques with high accuracy for the disease. The weakness can be caused by edemas, fatty muscle degeneration, and muscle atrophy or a combination of these pathologies.
Muscle histology. Typically, a muscle biopsy is not performed in the evaluation for hypoPP and histologic findings associated with the myopathy in hypoPP may depend on the specific pathogenic variant. In individuals with the p.Arg528His CACNA1S variant, the usual finding is vacuoles. Two male members of a family with the p.Arg672Gly SCNA4 variant presented only with tubular aggregates [Sternberg et al 2001].
Weakness without paralytic attacks. In some individuals, hypoPP may present solely with long-lasting weakness due to a progressive limb-girdle myopathy with no history of paralytic episodes.
Additional symptoms that can co-occur with hypokalemic periodic paralysis. People with genetically proven hypoPP often have other symptoms in addition to the paralysis:
Among the 30% of people who appear to have hypoPP but do not have pathogenic variants in either of the genes known to be associated with hypoPP, the following are often noted:
Migraines
Heart rhythm abnormalities
Attention deficit disorder (ADD, ADHD)
Relative insensitivity to the local anesthetic lidocaine and "dental anxiety"
Severe premenstrual syndrome
Genotype-Phenotype Correlations
There are no clear cut genotype-phenotype correlations for either of the genes known to be associated with hypoPP.
Penetrance
In general, the penetrance of this disorder is high (≥90%) in males and reduced in females. An exception to this occurs with pathogenic variants with an arginine-to-glycine substitution for which high penetrance in males and females is noted [Kim et al 2005, Wang et al 2005, Winczewska-Wiktor et al 2007].
Nomenclature
Names for hypokalemic periodic paralysis no longer in use include the following:
Hypokalemic periodic paralysis was formerly most often known as Westphal's disease, as Karl Friedrich Otto Westphal (1833-1890) first described extensively and convincingly the main characteristics of the disease, which had previously been described as "periodic palsy" by Musgrave in 1727, Cavaré in 1853, and Romberg in 1857. Hartwig reported a case of palsy with muscle inexcitability provoked by rest after exercise in 1875. Westphal described a simplex case (i.e., single occurrence in a family); it was not until 1887 that a dominant pedigree was described by Cousot.
Familial versus sporadic hypoPP. Hypokalemic periodic paralysis may be familial (f-hypoPP) or sporadic (s-hypoPP), meaning that the affected individual has no known family history of hypoPP. Sporadic cases may be the result of a de novo variant, of pathogenic variants with incomplete penetrance in other family members with the variant, or of other as-yet unexplained factors. Pathogenic variants in the same genes may lead to either f-hypoPP or s-hypoPP.
HypoPP1 versus hypoPP2. HypoPP1 and hypoPP2 are hypokalemic periodic paralysis linked respectively to CACNA1S and SCN4A pathogenic variants. Some authors use the terms "hypoPP type 1" and "hypoPP type 2." However, this could wrongly suggest that there are two clinical types of hypoPP. In fact, hypoPP1 and hypoPP2 are not distinct clinically.
Prevalence
The prevalence of hypoPP is unknown but thought to be approximately 1:100,000 [Statland et al 2018]. However, a demographic survey in England, relying on the data of the national specialist channelopathy service, reported a minimum point prevalence of 0.13:100,000 (95% CI 0.10-0.17) [Horga et al 2013].
Differential Diagnosis
The following signs and symptoms suggest a diagnosis other than hypokalemic periodic paralysis (hypoPP):
Associated sensory symptoms, including pain or tenderness
Sensory loss could suggest polyneuropathy such as Guillain-Barré syndrome.
Pain could suggest myositis; however, some individuals with hypoPP report paralytic episodes as painful.
Urinary retention or constipation, which may be observed in other causes of acute or subacute paralysis, but can occur rarely in hypoPP
Associated symptoms that suggest myasthenia or involvement of the neuromuscular junction, including:
Ptosis
Diplopia
Dysphagia
Dysarthria
Alteration or loss of consciousness
Abnormal movement
History of fever days before an attack, which could suggest poliomyelitis or other virus-caused paralysis
History of back pain days before an attack, which could suggest acute transverse myelitis
History of tick bite, which could suggest tick paralysis
HypoPP is the most common cause of periodic paralysis. The four major differential diagnoses are normokalemic potassium-sensitive periodic paralysis (normoPP), hyperkalemic periodic paralysis (hyperPP), thyrotoxic periodic paralysis (TPP), and Andersen-Tawil syndrome (ATS) (see Table 3).
Table 3.
The Different Categories of Periodic Paralyses (PP) with Membrane Excitability Disorder and Associated Findings
View in own window
| HypoPP | NormoPP | HyperPP | TPP 1 |
ATS
|
|---|
|
Main clinical features
| Weakness episodes lasting hrs to days w/concomitant hypokalemia | Weakness episodes lasting hrs to days w/concomitant normokalemia | Weakness episodes lasting mins to hrs w/concomitant normo- or hyperkalemia | Identical to that of the paralytic episodes of hypoPP | Episodic, periodic paralysis, ventricular arrhythmias, prolonged QT interval, characteristic anomalies 2 |
|
Age at first attacks
| Late in 1st decade or in 2nd decade | Late in 1st decade or in 2nd decade | 1st years of life | Variable, dependent on onset of thyrotoxicosis | Late in 1st decade or in 2nd decade (usually after cardial events) |
|
Main triggers
| Rest after exercise, carbohydrate rich meal, salt intake, stress, cold | Rest after exercise, carbohydrate rich meal, salt intake, stress, cold | Cold, rest after exercise, stress, fatigue, alcohol, hunger, changes in activity level, potassium in food, specific foods | Thyrotoxicosis | Prolonged rest, rest after exertion |
|
EMG: myotonic discharges
| No | Some | Some | No | No |
|
EMG tests
| Late decrement w/LET
(Pattern IV, V) | Late decrement w/LET
(Pattern IV, V) | Pattern IV, V | Initial CMAP increase + decline | Variable (CMAP increase + decline, normal CMAP + decline etc) |
|
Extramuscular expression
| None | None | None | Possible manifestations of thyrotoxicosis | Cardiac arrhythmia; Dysmorphy |
|
Prevention of paralysis attacks
| ACZ, DCP | ACZ, DCP | ACZ, DCP | Normal thyroid function | ACZ, DCP |
|
Curative treatment
| None | None | None | Treatment of thyroid disorder | None |
|
Known causal or susceptibility gene(s)
| CACNA1S;
SCN4A |
SCN4A
|
SCN4A
|
KCNJ18
|
KCNJ2
|
|
Defective ion channel(s)
| Cav 1.1;
Nav 1.4;
Kir 6.2 | Nav 1.4 | Nav 1.4 | Kir 6.2 | Kir 2.1 |
ACZ = acetazolamide; CMAP = compound muscle action potential; DCP = dichlorphenamide; LET = long exercise test; SET = short exercise test
- 1.
- 2.
ATS anomalies include low-set ears, widely spaced eyes, small mandible, fifth-digit clinodactyly, syndactyly, short stature, and scoliosis.
Normo- and
hyperkalemic paralysis
(normo/hyperPP) differ in several ways from hypoPP:
Serum concentration of potassium during the paralytic attacks is normal or elevated.
Some triggering factors for hypoPP attacks (e.g., carbohydrate-rich meals) are not found.
Age of onset of paralytic attacks is lower.
Duration of attacks is assumed to be shorter. However, this is questionable, according to surveys of affected individuals.
Electromyography shows myotonic discharges in most individuals between attacks; however, the response patterns for short exercise test (SET) and long exercise test (LET) may be indiscernible; i.e., pattern IV or V defined by
Fournier et al [2004] may be caused by both hypokalemic and normo/hyperkalemic periodic paralysis.
In normokalemic PP, the reaction to oral potassium administration may be different than for hypoPP – anything from amelioration to worsening of the weakness [
Jurkat-Rott et al 2012].
Usually, the distinction between hypoPP and normo/hyperPP can be made on the basis of clinical, biologic (i.e., kalemia during an attack), and EMG findings and confirmed by molecular genetic testing [Miller et al 2004, Vicart et al 2004, Fan et al 2013].
Thyrotoxic periodic paralysis (TPP) (OMIM 613239) is most often not familial, but in some instances there may be a familial predisposition. The clinical and biologic picture of TPP is identical to that of the paralytic episodes of hypoPP. Furthermore, the EMG response patterns for SETs and LETs (i.e., patterns IV or V defined by Fournier et al [2004]) for familial genetic hypoPP and TPP are identical when thyrotoxicosis is present. Males of Asian origin, and possibly people of Latin American and African American origin, are assumed to be at greater risk than people of other ethnic/racial origins for developing periodic paralysis as a consequence of thyrotoxicosis.
Although TPP is not usually caused by classic hypoPP-causing pathogenic variants [Dias da Silva et al 2002, Ng et al 2004], the association of TPP with genetically defined hypoPP and normoPP has been reported [Lane et al 2004, Vicart et al 2004]. An association with CACNA1S 5'UTR and intronic SNPs has been suggested but not confirmed [Kung et al 2004]. A pathogenic variant in an inwardly rectifying potassium (Kir) channel (encoded by KCNJ18) was identified in approximately one third of affected individuals in one series [Ryan et al 2010].
Because thyrotoxicosis may be a precipitating factor of genetically defined hypokalemic or normokalemic periodic paralysis [Lane et al 2004, Vicart et al 2004], the following should be measured in anyone with weakness and hypokalemia:
Plasma thyroid-stimulating hormone (TSH) (reference range: 0.45-4.5 µU/mL)
Free thyroxine (FT4) (reference range: 8.0-20.0 pg/mL)
Free triiodothyronine (FT3) (reference range: 1.4-4.0 pg/mL)
Note: (1) Low TSH together with high FT3 and FT4 are diagnostic of hyperthyroidism. Treatment of hyperthyroidism cures TPP. (2) TPP is distinct from hypokalemic periodic paralysis (hypoPP); however, at least two instances of genetically diagnosed familial hypoPP for which hyperthyroidism was an additional trigger for hypokalemic paralytic episodes have been reported [Lane et al 2004, Vicart et al 2004].
Andersen-Tawil syndrome
(ATS) is characterized by a triad of episodic flaccid muscle weakness (i.e., periodic paralysis), ventricular arrhythmias and prolonged QT interval, and anomalies including low-set ears, widely spaced eyes, small mandible, fifth-digit clinodactyly, syndactyly, short stature, and scoliosis. Affected individuals present in the first or second decade with either cardiac symptoms (palpitations and/or syncope) or weakness that occurs spontaneously following prolonged rest or following rest after exertion. Long-lasting interictal weakness is common. Mild learning difficulties and a distinct neurocognitive phenotype (i.e., deficits in executive function and abstract reasoning) have been described. Incomplete clinical presentations are possible: Andersen-Tawil syndrome may express itself as pure hypoPP. An electrocardiogram or a Holter-EKG recording between attacks of weakness is necessary to evaluate for the possibility of Andersen-Tawil syndrome. The EMG response patterns for short and long exercise tests may be identical; i.e., patterns IV or V defined by Fournier et al [2004] may be caused by Andersen-Tawil syndrome as well as hypoPP. EKG should be performed again in an interictal period in order to evaluate for a U wave, which is observed in Andersen-Tawil syndrome. Pathogenic variants in KCNJ2 are causative [Plaster et al 2001]. Inheritance is autosomal dominant with reduced penetrance and variable expressivity.
Hypokalemia caused by reduced potassium intake, enhanced renal excretion, or digestive loss. Because the main clinical manifestation of hypokalemia is muscle weakness, it may be difficult in some cases to discriminate between a paralytic attack of hypoPP and an episode of weakness associated with chronic hypokalemia of another cause. In such cases the diagnosis relies on the correct interpretation of findings such as blood pressure, urinary concentration of potassium, and blood concentration of bicarbonate (see Table 4).
See also Clinical Testing Available, Transtubular potassium concentration gradient and potassium-creatinine ratio during paralytic attack (pdf).
Table 4.
Identifying the Cause of Secondary Hypokalemia
View in own window
If blood
pressure
is: | & urinary
potassium
is: | & blood
bicarbonate
is: | Diagnostic explanations |
|---|
| High | | | Primary or secondary inappropriate (pseudo) hyperaldosteronism Secondary hyperaldosteronism (↑ renin blood concentration): renin secreting tumor, renal artery stenosis, malignant hypertension Hyperglucocorticism (normal renin blood concentration) Licorice (normal renin blood concentration)
|
| >25 mmol/L | High | Liddle syndrome (tubulopathy) |
| Normal | <25 mmol/L | High | Past treatment w/diuretics |
| Low or normal |
|
| >25 mmol/L | High | Vomiting Present treatment w/diuretics Bartter syndrome (tubulopathy w/normo- or hypercalcuria, normomagnesemia) Gitelman syndrome (tubulopathy w/hypocalciuria, hypomagnesemia)
|
| Low |
|
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with hypokalemic periodic paralysis (hypoPP), the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended.
During an acute paralytic attack
Assessment of respiratory status to detect those individuals who may have early respiratory failure
Measurement of serum potassium concentration
Cardiac electrophysiologic testing (EKG) to assess for life-threatening cardiac consequences of hypokalemia
Assessment for swallowing difficulty
Between attacks or in a currently asymptomatic individual
Neurologic examination to assess muscle strength in the legs
Measurement of the following thyroid functions (see Note):
In those with long-lasting interictal weakness, consideration of muscle sonography or MRI scan of muscles (e.g. thigh) to evaluate the extent of myopathy [
Weber et al 2019]
Consultation with a clinical geneticist and/or genetic counselor
Note: (1) Hyperthyroidism may be a trigger for a hypokalemic episode in individuals with hypoPP; (2) thyroid function tests may help to distinguish between hypoPP and TPP in those who have a pathogenic variant in KCNJ18.
Treatment of Manifestations
For a comprehensive summary of the management of hypokalemic periodic paralysis, see Levitt [2008] (full text). The principles of treatment are summarized in Table 5 [Jurkat-Rott & Lehmann-Horn 2010, Greig 2016.]
Table 5.
Principles of Treatment for Individuals with HypoPP
View in own window
| Goal | Means | Practical Details |
|---|
| To avoid triggering or aggravating factors for paralytic attacks | Avoid:
Strenuous effort; Prolonged immobility; Carbohydrate-rich diet; High sodium diet.
|
|
| Treatment of paralytic attack:
|
| Do not use slow-release forms of potassium. Oral potassium: initially, 1 mEq/kg; add 0.3 mEq/kg after 30 min if no improvement IV potassium: 0.3 mEq/kg/h
|
| Preventive treatment for paralytic attacks | Daily potassium supplementation | Slow-release forms of potassium may be used. |
| Acetazolamide | |
| Dichlorphenamide | |
| Potassium-sparing diuretics | |
| Preventive treatment for late-onset myopathy | Acetazolamide? | |
| Medical precautions | Avoid corticosteroids if possible. Use alpha- or beta adrenergic drugs w/caution, even in local anesthesia or ophthalmology.
| |
| Other elements of management |
| |
Paralytic crisis. Treatment of the paralytic crisis is far from perfect, as the only tool is the administration of potassium by mouth or IV, which addresses the hypokalemia directly, but the weakness only indirectly. Resolution of muscle weakness and normalization of the serum concentration of potassium are not strictly parallel. The serum concentration of potassium may normalize before weakness begins to resolve.
During a paralytic attack, there is usually no true potassium depletion in the body (unless there are associated digestive or renal losses from another cause), but there is a reversible transfer of potassium from the extracellular to the intracellular space. The global potassium pool (and especially the intracellular pool of the body) is conserved, and the goal of the treatment is to raise the blood potassium level just enough to trigger the shift to repolarization of skeletal muscle membrane, and the liberation of intracellularly sequestered potassium.
Mild to moderate paralytic episodes
Treatment may occur in a familial or non-medical setting if the diagnosis is well-established and the affected individual is able to manage paralytic episodes.
Rapid recovery is typically possible with oral intake of chloride potassium salts, either as capsules or liquid-containing vials. Aqueous potassium contained in vials may act more rapidly.
An initial intake of 1mEq/kg potassium chloride is often used (60 mEq; i.e., 4.5 g of potassium chloride for a 60-kg person).
A response (at least partial) should be seen after 30 minutes. If no improvement occurs after 30 minutes, an additional 0.3 mEq/kg can be administered (20 mEq; i.e., 1.5 g of potassium chloride for a 60 kg person).
Note: (1) Slow-release forms of potassium should be avoided during a paralytic attack. (2) Potassium ingesting should be followed by oral intake of water (e.g., 100 mL (4 oz) of water for each 20 mEq of potassium). (3) Liquids containing high sugar or sodium content should be avoided.
Severe paralytic episodes. If an affected individual has followed the instructions above for a mild to moderate attack and either no improvement or further aggravation is observed after an additional 30 minutes, medical supervision with measurement of serum potassium level should be considered.
The total dose of potassium taken over a 24-hr period for the treatment of an acute attack should not exceed 200 mEq.
During severe paralytic episodes or attacks associated with respiratory, swallowing, or speaking difficulties, or with signs of arrhythmia, the affected individual must be transferred to hospital.
In the case of very low serum potassium and severe symptoms (airway compromise with ictal dysphagia, accessory respiratory muscle paralysis, arrhythmia associated with hypokalemia), intravenous potassium treatment should be initiated. Note the following critical points:
The concentration of intravenous potassium chloride solution should not exceed 40 mEq/L because of the risk for thrombophlebitis (the use of a central catheter is rarely necessary).
The solution should be given as a continuous infusion that does not exceed 0.3 mEq/kg/h of potassium (i.e., 18 mEq/h for a 60-kg person) because of the high risk for arrhythmia or cardiac arrest associated with faster infusions.
The physician should be aware that hyperkalemia may occur, as there is no true potassium depletion; a reverse shift of potassium from the intracellular to the extracellular space occurs during the resolution of paralytic episode.
A Y-branched peripheral venous line containing potassium chloride should be branched to a perfusion of mannitol or normal saline (avoid glucose-containing solutions, which may enhance hypokalemia).
To prevent cardiac arrhythmias, it is important to monitor the EKG before, during, and after treatment and to perform repeat assessments of blood potassium concentration:
A prominent increase in the amplitude of the U wave, triggered by hypokalemia, is associated with a higher susceptibility to the ventricular arrhythmia known as torsades de pointes. Some individuals exhibit serious arrhythmias even in the presence of mild hypokalemia.
Large and sharp T waves are a marker of hyperkalemia and may occur during and after recovery; they are associated with a risk for cardiac arrest.
Monitoring of EKG and blood potassium concentration must be continued some hours after normalization of the serum potassium concentration, in order to detect a relapse of hypokalemia or the development of hyperkalemia secondary to excessive potassium load.
Administration of supplemental potassium must be discontinued when the serum potassium concentration is normalized, even if weakness persists.
Attempting to abort paralytic attacks when they begin. Affected individuals are advised to keep a sufficient dose of potassium in various places (at the bedside, in pockets or handbags, in the car) so that when warning symptoms appear, the person can take potassium and possibly avoid a full-blown attack, which would usually occur within minutes [Levitt 2008]. It is also acknowledged that maintaining mild physical activity may abort attacks in some cases.
Myopathy. No curative treatment is known for fixed myopathy in hypoPP. The effects of muscle weakness are managed as in other disorders with similar manifestations.
Prevention of Primary Manifestations
Preventive treatment is intended to decrease the frequency and intensity of paralytic attacks. Triggering factors need to be identified and, if possible, avoided (see Table 5).
A diet rather low in sodium and carbohydrate and rich in potassium is recommended.
Potassium supplementation for prevention and treatment of attacks is an empiric but effective treatment for shortening attacks and sometimes preventing their occurrence (see Treatment of Manifestations).
Oral intake of potassium salts (10-20 mmol/dose, 3 doses/day) can prevent attacks, especially if the dose of potassium is taken some hours before the usual time of the attack (i.e., a nocturnal dose if crises occur at awakening).
For individuals receiving chronic potassium supplementation for hypoPP, magnesium might be added, which can be helpful to promote renal retention of potassium and, therefore, reduce the potassium dose [
Statland et al 2018].
Carbonic anhydrase inhibitors (in particular acetazolamide and dichlorphenamide) have been used for almost 50 years as empiric treatment for both hypoPP and hyperPP [Matthews et al 2011]. The mechanism of action in periodic paralyses is incompletely understood. Carbonic anhydrase inhibitors promote kaliuresis and a nonanion gap acidosis by increasing urinary bicarbonate excretion. The systemic acidosis may reduce the susceptibility to periodic paralysis [Matthews & Hanna 2010]. An alternative proposal is enhanced opening of calcium-activated K+ channels [Tricarico et al 2004]. In addition, carbonic anhydrase inhibitors also may be effective for treating long-lasting interictal weakness in hypoPP by reducing intracellular sodium accumulation (vide supra) [Statland et al 2018].
Acetazolamide is generally considered the additive treatment of choice for prevention of paralytic attacks and myopathy in hypoPP.
However, there is no standardized treatment regimen and no consensus as to when to start treatment with acetazolamide.
Typical dosage for acetazolamide in adults is between 125 mg/day and 1000 mg/day (usually 250-500 mg/day), divided into three doses and taken with meals; in children a dose of 5-10 mg/kg/day, divided into three doses and taken with meals, is used.
Acetazolamide treatment:
Is beneficial in approximately 50% of individuals with hypoPP
Has no effect in 30% of affected individuals
May exacerbate hypoPP in individuals with who have a pathogenic variant in SCN4A
In some affected persons, long-lasting interictal weakness may be partly reversed and muscle strength may be improved by acetazolamide treatment [
Lehmann-Horn et al 2008]
Whether acetazolamide treatment prevents or treats myopathy and the resulting fixed weakness that occurs with age is unknown.
Further studies are needed to evaluate the effect of preventive acetazolamide treatment on attack rate, severity-weighted attack rate, long-lasting interictal weakness, and myopathy.
Common side effects of carbonic anhydrase inhibitors include paresthesia, fatigue, mild, reversible cognitive disturbances and an increased risk of nephrolithiasis.
Dichlorphenamide was recently approved by the FDA for the treatment of PP. Dichlorphenamide has been evaluated in four randomized, placebo- controlled studies, two each in patients with hypoPP and hyperPP [Sansone et al 2016].
While randomized controlled trials of dichlorphenamide were performed in adults, the same approach is taken for children; dose adjustments may be required based on age.
These studies demonstrated a significant reduction in the frequency and severity of the attacks. During a 52-week extension, in which all remaining participants received open-label dichlorphenamide, continued improvement in outcomes was observed in both placebo and dichlorphenamide groups.
The dose of dichlorphenamide was 50 mg twice daily for treatment-naıve patients.
Individuals already on dichlorphenamide before the study continued on the same dose during the study.
In those taking acetazolamide before the study, the dose of dichlorphenamide was set at 20% of the acetazolamide dose.
Dose reduction for tolerability was permitted.
The mean dose of dichlorphenamide at week 9 was 82 mg/day.
The most common side effects with dichlorphenamide were paresthesias, cognitive disorder, dysgeusia, headache, fatigue, hypoesthesia, and muscle spasms, generally not requiring discontinuation of dichlorphenamide, and reversible with drug discontinuation.
In the recent study of diclorphenamide [Sansone et al 2016], quality of life was assessed at 9 weeks and significant improvement was reported for the physical component and physical functioning, working time, bodily pain, vitality, and social functioning in those with hypoPP.
Alternatives to carbonic anhydrase inhibitors. If carbonic anhydrase inhibitors are not tolerated or not effective after prolonged use, alternatives include potassium sparing diuretics like triamterene 50–150 mg/day, spironolactone 25–100 mg/day or eplerenone 50– 100mg daily.
Because spironolactone is associated with a long half-life for substrate degradation, the individual can become hyperkalemic and weaker, develop cardiac arrhythmias, and suffer from hair loss. Additionally, spironolactone has androgenic side effects.
The modern spironolactone derivate Eplerenone may be preferred because it causes fewer androgenic side effects. In addition, it has a very high repolarizing power, the parameter considered as most relevant for a beneficial effect.
For individuals with hypoPP, potassium supplementation and a potassium-sparing diuretic may be used concomitantly, but potassium levels should be routinely monitored.
Prevention of Secondary Complications
Creating a safe environment, getting help in case of paralytic attack, and preventing falls and accidents are critical [Levitt 2008].
An affected person experiencing a paralytic attack must have access to potassium as well as physical assistance. Thus, those with hypoPP should inform their companions or acquaintances of their risk for paralytic attack, especially in a sports or school context, so that they can access appropriate help rapidly in case of an attack.
Falls and injuries from falls are frequent in those with hypoPP (67% of affected individuals age >40 years report such falls and injuries) [
Cavel-Greant et al 2012].
Pre- or postoperative paralysis. Because of the risk for paralysis preceding or following anesthesia, precautions should be taken during administration of anesthesia to individuals with hypoPP. Individuals with hypoPP should be considered possibly susceptible to malignant hyperthermia and managed with a non-triggering anesthetic technique – although general anesthesia using volatile anesthetics and succinylcholine has been reported as safe in a small number of individuals with hypoPP.
General guidelines for perioperative care include the following:
Strict control of serum potassium concentration
Avoidance of large glucose and salt loads
Low-carbohydrate diet
Maintenance of body temperature and acid-base balance
Careful use of neuromuscular blocking agents and no depolarizing muscle relaxants
Late-onset myopathy with fixed muscle weakness. It is not known whether the prevention of paralytic attacks also prevents the development of myopathy. Individuals with known pathogenic variants who developed myopathy without having experienced episodes of weakness have been reported. Therapy with spironolactone or eplerenone might be beneficial.
Surveillance
The frequency of consultations needs to be adapted to the individual's signs and symptoms and response to preventive treatment. Neurologic examination with attention to muscle strength in the legs should be performed, in order to detect long-lasting interictal weakness associated with myopathy.
For those individuals who take acetazolamide the following parameters should be evaluated every three months: complete blood count, electrolytes, glucose, uric acid, and liver enzyme levels. Renal ultrasound should be performed annually.
Agents/Circumstances to Avoid
Avoid anything that can trigger paralytic attacks in the individual case, including the following:
Unusually strenuous effort
Excess of carbohydrate-rich meals or sweets
Cold
Stress/excitement/fear
High salt intake
Prolonged immobility
Oral or intravenous glucosteroids
Use of cooling, glucose and/or mannitol infusion, excessive sodium- containing fluids and certain anesthetics such as succinylcholine during anesthesia
Use of alcohol
Evaluation of Relatives at Risk
It is appropriate to evaluate apparently asymptomatic older and younger at-risk relatives of an affected individual in order to identify as early as possible those at risk for unexpected acute paralysis and/or possibly malignant hyperthermia and who would benefit from prompt initiation of treatment and preventive measures.
Evaluations include:
Molecular genetic testing if the pathogenic variant in the family is known;
Detailed history (with attention to full or incomplete paralytic crises in the past and/or adverse response to glucose infusion, surgery, or general anesthesia) and neurologic examination.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
In general, there is no increased risk for paretic attacks in pregnancy. While in utero acetazolamide exposure is reported to cause limb defects in rodents, acetazolamide therapy during human pregnancy does not appear to increase the risk for fetal malformations [Heinonen et al 1977]. Eplerenone, if taken during pregnany, may cause hirsutism in the newborn.
See MotherToBaby for further information on medication use during pregnancy.
Therapies Under Investigation
Recent studies in mouse models of hypoPP with both SCN4A pathogenic variants and CACNA1S pathogenic variants show that maneuvers to reduce the activity of the Na-K-2Cl (NKCC) co-transporter can reverse an acute attack of hypoPP and protect against an attack triggered by low K+ exposure [Wu et al 2013]. The beneficial effect is the result of biasing intracellular chloride to be low, which promotes hyperpolarization of the resting potential. The NKCC co-transporter is activated by hyperosmolarity (hence the importance of avoiding high sodium diet, dehydration, hyperglycemia and mannitol) and is inhibited by loop diuretics such as bumetanide [Wu et al 2013, Statland et al 2018].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
Hypokalemic periodic paralysis (hypoPP) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
Most individuals diagnosed with
CACNA1S or
SCN4A-related hypoPP inherited a pathogenic variant from a heterozygous parent who may or may not be affected (the autosomal dominant inheritance pattern may be masked by reduced penetrance or failure to recognize the disorder in preceding generations). Less frequently, a proband with
CACNA1S or
SCN4A-related hypoPP has the disorder as the result of a
de novo pathogenic variant [
Sung et al 2012].
The overall proportion of hypoPP cases caused by a de novo pathogenic variant is unknown.
Recommendations for the evaluation of parents of an individual with an apparent de novo pathogenic variant (i.e., no known family history of hypoPP) include:
A search for a history of full or incomplete paralytic crises in the past and/or adverse response to glucose infusion, surgery, or general anesthesia
Neurologic examination with evaluation of muscle strength
Molecular genetic testing if the hypoPP-related pathogenic variant has been identified in the proband.
If the pathogenic variant found in the proband cannot detected in leukocyte DNA of either parent, possible explanations include a de novo pathogenic variant in the proband or parental germline mosaicism (though theoretically possible, no instances of germline mosaicism have been reported).
If clinical testing of both parents is inconclusive and predictive molecular genetic testing is not possible (i.e., if a pathogenic variant has not been identified in the proband), both parents may need to be considered at risk for complications, including unexpected acute paralysis and hypokalemia, and possibly malignant hyperthermia associated with anesthesia.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband’s parents:
If a parent is affected and/or is known to have the pathogenic variant identified in the proband, the risk to each sib of inheriting the CACNA1S, or SCN4A pathogenic variant is 50%. Reduced penetrance has been observed.
If the
CACNA1S or
SCN4A pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [
Rahbari et al 2016].
If the parents are clinically unaffected but their genetic status is unknown, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for hypoPP because of the possibility of reduced penetrance in a parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with hypoPP has a 50% risk of inheriting the hypoPP-related pathogenic variant.
Other family members. The risk to other family members depends on the genetic status of the proband's parents: if a parent is affected and/or has a pathogenic variant in CACNA1S or SCN4A, his or her family members are at risk.
Prenatal Testing and Preimplantation Genetic Testing
Once the CACNA1S or SCN4A pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and in families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Hypokalemic Periodic Paralysis: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
114208 | CALCIUM CHANNEL, VOLTAGE-DEPENDENT, L TYPE, ALPHA-1S SUBUNIT; CACNA1S |
|
170400 | HYPOKALEMIC PERIODIC PARALYSIS, TYPE 1; HOKPP1 |
|
603967 | SODIUM VOLTAGE-GATED CHANNEL, ALPHA SUBUNIT 4; SCN4A |
Molecular Pathogenesis
The two genes associated with primary hypoPP, CACNA1S and SCN4A, encode subunits of voltage-gated ion channels that are primarily expressed in skeletal muscle cells:
CACNA1S encodes Cav1.1
SCN4A encodes Nav1.4
The common etiology for weakness in an episode of hypoPP is a failure to maintain the resting potential in low K+. The clinical hypoPP phenotype is identical, regardless of whether the underlying pathogenic variant is in CACNA1S or SCN4A. HypoPP-causing variants result in an anomalous gating pore current.
In all forms of periodic paralysis, ictal paresis is caused by depolarization of the muscle sarcolemma, which in turn causes sodium channel inactivation and reduced fiber excitability. This depolarization is caused by a pathogenic variant in voltage-gated cation channels, i.e. most often CACNA1S or SCN4A. Weakness in an episode of hypoPP is a failure to maintain the resting potential in low K+. Pathogenic variants tend to result in the neutralization of positively charged residues in the voltage-sensing S4 transmembrane segments, impeding its movement. In addition to abnormal gating of the ion-conducting alpha pore, pathogenic variants can also cause omega currents or gating pore currents, facilitated by the movement of mutated S4 segments, which also contribute to symptoms. These omega currents conduct cations by aqueous cavities with varying ion selectivity and are activated in either a hyperpolarized or depolarized voltage range. This omega current is of relatively small amplitude and difficult to distinguish from a nonspecific leakage current.
Both hypoPP and normoPP are caused by single pathogenic variants in positively charged residues (i.e., gating charges) in the S4 transmembrane segment of the voltage sensor of Nav 1.4 or Cav 1.1. Pathogenic variants of the outermost gating charges (R1 and R2) cause hypoPP by creating a pathogenic gating pore in the voltage sensor through which cations leak in the resting state. Pathogenic variants of the third gating charge (R3) cause normoPP by a cation leak both in activated and inactivated states (see and ).
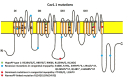
Cav 1.1 Sketch of the voltage-gated calcium channel of skeletal muscle, Cav1.1 enocoded by CACNA1S. Although Cav1.1 strikingly resembles Nav1.4, the various channel parts (e.g. the DIII–DIV linker) may have a different function. The channel scheme (more...)
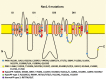
NaV 1.4 The voltage-gated sodium channel of skeletal muscle, NaV1.4 encoded bySCN4A, is composed of four highly homologous domains (DI–DIV) each consisting of six transmembrane segments (S1–S6). When inserted in membrane, the four domains (more...)
The protein domain location of pathogenic variants associated with disorders correlates between Cav1.1 and Nav1.4 (see and ) [Matthews et al 2011, Jurkat-Rott et al 2012, Groome et al 2014, Cannon 2017, Jiang et al 2018]:
HypoPP
Cav1.1: outermost gating charges R1 or R2 of domains II, III, and IV
Nav1.4: outer arginine residues of S4 (R1 or R2) in domains I, II, or III
NormoPP
CACNA1S
Gene structure.
CACNA1S has 44 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. See Table 6. Pathogenic variants in CACNA1S proven to cause hypoPP are almost exclusively nucleotide substitutions causing an amino acid change that turns positively charged arginines of voltage sensor S4 segments into other amino acids (most often histidine; sometimes glutamine, serine, glycine, cysteine; see . Additional dominant and recessive variants are shown in .
Table 6.
CACNA1S Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.1583G>A | p.Arg528His |
NM_000069.2
NP_000060.2
|
| c.1582C>G | p.Arg528Gly |
| c.1582C>T | p.Arg528Cys |
| c.2627T>A | p.Val876Glu |
| c.2691G>T | p.Arg897Ser |
| c.2700G>T | p.Arg900Ser |
| c.3716G>A | p.Arg1239His |
| c.3715C>G | p.Arg1239Gly |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
CACNA1S codes for the voltage-dependent L-type calcium channel alpha-1S subunit (Cav 1.1).
Abnormal gene product. HypoPP-causing variants are found in the S4 segment of domain II, II and IV ().
SCN4A
Gene structure.
SCN4A has 24 exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. See Table 7. Pathogenic variants in SCN4A proven to cause hypoPP are almost exclusively nucleotide substitutions causing an amino acid change that turns positively charged arginines of voltage sensor S4 segments into other amino acids (most often histidine; sometimes glutamine, serine, glycine, cysteine) (see and ).
Table 7.
SCN4A Pathogenic Variants Causing Hypokalemic Periodic Paralysis Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.664C>G | p.Arg222Gly |
NM_000334.4
NP_000325.4
|
| c.664C>T | p.Arg222Trp |
| c.2006G>A | p.Arg669His |
| c.2014C>A | p.Arg672Ser |
| c.2014C>G | p.Arg672Gly |
| c.2014C>T | p.Arg672Cys |
| c.2015G>A | p.Arg672His |
| c.3386G>A | p.Arg1129Gln |
| c.3395G>A | p.Arg1132Gln |
| c.3404G>A | p.Arg1135His |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
Normal gene product.
SCN4A encodes the skeletal muscle sodium channel protein type 4 subunit alpha (Nav 1.4), which is activated by membrane depolarization and is responsible for the upstroke of the action potential. It therefore plays a key role in muscle contraction, allowing a proper propagation of the action potential along the muscle membrane.
Abnormal gene product. Pathogenic variants in SCN4A causing hypoPP occur within the voltage-sensitive segment S4 of domain II of the sodium channel alpha subunit. Disease-causing variants change positively charged arginines to non-charged amino acid residues () enhancing fast [Jurkat-Rott et al 2000, Kuzmenkin et al 2002] and/or slow [Struyk et al 2000, Kuzmenkin et al 2002] inactivation and reducing current density [Jurkat-Rott et al 2000]. These variants act in a dominant-negative manner, thereby reducing the fraction of available non-inactivated sodium channels at the resting potential.
HypoPP-causing variants in Nav 1.4 S4 segments create an abnormal gating pore current; i. e., an accessory ionic transmembrane permeation pathway through the aqueous environment of S4 segment [Sokolov et al 2005, Sokolov et al 2007, Struyk & Cannon 2007, Sokolov et al 2008, Struyk et al 2008, Francis et al 2011, Jurkat-Rott et al 2012]. This current is a low-amplitude inward current at the resting potential, and varies in its amplitude and selectivity according to the precise pathogenic variant and to the physiologic or pathologic conditions, thus contributing to membrane depolarization in pathologic circumstances [Struyk et al 2008].
Chapter Notes
Acknowledgments
This work was supported by: the German Research Foundation (DFG), a network of the IHP Program funded by the European Community; the non-profit Hertie Foundation; the IonNeurONet of German Ministry of Research (BMBF); the German Muscle Society (DGM); and the Else-Kröner-Fresenius Foundation.
Author History
Marianne Arzel-Hézode, MD; Groupe Hospitalier Pitié-Salpêtrière (2014-2018)
Saïd Bendahhou, PhD; Université Nice Sophia Antipolis (2014-2018)
Bertrand Fontaine, MD, PhD; Groupe Hospitalier Pitié-Salpêtrière (2002-2018)
Emmanuel Fournier, MD, PhD; Groupe Hospitalier Pitié-Salpêtrière (2014-2018)
Jérôme Franques, MD; Hôpital de la Timone (2014-2018)
Bernard Hainque, PharmD, PhD; Groupe Hospitalier Pitié-Salpêtrière (2002-2018)
Frank Lehmann-Horn, MD, PhD, MS (2018 *)
Philippe Lory, PhD; Université de Montpellier (2014-2018)
Sophie Nicole, PhD; Groupe Hospitalier Pitié-Salpêtrière (2014-2018)
Damien Sternberg, MD, PhD; Groupe Hospitalier Pitié-Salpêtrière (2002-2018)
Nacira Tabti, MD, PhD; Assistance Publique - Hôpitaux de Paris (2002-2014)
Savine Vicart, MD; Groupe Hospitalier Pitié-Salpêtrière (2014-2018)
Frank Weber, MD, PhD (2018-present)
* Professor Lehmann-Horn died in May 2018 after a long illness.
Revision History
26 July 2018 (ha) Comprehensive update posted live
31 July 2014 (me) Comprehensive update posted live
28 April 2009 (me) Comprehensive updated posted live
4 March 2008 (cd) Revision: mutation scanning for CACNA1S no longer available on a clinical basis
4 August 2006 (me) Comprehensive update posted live
19 May 2004 (me) Comprehensive update posted live
30 April 2002 (me) Review posted live
20 November 2001 (bf) Original submission