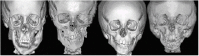
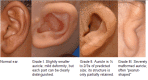
Craniofacial microsomia (CFM) includes a spectrum of malformations primarily involving structures derived from the first and second branchial arches.
No diagnostic criteria have been established; thus, CFM is often a diagnosis of exclusion (see Differential Diagnosis).
Note: Individuals with features of CFM have been classified under a number of different diagnoses. It has not yet been established whether these diagnoses are distinct entities or represent the phenotypic continuum of CFM. In this GeneReview, the term CFM includes all of the following terms (listed from most common to least common):
Embryology
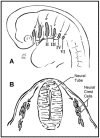
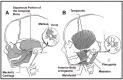
The head and neck originate from six embryonic structures called the pharyngeal apparati (see ), which resemble the branchial apparatus in fish [Moore & Persaud 2003]. Each pharyngeal apparatus comprises a pouch, an arch, a groove, and a membrane. In the fourth week of gestation neural crest cells migrate from the neural tube to begin the development of the pharyngeal arch ectomesenchyme (see ). Each arch has three layers (endoderm, mesenchyme from ectomesenchyme and mesoderm, and ectoderm), which produce the four primordial components: muscle, artery, nerve, and cartilage. The craniofacial structures most commonly affected in CFM develop from the first and second pharyngeal (branchial) arches (see , , , and ).
A. Lateral view of the six embryonic pharyngeal arches (I-VI) B. Coronal view through the first pharyngeal arch showing the migration of cells from the neural tube to the neural crest where they will develop into the head, neck, and body
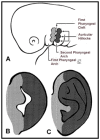
Origin of the external ear structures from the first and second pharyngeal arches Adapted from Sze et al [2002], published with permission from AJR Am J Roentgenol
Bones (A) and muscles (B) derived from the first pharyngeal arch Adapted from Sze et al [2002], published with permission from AJR Am J Roentgenol
Bones (A) and muscles (B) derived from the second pharyngeal arch Adapted from Sze et al [2002], published with permission from AJR Am J Roentgenol
The spectrum of anomalies involved in CFM may result from an embryonic "developmental field" functioning as a unit that responds in a similar manner to different insults such as chromosome abnormalities, mutation in a single gene, vascular disruption, and teratogens [Opitz 1985, Opitz & Lewin 1987, Cousley & Wilson 1992]. In support of this theory, CFM is causally heterogeneous (see Causes). In addition, malformations identical to CFM are observed in different vertebrate species, suggesting a developmental commonality.
The clinical findings in individuals with craniofacial microsomia can overlap with those observed in syndromes, developmental anomaly associations, and sequences. Examples include:
VATER (expanded to VACTERL: vertebral anomalies, anal atresia, cardiac anomalies, tracheoesophageal atresia, renal anomalies, and limb anomalies);
CHARGE (coloboma, heart, atresia choanae, retardation of growth and development, and genitourinary and ear anomalies);
MURCS (variable developmental anomalies of the müllerian ducts, unilateral renal, cervicothoracic, and somite structures or their derivatives); and
OEIS (omphalocele, exstrophy of the cloaca, imperforate anus, and spinal anomalies).
This overlap has led investigators to hypothesize that these conditions may represent developmental abnormalities which result in anomalies that may be a part of a broad spectrum, such as the axial mesodermal dysplasia spectrum [Hartsfield 2007].
Differential Diagnosis of CFM
As discussed in Embryology, the clinical features observed in individuals with CFM can also be associated with syndromes, developmental anomaly associations, and sequences. In this review, the diagnosis of CFM is only considered for those individuals who do not have features unique to these other conditions. Unlike CFM, individuals with the diagnoses listed below (in alphabetical order) typically have symmetric facial malformations. The similarities among the conditions are also illustrated in Table 2.
Auriculocondylar syndrome (OMIM 602483) is typically an autosomal dominant condition with variable mandibular hypoplasia frequently with temporomandibular joint ankylosis, cleft palate, and distinctive question mark appearance of the ears due to separation of the lobule from the external ear. Although findings are typically bilateral and symmetric, the occasional presence of preauricular tags and mandibular hypoplasia can overlap with CFM. Additional features include hearing loss, prominent cheeks, and microstomia. Heterozygous pathogenic variants have been noted in two genes in the endothelin receptor pathway, PLCB4 and GNAI3 [Rieder et al 2012].
Bixler syndrome (hypertelorism-microtia-clefting) (OMIM 239800) is a rare condition with presumed autosomal recessive inheritance based on recurrences in sibs and parental consanguinity. Clinical features include: hypertelorism, cleft lip and palate, broad nasal tip, and microtia. Etiology is not known [Amiel et al 2001].
Branchiootorenal (BOR) syndrome is characterized by malformations of the outer, middle, and inner ear associated with conductive, sensorineural, or mixed hearing impairment, branchial fistulae and cysts, and renal malformations. The diagnosis is based on clinical findings. A heterozygous pathogenic variant in any one of three genes is known to cause BOR: EYA1 (locus name: BOR1) (approximately 40% of affected individuals), SIX5 (locus name: BOR2) in 5%, and SIX1 in 4%. Inheritance is autosomal dominant.
CHARGE syndrome is a mnemonic for coloboma, heart defects, choanal atresia, retarded growth and development, genital abnormalities, and ear anomalies. CHARGE syndrome typically involves unilateral or bilateral coloboma of the iris, retina-choroid, and/or disc, with or without microphthalmos; unilateral or bilateral choanal atresia or stenosis; cranial nerve dysfunction; abnormal ears (including abnormal external ear shape, middle ear anatomy, and inner ear findings including Mondini defect of the cochlea, and absent or hypoplastic semicircular canals); cryptorchidism in males and hypogonadotropic hypogonadism in males and females; developmental delay; cardiovascular malformation; and growth deficiency. Orofacial clefts and tracheoesophageal fistula are seen in a subset of individuals. A heterozygous pathogenic variant in CHD7 is identified in 65%-70% of individuals with CHARGE syndrome. For those with a CHD7 pathogenic variant, inheritance is autosomal dominant.
Hemifacial myohyperplasia sequence (OMIM 606773) is a rare craniofacial condition characterized by unilateral hypertrophy of the facial muscles and ipsilateral hypoplasia of the facial bones of unknown cause. The facial asymmetry is progressive after birth [Pereira-Perdomo et al 2010].
Mandibulofacial dysostosis with microcephaly is characterized by malar and mandibular hypoplasia; congenital- or postnatal-onset microcephaly; malformations of the outer ear, auditory canal, and/or middle ear (ossicles and semi-circular canals) with hearing loss; and facial features that include: prominent glabella, broad nasal bridge, bulbous nasal tip, and everted lower lip. Individuals may also have cleft palate, choanal atresia, and facial asymmetry. Intellectual disability is a prominent feature. Extracranial malformations include: esophageal atresia, congenital heart disease, and thumb abnormalities. Presence of a heterozygous pathogenic variant in EFTUD2 is causative. Inheritance is autosomal dominant [Lines et al 2012, Lehalle et al 2014]. In one patient with a de novo heterozygous pathogenic variant in EFTUD2, clinical features included a unilateral epibulbar dermoid similar to findings seen in CFM [Luquetti et al 2013].
Miller syndrome (postaxial acrofacial dysostosis) (OMIM 263750) is characterized by postaxial limb deficiency (hypoplasia, syndactyly, absence of postaxial digits [the 5th digits and in some cases the 4th and 3rd digits], and ulnar hypoplasia that causes shortening of the forearms) and distinctive facial features (malar hypoplasia, micrognathia, cleft palate, rarely cleft lip, small "cup-shaped" ears, and colobomas and/or ectropion [drooping] of the lower eyelids). Miller syndrome is rare. Biallelic pathogenic variants in DHODH are causative. Inheritance is autosomal recessive [Chrzanowska & Fryns 1993].
Nager syndrome (preaxial acrofacial dysostosis) (OMIM 154400) is characterized by preaxial limb anomalies (hypoplasia or absence of radius, hypoplasia/absence of the thumbs, triphalangeal thumbs, radioulnar synostosis) and facial anomalies (malar hypoplasia with downward slanting palpebral fissures, lower eyelid coloboma, severe micrognathia). Nager syndrome is rare, most cases are simplex (i.e., a single occurrence in a family); however, families with autosomal dominant and autosomal recessive inheritance have been reported [Hecht et al 1987, Bonthron et al 1993]. Heterozygous pathogenic variants in SF3B4 have been reported in 58%-64% of individuals with clinical features of Nager syndrome [Bernier et al 2012, Czeschik et al 2013, Petit et al 2014].
Oculoauriculofrontonasal syndrome (OAFNS) (OMIM 601452) is a rare craniofacial condition with features of both frontonasal dysplasia and oculo-auriculo-vertebral spectrum [Gabbett et al 2008]. Characteristic findings include: ocular hypertelorism, notched or bifid nasal tip, microtia, ear and facial skin tags, eyelid colobomas, epibulbar dermoids, mandibular hypoplasia, and facial asymmetry. Extracranial findings are similar to those noted in individuals with CFM and include: cardiac (44%), renal (23%), and vertebral segmentation (37%). Cranial CT imaging showed distinctive nasal ossification in five of seven individuals studied [Evans et al 2013]. Most cases are simplex (i.e., a single occurrence in a family) and genetic cause is not known.
Parry Romberg syndrome (progressive hemifacial atrophy) (OMIM 141300) is characterized by slowly progressive unilateral atrophy of the skin and soft tissues of the face [Grippaudo et al 2004]. It is more common in females than in males. Initial facial changes usually involve the tissues above the maxilla or near the nasolabial fold and later progress to involve the areas around the eye, lower face, and neck. The tongue and gums may also atrophy. The skin overlying affected areas may become darkly pigmented. Neurologic abnormalities include seizures and trigeminal neuralgia. In Parry-Romberg syndrome atrophy typically begins between ages five and 15 years, whereas in CFM the facial malformations are congenital (i.e., present at birth).
Townes-Brocks syndrome (TBS) is characterized by a triad of clinical findings: imperforate anus, dysplastic ears (overfolded superior helices andpreauricular tags) frequently associated with sensorineural and/or conductive hearing impairment, and thumb malformations (triphalangeal thumbs, duplication of the thumb, and rarely hypoplasia of the thumbs). Associated findings include: renal impairment, including end-stage renal disease, may occur with or without structural abnormalities (mild malrotation, ectopia, horseshoe kidney, renal hypoplasia, polycystic kidneys, vesicoureteral reflux), congenital heart disease, foot malformations, and genitourinary malformations. Intellectual disability occurs in approximately 10% of individuals. Most individuals with CFM do not have thumb and/or anal malformations; however, the overlap of the ear, renal, and cardiac findings in CFM and TBS is significant. Presence of a heterozygous pathogenic variant in SALL1 is causative. Inheritance is autosomal dominant.
At least three individuals with predominant features of CFM had mutation of SALL1 [Kohlhase et al 1999, Keegan et al 2001, Kosaki et al 2007]; however, each also had anterior displacement of the anus and/or preaxial polydactyly.
Treacher Collins syndrome (TCS) is characterized by malar (zygoma) and mandibular hypoplasia, external ear abnormalities (microtia), coloboma of the lower eyelid with deficient eyelashes, and a distribution of the scalp hair in the preauricular region (i.e., sideburn). An estimated 40%-50% of individuals with TCS have conductive hearing loss resulting from abnormalities/hypoplasia of middle ear structures. Extracranial malformations are rare and intellect is typically normal. Mutation in one of three genes – TCOF1 (78%-93%); POLR1C and POLR1D (8%) – is causative. Inheritance is most often autosomal dominant (TCOF1 and POLR1D) with rare reports of autosomal recessive inheritance as a result of biallelic pathogenic variants in POLR1C or POLR1D [Schaefer et al 2014].
Of note, one individual with a diagnosis of oculo-auriculo-vertebral spectrum (unilateral microtia, aural atresia, and unilateral mandibular hypoplasia with an absent zygomatic arch) had a TCOF1 missense variant (1084G>A) resulting in the substitution Ala362Thr [Su et al 2007].
In another study, two individuals diagnosed with Goldenhar syndrome had silent (Glu621Glu; Gln54Gln) TCOF1 sequence variants not noted in 150 controls [Splendore et al 2002].
Table 2.
Comparison of Phenotypic Characteristics Among Conditions in the Differential Diagnosis of CFM
View in own window
| Condition | Gene(s) | Phenotypic Characteristics |
|---|
Maxillary
&/or
mandibular
hypoplasia | Facial
asymmetry | Microtia | Facial
/ ear
tags | Vertebral
segmentation
anomalies | Cardiac
anomalies | Renal
anomalies |
|---|
| Auriculocondylar syndrome 1 |
PLCB4, GNAI3
| X | X | X | | | | |
| Axial mesodermal dysplasia complex 2 | Unknown | X | X | X | X | X | X | X |
| Bixler (hypertelorism -microtia-clefting) syndrome 3 | Unknown | | | X | | | | |
|
Branchiootorenal spectrum disorders
|
EYA1, SIX1, SIX5
| X | X | X | X | | | X |
| Cat-eye syndrome 4 | Chromosome 22 marker | X | X | X | X | X | X | X |
|
CHARGE syndrome
|
CHD7
| X | X | X | | | X | X |
| Hemifacial myohyperplasia 5 | Unknown | X | X | | | | | |
| Mandibulofacial dysostosis with microcephaly 6 |
EFTUD2
| X | X | X | X | X | X | |
| Miller syndrome 7 |
DHODH
| X | | X | | | | |
| Nager syndrome 8 |
SF3B4
| X | | X | X | | X | X |
| Parry Romberg syndrome 9 | Unknown | X | X | | | | | |
|
Townes-Brocks syndrome
| SALL1 (SALL4) | X | X | X | X | X | X | X |
|
Treacher Collins syndrome
|
TCOF1, POLR1C, POLR1D
| X | | X | | | | |
| VACTERL association 10 | Unknown | | | | | X | X | X |
X indicates presence of phenotypic characteristic.
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.