Molecular Pathogenesis
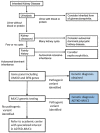
Mucin-1, encoded by MUC1, a membrane-anchored mucoprotein found in breast, respiratory tract, sebaceous gland, salivary gland, and kidney tubules, is important in signal transduction. Mucin-1 provides (among other functions) a protective barrier that prevents pathogens from accessing the cell surface.
MUC1 pathogenic variants causing ADTKD-MUC1 always result in the creation of the same frameshift protein (MUC1fs) (see MUC1-specific
laboratory technical considerations). This protein accumulates within the endoplasmic reticulum Golgi intermediate compartment (ERGIC) [Dvela-Levitt et al 2019]. In individuals with ADTKD-MUC1, MUC1fs is found in all cell types normally expressing MUC1 (including breast, gastric mucosa, and lung); however, clinical manifestations of disease only occur in the kidney, where its deposition leads to accelerated apoptosis [Staubach et al 2018], tubular cell death, nephron dropout, and progressive chronic kidney disease.
Mechanism of disease causation. Gain of function. All affected individuals harbor pathogenic variants creating the same MUC1fs protein; no person with this disorder has had truncating variants or other variants that would result in loss of function [Olinger et al 2020].
MUC1-specific laboratory technical considerations
MUC1 contains within the coding region a domain of multiple variable-number tandem repeats (VNTRs), called the "VNTR domain."
Each VNTR is an oligonucleotide comprising 60 nucleotides (also referred to as a "60-mer"). Each 60-nucleotide repeat includes a sequence of seven cytosines (also called "a 7-cytosine tract").
The normal number of 60-mer VNTRs comprising a VNTR domain ranges from 20 to 125.
In most MUC1 disease-causing variants an additional nucleotide inserted within a seven-cytosine sequence causes a frameshift that encodes an abnormal MUC1 protein called a "frameshift protein," known as MUC1fs. The most commonly inserted nucleotide is cytosine; less commonly, it is a different nucleotide (e.g., adenosine).
The additional nucleotide can either be within the VNTR [Kirby et al 2013] or immediately before the VNTR [Yamamoto et al 2017]; however, in approximately 95% of cases, MUC1fs results from insertion of a cytosine in the seven-cytosine sequence within the VNTR [Authors, personal observations based on 196 families with ADTKD-MUC1]. See .
Illustration of the mechanism by which the MUC1fs protein is encoded within a 60-mer block of repeated sequence that forms a MUC1 variable-number tandem repeat (VNTR). In this figure of a hypothetic coding sequence of ten 60-mer blocks of repeated sequence, (more...)
Clinical testing issues. Due to the high guanosine-cytosine content and the repetitive nature of the 60-mer VNTR sequences, variants within a VNTR cannot be identified by commonly used sequencing methods (e.g., exome sequencing, Sanger sequencing).
The CLIA-certified Broad Institute of Harvard and MIT laboratory performs targeted analysis for the common MUC1 frameshift variant (i.e., insertion of a cytosine within the 7-cytosine sequence) and the less common frameshift variant (insertion of an adenine in a 7-cytosine sequence). (See Author Notes regarding testing available at no cost to families with autosomal dominant tubulointerstitial kidney disease.)
Other published test methods for detecting variants not identified by standard genetic sequencing techniques include the following:
Long-read single-molecule real-time (SMRT) sequencing can identify the insertion of a cytosine within the seven-cytosine sequence as well as many (but not all) other disease-causing
MUC1 variants. This method also determines the length and structure of the
MUC1 VNTR and the exact position of the variant within the VNTR [
Wenzel et al 2018,
Knaup et al 2018,
Wang et al 2020].
Illumina-based sequencing of
MUC1 VNTR amplicons can identify the cytosine insertion as well as many (but not all) other disease-causing variants [
Živná et al 2018].
Table 4.
Notable MUC1 Pathogenic Variants
View in own window
Reference
Sequences | DNA Nucleotide Change | Predicted
Protein Change | Comment [Reference] |
|---|
NM_002456.5
NP_002447.4
| c.253C>T | p.Gln85Ter | Presence of an adenine in the 7-cytosine sequence w/in a VNTR results in encoding of MUC1fs protein.1 Exact position of this VNTR w/in the VNTR domain is unknown & may vary between families [Živná et al 2018]. |
|
NC_000001.10
| g.(155160963_155162030)insC 2 | Frameshift resulting in early termination | Insertion of a cytosine w/in 7-cytosine sequence in a VNTR is most common variant encoding MUC1fs protein. Exact position of this VNTR in VNTR domain is unknown & may vary between families [Kirby et al 2013]. |
| g. 155161911_155161912delAG | Frameshift resulting in early termination | Occurs before VNTR domain but nonetheless encodes MUC1fs protein [Yamamoto et al 2017] |
| g.(155160963_155162030)insG 2 | Frameshift resulting in early termination | Variant (not detected by clinically available MUC1 genetic testing) encodes MUC1fs protein [Živná a et al 2018]. |
| g.(155160963_155162030)delinsAT 2 | Frameshift resulting in early termination | Variant (not detected by clinically available MUC1 genetic testing) encodes the MUC1fs protein [Živná et al 2018]. |
| g.(155160963_155162030)dupGCCGGCCCCGGGTCC 2 | Frameshift resulting in early termination | Variant (not detected by clinically available MUC1 genetic testing) encodes MUC1fs protein [Živná et al 2018]. |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
VNTR = variable-number tandem repeat
- 1.
illustrates a hypothetic example.
- 2.
Variant designation that does not conform to current naming conventions