Summary
Clinical characteristics.
Spinocerebellar ataxia type 17 (SCA17) is characterized by ataxia, dementia, and involuntary movements, including chorea and dystonia. Psychiatric symptoms, pyramidal signs, and rigidity are common. The age of onset ranges from three to 55 years. Individuals with full-penetrance alleles develop neurologic and/or psychiatric symptoms by age 50 years. Ataxia and psychiatric abnormalities are frequently the initial findings, followed by involuntary movement, parkinsonism, dementia, and pyramidal signs. Brain MRI shows variable atrophy of the cerebrum, brain stem, and cerebellum. The clinical features correlate with the length of the polyglutamine expansion but are not absolutely predictive of the clinical course.
Diagnosis/testing.
The diagnosis of SCA17 is established in a proband by identification of an abnormal CAG/CAA repeat expansion in TBP. Affected individuals usually have more than 41 repeats. The CAA and CAG codons both encode glutamine residues resulting in a pathogenic polyglutamine expansion.
Management.
Treatment of manifestations: Psychotropic medications for psychiatric issues, anti-seizure medication for seizures (ASM); botulinum toxin injections for dystonia; adaptation of the environment to accommodate dementia.
Prevention of secondary complications: Side effects of psychotropic medications and ASMs may require total or intermittent discontinuation of the treatment or reduction in dose.
Surveillance: Annual or semiannual evaluation by a neurologist or more frequently if symptoms are progressing rapidly.
Agents/circumstances to avoid: Sedative/hypnotic agents, such as ethanol or certain medications, may exacerbate incoordination.
Genetic counseling.
SCA17 is inherited in an autosomal dominant manner. Offspring of affected individuals are at a 50% risk of inheriting the expanded TBP allele. The age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be precisely predicted by family history or size of expansion. Prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible if the diagnosis has been established in an affected family member by molecular genetic testing.
Diagnosis
Suggestive Findings
Spinocerebellar ataxia type 17 (SCA17) should be suspected in individuals with the following:
- Ataxia
- Dementia
- Involuntary movements – e.g., chorea and dystonia (blepharospasm, torticollis, writer's cramp, foot dystonia)
- Psychiatric symptoms
Establishing the Diagnosis
The diagnosis of SCA17 is established in a proband by identification of a heterozygous pathogenic variant of CAG (and sometimes CAA) repeats in TBP by molecular genetic testing (see Table 1). Because both codons CAA and CAG encode glutamine residues, the resulting proteins will have variable tracts of glutamine residues.
Allele sizes. The structure of the repeat sequence in a normal, stably transmitted allele is variable but typically consists of series of CAG repeats interrupted by CAA repeats – e.g., (CAG)3 (CAA)3 (CAG)9 CAA CAG CAA (CAG)16 CAA CAG. Allele size (sometimes expressed as length or number of repeats) is determined by counting all triplet repeats; the total number of CAG/CAA repeats in the example above would be 36, which would translate to 36 contiguous glutamine residues in the protein.
- Normal alleles. 25 to 40 CAG/CAA repeats
- Mutable normal alleles. Not reported to date
- Reduced-penetrance alleles. 41 to 48 CAG/CAA repeats. An individual with an allele in this range may or may not develop symptoms. The significance of alleles of 41 and 44 repeats is particularly controversial because penetrance is estimated to be 50%, making genotype-phenotype correlations difficult. One symptomatic individual having 41 repeats and four symptomatic persons having 42 repeats have been reported [Nanda et al 2007, Nolte et al 2010]. Heterozygous STUB1 pathogenic variants may contribute to the variable penetrance of short TBP CAG/CAA expansions. A phenotype consistent with SCA17 has been reported in individuals who are double heterozygotes for a TBP allele with 41 to 46 CAG/CAA repeats and a STUB1 pathogenic variant [Magri et al 2022, Reis et al 2022]. This novel pathomechanism is referred to as digenic TBP/STUB1-related SCA17 or SCA17-DI.
- Full-penetrance alleles. 49 or greater CAG/CAA repeats. The largest repeat size reported to date is 66 [Maltecca et al 2003].
CAA CAG CAA interruption. The CAA CAG CAA interruption between (CAG)x and (CAG)y is present in all expanded alleles that are stably transmitted (i.e., the allele size is unchanged during meiosis).
The CAA CAG CAA interruption between (CAG)x and (CAG)y was absent in two families with allele size instability (i.e., change in allele size) during transmission [Zühlke et al 2001, Maltecca et al 2003]. Thus, loss of this interruption may be a prerequisite of instability in SCA17 as in other disorders caused by repeat expansions [Maltecca et al 2003, Zühlke et al 2003b, Zühlke et al 2005].
Molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Targeted analysis for a heterozygous CAG/CAA repeat number in TBP should be performed first.
- A multigene panel that includes TBP CAG/CAA repeat analysis and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in Spinocerebellar Ataxia Type 17
Clinical Characteristics
Clinical Description
Spinocerebellar ataxia type 17 (SCA17) is characterized by ataxia (95%), dementia (~90%), and involuntary movements (~70%), including chorea and dystonia (blepharospasm, torticollis, writer's cramp, foot dystonia) [Cellini et al 2004, Toyoshima et al 2004]. Psychiatric symptoms, pyramidal signs, and rigidity are common.
Onset ranges from age three to 75 years (mean: 34.6 years) [Stevanin & Brice 2008]. All individuals with full-penetrance alleles develop neurologic and/or psychiatric symptoms by age 50 years [Koide et al 1999; Fujigasaki et al 2001; Nakamura et al 2001; Zühlke et al 2001; Silveira et al 2002; Maltecca et al 2003; Stevanin et al 2003; Zühlke et al 2003b; Bauer et al 2004; Hagenah et al 2004; Oda et al 2004; Toyoshima & Takahashi 2018; Toyoshima, personal observation].
Although the disease course is variable, ataxia and psychiatric abnormalities are frequently the initial findings, followed by involuntary movement, parkinsonism, dementia, and pyramidal signs.
Brain MRI shows variable atrophy of the cerebrum, brain stem, and cerebellum (Figure 1). Most people present with cerebellar atrophy. The age of the individual and the length of CAG/CAA repeat influence the degree of atrophy. For example, in older individuals – even those with a small full-penetrance allele – severe atrophy is present on brain MRI. High-intensity T2-weighted images and selective atrophy on caudate nucleus are not observed. Some correlation of region of brain atrophy with clinical characteristics is seen [Lasek et al 2006].
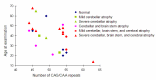
Figure 1.
Number of CAG/CAA repeats versus age of individuals with SCA17
Neuropathology. The brain shows atrophy of the striatum (more apparent in the caudate nucleus) and cerebellum. Histologically, neuronal loss is observed in the striatum and Purkinje cell layer. Loss of cerebral cortical neurons is seen in some individuals.
Immunohistochemistry for the expanded polyglutamine (polyQ) tracts shows diffuse labeling of the neuronal nucleoplasm.
Note: Intranuclear inclusions are a much less common finding than diffuse labeling. No labeling is detectable in the cytoplasm or in the neuropil. Glial cell involvement is occasionally seen.
In individuals who are homozygous for an expanded allele in the full-penetrance range, nuclear polyQ pathology involves other CNS regions including the cerebral cortex, thalamus, and brain stem [Toyoshima et al 2004]. The abundant nuclear accumulation of polyQ in the cerebral cortices and subcortical nuclei (e.g., dorsomedian thalamic nucleus) are possibly associated with the prominent cognitive and behavioral decline in affected individuals.
Genotype-Phenotype Correlations
Heterozygotes
Clinical features. The length of the CAG/CAA repeat in TBP correlates with the clinical features based on data available from 52 individuals (50 from the literature and 2 unreported) (Table 2, Figure 2). As the information reported in the literature was incomplete, the frequencies listed for symptom occurrence may be underestimated [Koide et al 1999, Fujigasaki et al 2001, Nakamura et al 2001, Zühlke et al 2001, Silveira et al 2002, Maltecca et al 2003, Rolfs et al 2003, Stevanin et al 2003, Zühlke et al 2003a, Bauer et al 2004, Hagenah et al 2004, Oda et al 2004]. Of note is the high proportion of individuals with psychiatric symptoms and chorea.
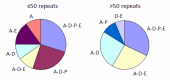
Figure 2.
The clinical features in SCA17 depend on the length of CAG/CAA repeats. The clinical features reported in affected individuals are denoted by letter. For example, A-E denotes affected individuals who have ataxia with parkinsonism. A = ataxia
- CAG/CAA repeat size from 41 to 50. More than 75% of individuals have intellectual deterioration; in some individuals, intellectual problems and involuntary movements are the only signs. Psychiatric symptoms or dementia, parkinsonism, and chorea – a clinical constellation resembling Huntington disease – are more frequently observed in individuals with CAG/CAA repeats in this range than in individuals with larger repeats [Stevanin et al 2003, Bauer et al 2004, Toyoshima et al 2004].
- CAG/CAA repeat size from 43 to 47. Individuals with an allele of 43-47 repeats tend to have a parkinsonian phenotype [Kim et al 2009, Chen et al 2010].
- CAG/CAA repeat size from 50 to 60. All individuals have ataxia and 75% have reduced intellectual function. Pyramidal signs (e.g., increased deep tendon reflexes) and dystonia are more common than in those with smaller repeats.
- CAG/CAA repeat size greater than 60. Two individuals with repeats in this size range have been reported. The largest CAG/CAA repeat is 66 repeats, observed in one familial case [Maltecca et al 2003]. The child developed gait disturbance at age three years followed by spasticity, dementia, and psychiatric symptoms. The other child, who had a de novo CAG repeat expansion of 63 repeats, developed ataxia and intellectual deterioration at age six years followed later by spasticity [Koide et al 1999]. Brain MRI showed severe atrophy in the cerebrum, cerebellum, and brain stem.
Table 2.
Frequency of Clinical Features in Spinocerebellar Ataxia Type 17 Correlated with TBP Repeat Size
Homozygotes
Four homozygous individuals and one compound heterozygous individual have been reported [Zühlke et al 2003b, Oda et al 2004, Toyoshima et al 2004, Hire et al 2011]. Four individuals who were homozygous for 47 or 48 CAG/CAA repeats had onset in the fourth decade, not unlike the age of onset predicted for heterozygotes [Zühlke et al 2003b, Toyoshima et al 2004]. Their symptoms were severe and rapidly progressive, and in one individual differed from those of his parents, suggesting that the presence of two expanded alleles influences the severity and rate of progression of symptoms.
Penetrance
The penetrance of alleles of 41-44 repeats is estimated at 50% and the penetrance of alleles of 45-48 repeats is estimated at greater than 80% [Toyoshima et al 2004].
- Individuals with 41 CAA/CAG repeats developed ataxia and mild dementia [Nanda et al 2007, Doherty et al 2014]. Two individuals who had 41 repeats developed parkinsonism and chorea [Herrema et al 2014, Park et al 2016]. One individual with 41 repeats developed late-onset chorea and psychiatric symptoms [Alibardi et al 2014].
- Four individuals with 42 CAG/CAA repeats developed a relatively benign phenotype consisting of mild gait ataxia, dysarthria, and dysdiadochokinesia [Nolte et al 2010].
- An individual with 43 CAG/CAA repeats developed ataxia with dementia at age 52 years [Silveira et al 2002]; six individuals diagnosed with parkinsonism were found to have 43 CAG/CAA repeats [Kim et al 2009]. An individual with 43 repeats developed severe dementia [Nielsen et al 2012].
- An individual with 46 CAG/CAA repeats developed symptoms at age 75 years, the latest onset observed to date [Wu et al 2005].
- Asymptomatic elderly individuals with 43-49 CAG/CAA repeats have also been reported [Nakamura et al 2001, Zühlke et al 2003a, Oda et al 2004, Zühlke et al 2005, Mariotti et al 2007].
- Heterozygous STUB1 pathogenic variants may contribute to the variable penetrance of TBP alleles with 41 to 46 CAG/CAA repeats. Individuals with 41 to 46 CAG/CAA repeats in TBP and a heterozygous STUB1 pathogenic variant developed a Huntington disease-like phenotype [Magri et al 2022, Reis et al 2022].
Age of onset. The correlation between the size of the CAG/CAA repeat and the age of onset in SCA17 (Figure 3) is not as strong as in other disorders (SCA1, SCA2, SCA3, SCA6, SCA7, Huntington disease, DRPLA, SBMA]) caused by expansion of a polyglutamine tract [Rolfs et al 2003, Toyoshima et al 2004,Toyoshima & Takahashi 2018].

Figure 3.
Correlation between age at onset and length of CAG/CAA repeat in individuals with SCA17 For references, see Penetrance, Age of onset.
Nomenclature
Bauer et al [2004] reported nine individuals with TBP alleles larger than 45 CAG/CAA repeats among 1,712 individuals with Huntington disease-like 2, and observed that CAG/CAA repeat expansions in TBP represented a more common monogenic cause for a Huntington disease-like phenotype than Huntington disease-like 1 [Xiang et al 1998] or Huntington disease-like 2 [Margolis et al 2001]. Therefore, SCA17 is also referred to as Huntington disease-like 4 [Stevanin et al 2003, Schneider et al 2007, Harbo et al 2009].
Anticipation
Instability of the TBP CAG repeat in germline transmission is not clear in SCA17 [Fujigasaki et al 2001, Nakamura et al 2001, Shatunov et al 2004]. CAG repeats in TBP have two distinct configurations, which are differentiated by the absence or presence of CAA trinucleotide repeat interruptions. The basic structure of the allele is (CAG)3 (CAA)3 (CAG)x CAA CAG CAA (CAG)y CAA CAG. If the basic structure is broken (i.e., CAA repeat interruptions are absent), repeat stability may be reduced. In German and Italian families, an absence of CAA interruptions resulting in longer pure tracts of CAG repeats was detected. It is of note that intergenerational instability and anticipation were documented in these families [Zühlke et al 2001, Maltecca et al 2003]. It has been proposed that CAA interruptions may serve as a limiting element for further expansion of CAG repeats in TBP [Gao et al 2008].
The phenomenon termed anticipation, a trend toward an earlier age at onset and more severe disease manifestations in offspring of an affected individual, is infrequently documented in families with SCA17. In addition, because of low penetrance of the intermediate alleles (41-48 repeats), the age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be predicted by family history or size of expansion.
Prevalence
Fewer than 100 families with SCA17 have been reported.
The prevalence of SCA17 in the Japanese population is estimated at 0.47:1,000,000. SCA17 accounts for approximately 0.3% of autosomal dominant SCA [Maruyama et al 2002].
The minimum prevalence of SCA17 in northeast England is 0.16:100,000 [Craig et al 2005].
In a study of the Yugoslav population, none of the 115 individuals with autosomal dominant cerebellar ataxia or simplex cases of adult-onset ataxia had SCA17 [Alendar et al 2004].
The prevalence of SCA17 may be underestimated because some individuals with SCA17 have a phenotype similar to that of Huntington disease.
Genetically Related (Allelic) Disorders
No other phenotypes are known to be associated with germline pathogenic variants in TBP.
Differential Diagnosis
Table 3.
Inherited Conditions to Consider in the Differential Diagnosis of Spinocerebellar Ataxia Type 17 (SCA17)
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with spinocerebellar ataxia type 17 (SCA17), the following evaluations (if not performed as part of the evaluation that led to the diagnosis) are recommended:
- Neuropsychological testing to evaluate for dementia and/or psychiatric disturbance
- Brain MRI to evaluate areas and degree of atrophy
- Neurology consultation, if not completed prior to initial diagnosis
- Consultation with a clinical geneticist and/or genetic counselor
Treatment of Manifestations
Table 4.
Treatment of Manifestations in Individuals with Spinocerebellar Ataxia Type 17
Prevention of Secondary Complications
The side effects of psychotropic medications and ASMs (e.g., depression, sedation, nausea, restlessness, headache, neutropenia, and tardive dyskinesia) can be major secondary complications in persons with SCA17. For some individuals, the side effects of certain therapeutics may be worse than the symptoms of the disease; such individuals may benefit from total or intermittent discontinuation of the treatment or reduction in dose.
Surveillance
Affected individuals should be followed annually or semiannually by a neurologist or more frequently if symptoms are progressing rapidly, as may happen in the advanced stages [Toyoshima et al 2004].
Agents/Circumstances to Avoid
Agents with sedative/hypnotic properties, such as ethanol or certain medications, may markedly increase incoordination.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Spinocerebellar ataxia type 17 (SCA17) is inherited in an autosomal dominant manner.
Note: A novel pathomechanism, referred to as digenic TBP/STUB1-related SCA17 or SCA17-DI, is suggested by individuals with a phenotype consistent with SCA17 who are double heterozygotes for a TBP allele with 41 to 46 CAG/CAA repeats (i.e., an allele in the reduced-penetrance range) and a STUB1 pathogenic variant [Magri et al 2022, Reis et al 2022]. SCA17-DI is not discussed further in this section.
Risk to Family Members
Parents of a proband *
- Approximately 50% of individuals diagnosed with SCA17 have an affected parent.
- A proband with SCA17 may have the disorder as a result of a de novo expansion in TBP. Although 38% of individuals with SCA17 represent simplex cases (i.e., only one affected person in the family), most of these families have not been evaluated sufficiently to determine if the pathogenic variant occurred de novo; therefore, the proportion of cases caused by a de novo expansion is unknown [Koide et al 1999, Shatunov et al 2004, Bech et al 2010].
- Molecular genetic testing is recommended for the parents of a proband with an apparent de novo expansion.
- If an expansion (>40 CAG/CAA repeats) of TBP cannot be detected in the DNA of either parent, possible explanations include a de novo expansion in the proband or, theoretically, germline mosaicism in a parent, or expansion from a mutable normal allele in the parent.
- The family history of some individuals diagnosed with SCA17 may appear to be negative because of failure to recognize the disorder in family members due to its extremely variable phenotype, early death of the parent before the onset of symptoms, late onset of the disease in the affected parent, or reduced penetrance in a parent. Therefore, an apparently negative family history cannot be confirmed unless appropriate clinical evaluation and/or molecular genetic testing has been performed on the parents of the proband.
* Based on the family history of the 59 reported families with affected individuals [Toyoshima & Takahashi 2018]
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If one of the parents of the proband has an expanded TBP allele, the risk to the sibs of inheriting the expanded CAG/CAA allele is 50%. The age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be precisely predicted by family history or size of expansion.
- If an expansion (>40 CAG/CAA repeats) of TBP cannot be detected in the DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the expanded TBP allele but are clinically unaffected, sibs are still presumed to be at increased risk for SCA17 because of the possibility of reduced penetrance in a parent or the theoretic possibilities of parental germline mosaicism or expansion from a mutable normal allele in the parent.
Offspring of a proband
- Each child of an individual with SCA17 has a 50% chance of inheriting the expanded TBP allele.
- The age of onset, severity, specific symptoms, and progression of the disease are variable and cannot be precisely predicted by family history or size of expansion.
- Compared with other SCA subtypes caused by expanded trinucleotide repeats, anticipation is rare in SCA17 because CAA interruptions within the TBP CAG repeat configuration stabilize the repeat in germline transmission (see Anticipation). However, if the proband has a mild phenotype and a short number of repeats (41-43), examination of the CAG repeat configuration to determine if CAA repeat interruptions are present can be useful in assessing the likelihood of severe anticipation in offspring.
Other family members. The risk to other family members depends on the genetic status of the proband's parents: if a parent is affected and/or is known to have an expanded TBP allele, the parent's family members are at risk.
Related Genetic Counseling Issues
Considerations in families with an apparent de novo pathogenic variant. When neither parent of a proband with an autosomal dominant condition has the pathogenic variant identified in the proband or clinical evidence of the disorder, the pathogenic variant is likely de novo. However, non-medical explanations including alternate paternity or maternity (e.g., with assisted reproduction) and undisclosed adoption could also be explored.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected or at risk.
Predictive testing (i.e., testing of asymptomatic at-risk individuals)
- Predictive testing for at-risk relatives is possible once the molecular diagnosis of SCA17 has been confirmed in an affected family member. Such testing is not useful in predicting age of onset, severity, type of symptoms, or rate of progression in asymptomatic individuals.
- Potential consequences of such testing (including, but not limited to, socioeconomic changes and the need for long-term follow up and evaluation arrangements for individuals with a positive test result) as well as the capabilities and limitations of predictive testing should be discussed in the context of formal genetic counseling prior to testing.
Predictive testing in minors (i.e., testing of asymptomatic at-risk individuals younger than age 18 years)
- For asymptomatic minors at risk for adult-onset conditions for which early treatment would have no beneficial effect on disease morbidity and mortality, predictive genetic testing is considered inappropriate, primarily because it negates the autonomy of the child with no compelling benefit. Further, concern exists regarding the potential unhealthy adverse effects that such information may have on family dynamics, the risk of discrimination and stigmatization in the future, and the anxiety that such information may cause.
- For more information, see the National Society of Genetic Counselors position statement on genetic testing of minors for adult-onset conditions and the American Academy of Pediatrics and American College of Medical Genetics and Genomics policy statement: ethical and policy issues in genetic testing and screening of children.
In a family with an established diagnosis of SCA17, it is appropriate to consider testing of symptomatic individuals regardless of age.
Prenatal Testing and Preimplantation Genetic Testing
Once a diagnosis of SCA17 has been established by molecular genetic testing in an affected family member, prenatal and preimplantation genetic testing for SCA17 are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- NCBI Genes and Disease
- Ataxia UKUnited KingdomPhone: 0800 995 6037; +44 (0) 20 7582 1444 (from abroad)Email: [email protected]
- euro-ATAXIA (European Federation of Hereditary Ataxias)United KingdomEmail: [email protected]
- National Ataxia FoundationPhone: 763-553-0020Email: [email protected]
- Spanish Ataxia Federation (FEDAES)SpainPhone: 601 037 982Email: [email protected]
- CoRDS RegistrySanford ResearchPhone: 605-312-6300
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Spinocerebellar Ataxia Type 17: Genes and Databases
Table B.
OMIM Entries for Spinocerebellar Ataxia Type 17 (View All in OMIM)
Molecular Pathogenesis
TATA-box-binding protein (TBP) is an important general transcription initiation factor and is the DNA-binding subunit of RNA polymerase II transcription factor D, the multi-subunit complex crucial for the expression of most genes. TBP has a long tract of glutamines in the N-terminus. This region is thought to modulate the DNA binding activity of the C terminus, which affects the rate of transcription complex formation and initiation of transcription.
Mechanism of disease causation
- Unknown, but the contiguous tract of polygutamine encoded by the pathogenic expanded TBP allele is generally considered to confer a gain of function.
- Because TBP is a fundamental transcription factor expressed ubiquitously in all organs, including the CNS, the question of whether loss of TBP function plays a role in the pathogenesis of spinocerebellar ataxia Type 17 (SCA17) remains to be addressed. In a homozygote, however, no abnormality was observed in growth, and pathologic examination showed no specific changes in the visceral organs [Toyoshima et al 2004]. Taking into consideration the ubiquitous presence of TBP, the selective neuronal degeneration suggests no significant loss of protein function in individuals with SCA17.
TBP-specific laboratory considerations
- The trinucleotide CAG repeats, sometimes interrupted by CAA repeats, are in TBP exon 3. Both CAG and CAA trinucleotide repeats encode the amino acid glutamine resulting in a contiguous tract of polyglutamine at the protein level.
- The molecular genetic testing laboratory most commonly reports the number (length) of trinucleotide repeats for each TBP allele; that number is the total of both CAG and CAA repeats of a TBP allele. The alleles may be annotated using the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org) for alleles with trinucleotide sequences (www.hgvs.org/mutnomen/recs-DNA.html#var; see Repeated sequences).
Chapter Notes
Revision History
- 28 July 2022 (sw) Revision: novel pathomechanism suggested: digenic TBP/STUB1-related SCA17
- 12 September 2019 (sw) Comprehensive update posted live
- 17 May 2012 (me) Comprehensive update posted live
- 1 August 2007 (me) Comprehensive update posted live
- 29 March 2005 (me) Review posted live
- 24 August 2004 (yt) Original submission
References
Published Guidelines / Consensus Statements
- Committee on Bioethics, Committee on Genetics, and American College of Medical Genetics and Genomics Social, Ethical, Legal Issues Committee. Ethical and policy issues in genetic testing and screening of children. Available online. 2013. Accessed 12-29-22.
- National Society of Genetic Counselors. Position statement on genetic testing of minors for adult-onset conditions. Available online. 2018. Accessed 12-29-22.
Literature Cited
- Alendar A, Euljkovic B, Savic D, Djarmati A, Keckarevic M, Ristic A, Dragasevic N, Kosic V, Romac S. Spinocerebellar ataxia type 17 in the Yugoslav population. Acta Neurol Scand. 2004;109:185–7. [PubMed: 14763955]
- Alibardi A, Squitieri F, Fattapposta F, Missori P, Pierelli F, Trompetto C, Curra A. Psychiatric onset and late chorea in a patient with 41 CAG repeats in the TATA-binding protein gene. Parkinsonism Relat Disord. 2014;20:678–9. [PubMed: 24698056]
- Bauer P, Laccone F, Rolfs A, Wullner U, Bosch S, Peters H, Liebscher S, Scheible M, Epplen JT, Weber BH, Holinski-Feder E, Weirich-Schwaiger H, Morris-Rosendahl DJ, Andrich J, Riess O. Trinucleotide repeat expansion in SCA17/TBP in white patients with Huntington's disease-like phenotype. J Med Genet. 2004;41:230–2. [PMC free article: PMC1735701] [PubMed: 14985389]
- Bech S, Petersen T, Norremolle A, Gjedde A, Ehlers L, Eiberg H, Hjermind LE, Hasholt L, Lundorf E, Nielsen JE. Huntington’s disease-like and ataxia syndromes: identification of a family with a de novo SCA17/TBP mutation. Parkinsonism Relat Disord. 2010;16:12–5. [PubMed: 19595623]
- Cellini E, Forleo P, Nacmias B, Tedde A, Bagnoli S, Piacentini S, Sorbi S. Spinocerebellar ataxia type 17 repeat in patients with Huntington's disease-like and ataxia. Ann Neurol. 2004;56:163–4. [PubMed: 15236416]
- Chen CM, Lee LC, Soong BW, Fung HC, Hsu WC, Lin PY, Huang HJ, Chen FL, Lin CY, Lee-Chen GJ, Wu YR. SCA17 repeat expansion: mildly expanded CAG/CAA repeat alleles in neurological disorders and the functional implications. Clin Chim Acta. 2010;411:375–80. [PubMed: 20004653]
- Craig K, Keers SM, Walls TJ, Curtis A, Chinnery PF. Minimum prevalence of spinocerebellar ataxia 17 in the north east of England. J Neurol Sci. 2005;239:105–9. [PubMed: 16223509]
- Doherty KM, Warner TT, Lees AJ. Late onset ataxia: MSA-C or SCA 17? A gene penetrance dilemma. Mov Disord. 2014;29:36–8. [PubMed: 24343983]
- Fujigasaki H, Martin JJ, De Deyn PP, Camuzat A, Deffond D, Stevanin G, Dermaut B, Van Broeckhoven C, Durr A, Brice A. CAG repeat expansion in the TATA box-binding protein gene causes autosomal dominant cerebellar ataxia. Brain. 2001;124:1939–47. [PubMed: 11571212]
- Gao R, Matsuura T, Coolbaugh M, Zülke C, Nakamura K, Rasmussen A, Siciliano MJ, Ashizawa T, Lin X. Instability of expanded CAG/CAA repeats in spinocerebellar ataxia type 17. Eur J Hum Genet. 2008;16:215–22. [PubMed: 18043721]
- Hagenah JM, Zühlke C, Hellenbroich Y, Heide W, Klein C. Focal dystonia as a presenting sign of spinocerebellar ataxia 17. Mov Disord. 2004;19:217–20. [PubMed: 14978680]
- Harbo HF, Finsterer J, Baets J, Van Broeckhoven C, Di Donato S, Fontaine B, De Jonghe P, Lossos A, Lynch T, Mariotti C, Schöls L, Spinazzola A, Szolnoki Z, Tabrizi SJ, Tallaksen C, Zeviani M, Burgunder JM, Gasser T. EFNS guidelines on the molecular diagnosis of neurogenetic disorders: general issues, Huntington's disease, Parkinson's disease and dystonias. Eur J Neurol. 2009;16:777–85. [PubMed: 19469830]
- Herrema H, Mikkelsen T, Robin A, LeWitt P, Sidiropoulos C. SCA 17 phenotype with intermediate triplet repeat number. J Neurol Sci. 2014;345:269–70. [PubMed: 25091452]
- Hire RR, Katrak SM, Vadya S, Radhakrishnan K, Seshadri M. Spinocerebellar ataxia type 17 in Indian patients: two cases of homozygous expansions. Clin Genet. 2011;80:472–7. [PubMed: 21108634]
- Kim JY, Kim SY, Kim JM, Kim YK, Yoon KY, Kim JY. Spinocerebellar ataxia type 17 mutation as a causative and susceptibility gene in parkinsonism. Neurology. 2009;72:1385–9. [PubMed: 19380697]
- Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, Saito M, Yamada M, Takahashi H, Tsuji S. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease? Hum Mol Genet. 1999;8:2047–53. [PubMed: 10484774]
- Laplanche JL, Hachimi KH, Durieux I, Thuillet P, Defebvre L, Delasnerie-Lauprêtre N, Peoc'h K, Foncin JF, Destée A. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain. 1999;122:2375–86. [PubMed: 10581230]
- Lasek K, Lencer R, Gaser C, Hagenah J, Walter U, Wolters A, Kock N, Steinlechner S, Nagel M, Zühlke C, Nitschke MF, Brockmann K, Klein C, Rolfs A, Binkofski F. Morphological basis for the spectrum of clinical deficits in spinocerebellar ataxia 17 (SCA17). Brain. 2006;129:2341–52. [PubMed: 16760196]
- Magri S, Nanetti L, Gellera C, Sarto E, Rizzo E, Mongelli A, Ricci B, Fancellu R, Sambati L, Cortelli P, Brusco A, Bruzzone MG, Mariotti C, Di Bella D, Taroni F. Digenic inheritance of STUB1 variants and TBP polyglutamine expansions explains the incomplete penetrance of SCA17 and SCA48. Genet Med. 2022;24:29–40. [PubMed: 34906452]
- Maltecca F, Filla A, Castaldo I, Coppola G, Fragassi NA, Carella M, Bruni A, Cocozza S, Casari G, Servadio A, De Michele G. Intergenerational instability and marked anticipation in SCA-17. Neurology. 2003;61:1441–3. [PubMed: 14638975]
- Margolis RL, O'Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, Callahan C, Hwang HS, Troncoso JC, Ross CA. A disorder similar to Huntington's disease is associated with a novel CAG repeat expansion. Ann Neurol. 2001;50:373–80. [PubMed: 11761463]
- Mariotti C, Alpini D, Fancellu R, Soliveri P, Grisoli M, Ravaglia S, Lovati C, Fetoni V, Giaccone G, Castucci A, Taroni F, Gellera C, Di Donato S. Spinocerebellar ataxia type 17 (SCA 17): oculomotor phenotype and clinical characterization of 15 Italian patients. J Neurol. 2007;254:1538–46. [PubMed: 17934876]
- Maruyama H, Izumi Y, Morino H, Oda M, Toji H, Nakamura S, Kawakami H. Difference in disease-free survival curve and regional distribution according to subtype of spinocerebellar ataxia: a study of 1,286 Japanese patients. Am J Med Genet. 2002;114:578–83. [PubMed: 12116198]
- Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edström L, Anvret M, Prusiner SB. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet. 2001;69:1385–8. [PMC free article: PMC1235549] [PubMed: 11593450]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–8. [PubMed: 11448935]
- Nanda A, Jackson SA, Schwankhaus JD, Metzer WS. Case of spinocerebellar ataxia type 17 (SCA 17) associated with only 41 repeats of the TATA-binding protein (TBP) gene. Mov Disord. 2007;22:436. [PubMed: 17149738]
- Nielsen TT, Mardosiene S, Lokkegaard A, Stokholm J, Ehrenfels S, Bech S, Friberg L, Nielsen JK, Nielsen JE. Severe and rapidly progressing cognitive phenotype in a SCA17-family with only marginally expanded CAG/CAA repeats in the TATA-box binding protein gene: a case report. BMC Neurol. 2012;12:73–7. [PMC free article: PMC3475097] [PubMed: 22889412]
- Nolte D, Sobanski E, Wissen A, Regula JU, Lichy C, Müller U. Spinocerebellar ataxia type 17 associated with an expansion of 42 glutamine residues in TATA-box binding protein gene. J Neurol Neurosurg Psychiatry. 2010;81:1396–9. [PubMed: 20587494]
- Oda M, Maruyama H, Komure O, Morino H, Terasawa H, Izumi Y, Imamura T, Yasuda M, Ichikawa K, Ogawa M, Matsumoto M, Kawakami H. Possible reduced penetrance of expansion of 44 to 47 CAG/CAA repeats in the TATA-binding protein gene in spinocerebellar ataxia type 17. Arch Neurol. 2004;61:209–12. [PubMed: 14967767]
- Park H, Jeon BS, Shin JH, Park SH. A patient with 41 CAG repeats in SCA17 presenting with parkinsonism and chorea. Parkinsonism Relat Disord. 2016;22:106–7. [PubMed: 26613966]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Reis MC, Patrun J, Ackl N, Winter P, Scheifele M, Danek A, Nolte D. A severe dementia syndrome caused by intron retention and cryptic splice site activation in STUB1 and exacerbated by TBP repeat expansions. Front Mol Neurosci. 2022;15:878236. [PMC free article: PMC9048483] [PubMed: 35493319]
- Rolfs A, Koeppen AH, Bauer I, Bauer P, Buhlmann S, Topka H, Schols L, Riess O. Clinical features and neuropathology of autosomal dominant spinocerebellar ataxia (SCA17). Ann Neurol. 2003;54:367–75. [PubMed: 12953269]
- Shatunov A, Fridman EA, Pagan FI, Leib J, Singleton A, Hallett M, Goldfarb LG. Small de novo duplication in the repeat region of the TATA-box-binding protein gene manifest with a phenotype similar to variant Creutzfeldt-Jakob disease. Clin Genet. 2004;66:496–501. [PubMed: 15521976]
- Silveira I, Miranda C, Guimaraes L, Moreira MC, Alonso I, Mendonca P, Ferro A, Pinto-Basto J, Coelho J, Ferreirinha F, Poirier J, Parreira E, Vale J, Januario C, Barbot C, Tuna A, Barros J, Koide R, Tsuji S, Holmes SE, Margolis RL, Jardim L, Pandolfo M, Coutinho P, Sequeiros J. Trinucleotide repeats in 202 families with ataxia: a small expanded (CAG)n allele at the SCA17 locus. Arch Neurol. 2002;59:623–9. [PubMed: 11939898]
- Stevanin G, Brice A. Spinocerebellar ataxia 17 (SCA 17) and Huntington’s disease-like 4 (HDL4). Cerebellum. 2008;7:170–8. [PubMed: 18418687]
- Stevanin G, Fujigasaki H, Lebre AS, Camuzat A, Jeannequin C, Dode C, Takahashi J, San C, Bellance R, Brice A, Durr A. Huntington's disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain. 2003;126:1599–603. [PubMed: 12805114]
- Schneider SA, Walker RH, Bhatia K. The Huntington’s disease-like syndromes: what to consider in patients with a negative Huntington’s disease gene test. Nat Clin Pract Neurol. 2007;3:517–25. [PubMed: 17805246]
- Toyoshima Y, Takahashi H. Spinocerebellar ataxia type 17 (SCA17). Adv Exp Med Biol. 2018;1049:219–31. [PubMed: 29427105]
- Toyoshima Y, Yamada M, Onodera O, Shimohata M, Inenaga C, Fujita N, Morita M, Tsuji S, Takahashi H. SCA 17 homozygote showing Huntington's disease-like phenotype. Ann Neurol. 2004;55:281–6. [PubMed: 14755733]
- Wu YR, Fung HC, Lee-Chen GJ, Gwinn-Hardy K, Ro LS, Chen ST, Hsieh-Li HM, Lin HY, Lin CY, Li SN, Chen CM. Analysis of polyglutamine-coding repeats in the TATA-binding protein in different neurodegenerative diseases. J Neural Transm. 2005;112:539–46. [PubMed: 15365789]
- Xiang F, Almqvist EW, Huq M, Lundin A, Hayden MR, Edstrom L, Anvret M, Zhang Z. A Huntington disease-like neurodegenerative disorder maps to chromosome 20p. Am J Hum Genet. 1998;63:1431–8. [PMC free article: PMC1377554] [PubMed: 9792871]
- Zühlke C, Dalski A, Schwinger E, Finckh U. Spinocerebellar ataxia type 17: report of a family with reduced penetrance of an unstable Gln49 TBP allele, haplotype analysis supporting a founder effect for unstable alleles and comparative analysis of SCA17 genotypes. BMC Med Genet. 2005;6:27. [PMC free article: PMC1177950] [PubMed: 15989694]
- Zühlke C, Gehlken U, Hellenbroich Y, Schwinger E, Bürk K. Phenotypical variability of expanded alleles in the TATA-binding protein gene. Reduced penetrance in SCA17? J Neurol. 2003a;250:161–3. [PubMed: 12574945]
- Zühlke C, Hellenbroich Y, Dalski A, Kononowa N, Hagenah J, Vieregge P, Riess O, Klein C, Schwinger E. Different types of repeat expansion in the TATA-binding protein gene are associated with a new form of inherited ataxia. Eur J Hum Genet. 2001;9:160–4. [PubMed: 11313753]
- Zühlke CH, Spranger M, Spranger S, Voigt R, Lanz M, Gehlken U, Hinrichs F, Schwinger E. SCA17 caused by homozygous repeat expansion in TBP due to partial isodisomy 6. Eur J Hum Genet. 2003b;11:629–32. [PubMed: 12891385]
Publication Details
Author Information and Affiliations
Brain Research Institute
Niigata University
Niigata, Japan
Department of Brain Disease Research
Shinshu University School of Medicine
Matsumoto, Japan
University of Tokyo Graduate School of Medicine
Tokyo, Japan
Niigata University
Niigata, Japan
Publication History
Initial Posting: March 29, 2005; Last Revision: July 28, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Toyoshima Y, Onodera O, Yamada M, et al. Spinocerebellar Ataxia Type 17. 2005 Mar 29 [Updated 2022 Jul 28]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


