Summary
Clinical description.
SETD2 neurodevelopmental disorders (SETD2-NDDs) represent a clinical spectrum that most commonly includes various degrees of intellectual disability and behavioral findings (most typically an autism spectrum disorder), macrocephaly with or without ventriculomegaly, brain malformations (including Chiari I malformation and syringomyelia), and obesity with generalized overgrowth and advanced bone age. A specific, somewhat different phenotype (denoted SETD2-NDD with multiple congenital anomalies [MCA]) has been reported in association with a particular pathogenic variant, c.5218C>T (p.Arg1740Trp), which leads to a higher frequency of multiple congenital anomalies compared to those without this genetic change. Individuals with SETD2-NDD with MCA may have microcephaly, congenital heart malformations, urogenital anomalies, eye findings (specifically Coats disease of the retina), severe failure to thrive, hypotonia, hyponatremia, respiratory issues (tracheomalacia, frequent aspiration, hypoventilation), epilepsy, profound intellectual disability with limited-to-no speech, and distinctive craniofacial features.
Diagnosis/testing.
The diagnosis of a SETD2-NDD is established in a proband with suggestive findings and a heterozygous pathogenic variant in SETD2 identified by molecular genetic testing.
Management.
Treatment of manifestations:
- SETD2-NDD with or without macrocephaly/overgrowth. Nutritional management of obesity to include diet/exercise; consideration of growth hormone therapy in those with poor growth; standard treatment for developmental delay / autistic features, seizures, hypothyroidism, precocious puberty, hypotonia/hypermobility, scoliosis, refractive error / strabismus, hearing loss, congenital heart defects, and cryptorchidism.
- SETD2-NDD with MCA. Feeding therapy to include consideration of a gastrostomy tube; supplemental oxygen therapy with consideration of tracheostomy in those with tracheomalacia/hypoventilation; sodium supplementation for those with hyponatremia; standard treatment for developmental delay, seizures, joint contractures, sensorineural/conductive hearing loss, Coats disease of the retina, congenital heart defects, cryptorchidism, dysplastic kidneys, and skeletal anomalies.
Surveillance:
- SETD2-NDD with or without macrocephaly/overgrowth. Monitor for psychiatric symptoms, seizures, changes in tone, movement disorders, and developmental progress at each clinic visit; weight checks at home for obesity prevention starting in the second year of life; annual thyroid-stimulating hormone and free T4; clinical evaluation for precocious puberty and scoliosis at each visit during childhood; annual (or as clinically indicated) ophthalmology and audiology evaluations.
- SETD2-NDD with MCA. Monitor for appropriate growth, evidence of aspiration, respiratory sufficiency, seizures, changes in tone, movement disorders, and developmental progress at each clinic visit; electrolyte panel to include sodium level to assess for hyponatremia at each visit during infancy; annual (or as clinically indicated) ophthalmology and audiology evaluations.
Genetic counseling.
SETD2-NDDs are inherited in an autosomal dominant manner, although most affected individuals represent simplex cases (i.e., a single occurrence in a family). To date, transmission of a SETD2 pathogenic variant from a parent to a child has been reported in one family. If a parent of the proband is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. Each child of an individual with a SETD2-NDD has a 50% chance of inheriting the SETD2 pathogenic variant. Once the SETD2 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
GeneReview Scope
Table
SETD2 Neurodevelopmental Disorders: Included Phenotypes
Diagnosis
No consensus clinical diagnostic criteria for SETD2 neurodevelopmental disorders (SETD2-NDDs) have been published. SETD2-NDD represents a clinical spectrum with the most common well-defined phenotype being SETD2-NDD with macrocephaly/overgrowth, although not everyone has overgrowth; a specific, somewhat different phenotype has been reported in association with a particular pathogenic variant, c.5218C>T (p.Arg1740Trp), which leads to a higher frequency of multiple congenital anomalies (MCA) compared to those without this genetic change (see GeneReview Scope). This chapter covers both of these recognized phenotypes.
Suggestive Findings
SETD2-NDD with or without macrocephaly/overgrowth can be considered in individuals with the following nonspecific findings:
- Macrocephaly with or without ventriculomegaly (Figure 1)
- Brain malformations including Chiari I malformation and/or syringomyelia
- Developmental delay, especially in the area of speech and language development
- Intellectual disability, usually moderate (ranging from mild to severe)
- Behavioral difficulties including autism spectrum disorder and/or outbursts of aggression
- Overgrowth, although some affected individuals have growth that falls within the normal range

Figure 1.
Clinical features of individuals with SETD2-NDD with macrocephaly/overgrowth syndrome a & b. Lateral and frontal views of an affected individual; note the prominent forehead with high frontal hairline, frontal bossing, and low-set ears
SETD2-NDD with multiple congenital anomalies (MCA) can be suspected in individuals with the following specific findings [Rabin et al 2020]:
- Microcephaly with head size often normal at birth and microcephaly developing in infancy
- Brain malformations including the triad of hypoplasia of the corpus callosum, pons, and cerebellum (Figure 2)
- Profound intellectual disability with no speech and no independent ambulation
- Severe failure to thrive in infancy typically accompanied by hypotonia and associated respiratory and feeding difficulties
- Multiple congenital anomalies
- Congenital heart defects
- Urogenital anomalies
- Ophthalmologic findings including Coats disease of the retina characterized by telangiectatic, tortuous, and sometimes leaky retinal vessels
- Distinctive craniofacial features (Figure 3) including:
- Low anterior hairline
- Biparietal narrowing
- Flat face with maxillary hypoplasia
- Arched eyebrows
- Widely spaced eyes
- Short palpebral fissures
- Wide nasal bridge
- Short nose with anteverted nares
- Broad nasal tip with low-hanging columella
- Micrognathia with mandibular hypoplasia


Figure 3.
Facial appearance of individuals with SETD2-NDD with multiple congenital anomalies Patient 2 at age seven years (A); at age ten years (B-C)
Family history is consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations). Absence of a known family history does not preclude the diagnosis.
Establishing the Diagnosis
The diagnosis of a SETD2-NDD is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in SETD2 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of a heterozygous SETD2 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing).
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings of SETD2-NDD with MCA described in Suggestive Findings could be diagnosed using single-gene testing (see Option 1), whereas those in whom the diagnosis of a SETD2-NDD has not been considered are more likely to be diagnosed using a multigene panel or genomic testing (see Option 2).
Option 1
Single-gene testing. Sequence analysis of SETD2 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Typically, if no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Option 2
An overgrowth/macrocephaly or autism / intellectual disability multigene panel that includes SETD2 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in SETD2 Neurodevelopmental Disorders
Epigenetic Signature Analysis / Methylation Array
A distinctive epigenetic signature (disorder-specific genome-wide changes in DNA methylation profiles) in peripheral blood leukocytes has been identified in individuals with SETD2-NDD syndrome [Levy et al 2021]. Epigenetic signature analysis of a peripheral blood sample or DNA banked from a blood sample can therefore be considered to clarify the diagnosis in individuals with: (1) suggestive findings of SETD2-NDD syndrome but in whom no pathogenic variant in SETD2 has been identified via sequence analysis or genomic testing; or (2) suggestive findings of SETD2-NDD syndrome and a SETD2 variant of uncertain clinical significance identified by molecular genetic testing. For an introduction to epigenetic signature analysis click here.
Clinical Characteristics
Clinical Description
To date, 30 individuals have been reported with a SETD2 pathogenic variant, excluding those who have deletions of the 3p21.31 region that includes SETD2 and other adjacent genes (see Genetically Related Disorders) [O'Roak et al 2012a, O'Roak et al 2012b, Luscan et al 2014, Lumish et al 2015, Tlemsani et al 2016, van Rij et al 2018, Marzin et al 2019, Rabin et al 2020, Suda et al 2021].
SETD2 neurodevelopmental disorder (SETD2-NDD) with macrocephaly/overgrowth, the most common phenotype, can also include developmental delay / intellectual disability, obesity, advanced bone age, and behavioral findings (most typically an autism spectrum disorder). This spectrum also includes three individuals (2 male and 1 female) with a heterozygous c.5219G>A (p.Arg1740Gln) pathogenic SETD2 variant who have normal growth (see Genotype-Phenotype Correlations).
SETD2-NDD with multiple congenital anomalies (MCA) presents with microcephaly, brain malformations, profound intellectual disability, severe failure to thrive, and multiple congenital anomalies including congenital heart defects, urogenital anomalies, and ophthalmic findings such as Coats disease of the retina. These individuals have a c.5218C>T (p.Arg1740Trp) pathogenic SETD2 variant (see Genotype-Phenotype Correlations).
Table 2.
SETD2 Neurodevelopmental Disorders: Phenotypes by Selected Distinguishing Features
SETD2-NDD with or without Macrocephaly/Overgrowth
Growth parameters are typically normal at birth; however, macrocephaly can be observed at birth. Obesity and tall stature usually become apparent in childhood, although stature may normalize with age. A subset of individuals have normal growth parameters throughout their lives, although height tends to be above the 50th centile (see Genotype-Phenotype Correlations). Bone age is frequently advanced [Marzin et al 2019; Author, personal observation].
Developmental delay (DD) and intellectual disability (ID) range from severe disability to normal intelligence with behavioral issues. Most affected individuals have cognitive impairment that falls within the moderate range, with two individuals having severe ID and several having mild ID. Developmental delays are usually apparent early in life, with speech being the most severely affected.
Other neurologic features
- Hypotonia may be present. This typically does not require feeding therapy or supplemental tube feeds, as seen with SETD2-NDD with MCA.
- Epilepsy has been rarely described.
- Generalized tonic-clonic seizures occurred in one individual at age ten years, but the affected individual remained seizure free on lamotrigine monotherapy for at least three years [Lumish et al 2015].
- Another individual experienced one seizure at age three years, and a third individual experienced several seizures that did not recur after ventriculoperitoneal shunt placement [O'Roak et al 2012a, Marzin et al 2019].
Neuroimaging may identify Chiari I malformation, syringomyelia, hydrocephaly, ventriculomegaly, and Dandy-Walker malformation.
Behavioral findings can include autism spectrum disorder, attention-deficit disorder, aggressive outbursts, self-mutilating behaviors, frustration intolerance, anxiety, hyperphagia, and stereotypies.
Endocrinologic findings may include the following [Marzin et al 2019; Author, personal observation]:
- Precocious puberty
- Polycystic ovarian syndrome
- Hypothyroidism
- Growth hormone deficiency
Sensory impairment
- Hearing loss is uncommon but has been observed. It is more commonly seen in those with the SETD2 pathogenic variant c.5218C>T.
- Strabismus has been observed; cortical visual impairment and optic nerve hypoplasia have been described but are uncommon.
Other associated features that may be present:
- Recurrent infections, including recurrent otitis media, sinus infections, and/or respiratory infections
- Gastroesophageal reflux disease
- Constipation
- Congenital heart defects
- Sleep apnea (type not well described in the literature)
- Hirsutism
- Scoliosis
- Large- and small-joint hypermobility
- Cryptorchidism
- Nevi
SETD2-NDD with MCA
Prenatal complications include preterm labor. Brain malformation may be apparent in the third trimester. Cardiac and kidney anomalies are also sometimes detected prenatally. Polyhydramnios and maternal preeclampsia are also common.
Growth
- Microcephaly can have prenatal onset (2/9) or develop by early infancy. Microcephaly is usually progressive and at least 2.5 standard deviations (SD) below the mean, with head circumference reported to be up to 5.5 SD below the mean.
- Severe failure to thrive is noted in infancy and is frequently accompanied by hypotonia, which contributes to feeding issues.
- All affected individuals had normal weight and length at birth.
- Weight usually remains below the 50th centile in infancy and childhood, whereas height is more variable.
Feeding issues. Most affected individuals require nasogastric tube feedings, which may be transitioned to gastrostomy tube for long-term nutritional support, particularly in those with frequent aspiration (see Respiratory issues).
Cleft palate with Pierre Robin sequence is common, observed in 10/12 individuals, and may contribute to both feeding and breathing issues.
Respiratory issues include tracheomalacia, frequent aspiration, hypoventilation, desaturations, and sleep apnea (both obstructive and central). Three of 12 affected individuals required tracheostomy; one affected individual without a tracheostomy required oxygen support at night, and another used CPAP at night.
Developmental delay and intellectual disability is severe to profound in all affected individuals. All affected individuals reported are nonverbal and nonambulatory. Two were able to take a few steps in late childhood and some were able to sit independently. No regression of developmental skills and no behavioral concerns have been reported.
Epilepsy. Seizures typically have onset in infancy and are usually difficult to control. Types of seizures observed include migrating focal seizures; infantile spasms; and apneic, absence, and generalized myoclonic seizures.
One individual had medically intractable seizures until treatment with phenobarbital and a ketogenic/modified Atkin's diet. Another had better seizure control while taking cannabidiol (CBD) oil.
Neuroimaging. Brain malformations have been identified in all individuals who have undergone brain imaging and are strikingly similar.
- A triad of findings include hypoplasia of the corpus callosum, pons, and cerebellum.
- Shallow sulci, ventriculomegaly, and mega cisterna magna can also be observed.
Sensory impairment
- Hearing loss of a conductive or mixed nature was reported to range from mild to severe; some affected individuals wear hearing aids for support. Some have had tympanostomy tubes placed due to middle ear effusion.
- Coats disease of the retina is reported in 8/10 individuals with telangiectatic vessels of the eyes. Additional ophthalmic abnormalities include optic nerve hypoplasia, glaucoma, and/or cataracts. Onset of ophthalmic abnormalities, including Coats disease, is typically in infancy.
Other associated features
- Congenital heart defects may include ventricular septal defect, atrial septal defect, patent ductus arteriosus, tetralogy of Fallot, double outlet right ventricle, pulmonary stenosis, persistent left superior vena cava, dysplastic pulmonary valve, pulmonary artery hypoplasia, hypoplastic aortic valve, transverse arch hypoplasia, and coarctation of the aorta. The majority of affected individuals have multiple congenital heart defects.
- Urogenital anomalies may include dilated or duplicated collecting system and multicystic dysplastic kidneys.
- One affected individual with multicystic dysplastic kidneys developed end-stage kidney disease.
- All males reported have cryptorchidism; micropenis and shawl scrotum have also been reported in some.
- Two females have anteriorly placed anus, and one also has a short vagina, absent cervix, and absent midline müllerian structures.
- Endocrinologic findings
- Hyponatremia is common in infancy, observed in 8/12 affected individuals.The hyponatremia was initially concerning for the syndrome of inappropriate antidiuretic hormone (SIADH) secretion, but hyponatremia ultimately resolved with sodium supplementation.
- Additional endocrine abnormalities have been observed including acquired hypothyroidism and hypothalamic hamartoma in one affected individual each.
- Skeletal abnormalities are observed in all individuals. These include:
- Hip dysplasia
- Contractures of digits, knees, and/or elbows
- Thoracic dysplasia
- Craniosynostosis involving sagittal and metopic sutures
- Neuromuscular scoliosis
- Abnormalities of the hands and feet
- Brachydactyly
- Camptodactyly
- Syndactyly
- Proximally implanted triphalangeal thumbs
- Broad proximally implanted halluces
- Hypoplastic distal phalanges and nails
- Rocker bottom feet
- Small hands and feet
- Persistent fetal fingertip pads
- Malignancy. Osteosarcoma was diagnosed in two individuals, age 12 and 15 years [personal communication with Francis Sansbury, MB, PhD, All Wales Medical Genomics Service and John A Bernat, MD, PhD, University of Iowa Division of Medical Genetics and Genomics].
Genotype-Phenotype Correlations
SETD2-NDD with normal growth and without macrocephaly. The c.5219G>A (p.Arg1740Gln) variant is the only SETD2 pathogenic variant known to be associated with this phenotype (see Molecular Genetics). Features of the three individuals with this finding include the following:
- Growth. Head circumference may drift toward the lower end of normal, but not within the microcephalic range.
- Developmental delay and intellectual disability. All three developed some speech by age two years.
- Behavioral problems. Autism spectrum disorder was not observed. One individual has anxiety, executive functioning impairment, and slow processing speed.
- Other associated features (each reported in 1 individual):
- Strabismus
- Myopia
- Laryngomalacia
- Constipation
SETD2-NDD with MCA. The c.5218C>T (p.Arg1740Trp) variant is the only SETD2 pathogenic variant known to be associated with this phenotype (see Table 2 and Molecular Genetics).
Prevalence
SETD2-NDDs appear to be very rare. Fewer than 40 affected individuals have been reported in the medical literature to date.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with a heterozygous germline pathogenic variant in SETD2.
Deletion of 3p21.31. Deletions of this chromosomal region are rare, although one report of a male age seven years with a deletion of 3p21.31 including SETD2 and 28 other genes has been published [Lovrecic et al 2016]. This individual had many features that overlapped with SETD2-NDD with macrocephaly/overgrowth (Figure 4).
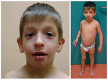
Figure 4.
Craniofacial characteristics in an individual with a 3p21.31 deletion. A long face, downslanted palpebral fissures, broad nasal tip, deep philtrum, and low-set, dysmorphic ears are shown. Reprinted with permission from Lovrecic et al [2016]
Sporadic tumors (including clear cell renal cell cancer, primary central nervous system tumors, leukemia) occurring as single tumors in the absence of any other findings of SETD2 neurodevelopmental disorders frequently contain somatic variants in SETD2 that are not present in the germline. In these circumstances predisposition to these tumors is not heritable. For more information, see Cancer and Benign Tumors.
Differential Diagnosis
Because the phenotypic features associated with SETD2 neurodevelopmental disorder (SETD2-NDD) without macrocephaly/overgrowth or multiple congenital anomalies (MCA) are not sufficient to diagnose this condition, all disorders with intellectual disability without other distinctive findings should be considered in the differential diagnosis. See OMIM Autosomal Dominant, Autosomal Recessive, Nonsyndromic X-Linked, and Syndromic X-Linked Intellectual Developmental Disorder Phenotypic Series. For the differential diagnosis of SETD2-NDD with macrocephaly/overgrowth, see Table 3. For the differential diagnosis of SETD2-NDD with MCA, see Table 4.
Table 3.
Differential Diagnosis of SETD2-NDD with Macrocephaly/Overgrowth
Table 4.
Differential Diagnosis of SETD2-NDD with Multiple Congenital Anomalies
Management
No clinical practice guidelines for SETD2 neurodevelopmental disorders have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with SETD2 neurodevelopmental disorders (SETD2-NDD), the evaluations summarized in Table 5 (SETD2-NDD w/or w/o macrocephaly/overgrowth) and Table 6 (SETD2-NDD w/MCA) ‒ if not performed as part of the evaluation that led to the diagnosis ‒ are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis: SETD2-NDD with or without Macrocephaly/Overgrowth
Table 6.
Recommended Evaluations Following Initial Diagnosis: SETD2-NDD with Multiple Congenital Anomalies (c.5218C>T Pathogenic Variant)
Treatment of Manifestations
Table 7 and Table 8 summarize the recommended treatment for individuals with SETD2-NDD with or without macrocephaly/overgrowth and those with SETD2-NDD with MCA, respectively.
Table 7.
Treatment of Manifestations: SETD2-NDD with or without Macrocephaly/Overgrowth
Table 8.
Treatment of Manifestations: SETD2-NDD with Multiple Congenital Anomalies (c.5218C>T Pathogenic Variant)
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Table 9 and Table 10 summarize the recommended surveillance for individuals with SETD2-NDD with or without macrocephaly/overgrowth and those with SETD2-NDD with MCA, respectively.
Table 9.
Recommended Surveillance for Individuals with SETD2-NDD with or without Macrocephaly/Overgrowth
Table 10.
Recommended Surveillance for Individuals with SETD2-NDD with Multiple Congenital Anomalies (c.5218C>T Pathogenic Variant)
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
SETD2 neurodevelopmental disorders (SETD2-NDDs) are inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- An individual diagnosed with a SETD2-NDD may have the disorder as the result of a SETD2 pathogenic variant inherited from a parent. To date, transmission of a SETD2 pathogenic variant from a parent to a child has been reported in one family; it is unknown whether the heterozygous parent had features of a SETD2-NDD [O'Roak et al 2012a].
- Molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the SETD2 pathogenic variant identified in the proband is not identified in either parent, the following possibilities should be considered:
- The proband has a de novo SETD2 pathogenic variant. Note: A pathogenic variant is reported as "de novo" if: (1) the pathogenic variant found in the proband is not detected in parental DNA; and (2) parental identity testing has confirmed biological maternity and paternity. If parental identity testing is not performed, the variant is reported as "assumed de novo" [Richards et al 2015].
- The proband inherited a SETD2 pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Mosaicism for a SETD2 pathogenic variant has been observed in one individual. This individual was mildly affected and presented with hearing loss, Chiari malformation, epilepsy, hypoglycemia, and normal intelligence [R Rabin, personal communication]. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.* A parent with somatic and germline mosaicism for a SETD2 pathogenic variant may be mildly/minimally affected.
- The family history of some individuals diagnosed with SETD2-NDD may appear to be negative because of failure to recognize the disorder in family members or reduced penetrance. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has demonstrated that neither parent is heterozygous for the pathogenic variant identified in the proband.
Sibs of a proband. The risk to sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is affected is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%. The penetrance of SETD2-NDD in a sib who inherits a familial pathogenic variant and the likelihood of intrafamilial clinical variability are unknown.
- If the proband has a known SETD2 pathogenic variant that cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If the parents have not been tested for the SETD2 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for a SETD2-NDD because of the possibility of reduced penetrance in a heterozygous parent or the theoretic possibility of parental germline mosaicism.
Offspring of a proband. Each child of an individual with a SETD2-NDD has a 50% chance of inheriting the SETD2 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the SETD2 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected.
Prenatal Testing and Preimplantation Genetic Testing
Once the SETD2 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal preimplantation genetic testing. While most health care professionals would consider use of prenatal and preimplantation genetic testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Luscan-Lumish SETD2 SupportEmail: [email protected]
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- Autism SocietyPhone: 800-328-8476Email: [email protected]
- National Center on Birth Defects and Developmental Disabilities (NCBDDD)Phone: 800-232-4636 (toll-free); 888-232-6348 (TTY)
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
SETD2 Neurodevelopmental Disorders: Genes and Databases
Table B.
OMIM Entries for SETD2 Neurodevelopmental Disorders (View All in OMIM)
Molecular Pathogenesis
SETD2 codes for a histone methyltransferase that trimethylates the lysine at position 36 of histone H3 (H3K36me3) [Edmunds et al 2008]. Deficiency of the SETD2 protein has been associated with loss of H3K36me3 and abnormal DNA methylation [Xu et al 2019]. SETD2 is a dual-function methyltransferase for histones and microtubules and plays an important role in transcriptional regulation, genomic stability, and cytoskeletal functions [Zhou et al 2011, Park et al 2016, McDaniel & Strahl 2017].
Loss-of-function variants in SETD2 lead to hypomethylation of H3 at K36, which has been associated with overgrowth [Weinberg et al 2019]. Evidence from hypermethylation of polycomb-regulated regions [Heyn et al 2019] and association with microcephalic dwarfism points to possible loss of function associated with the variant c.5218C>T (p.Arg1740Trp) in SETD2 neurodevelopmental disorder with multiple congenital anomalies.
Mechanism of disease causation. Loss of function
Table 11.
Notable SETD2 Pathogenic Variants
Cancer and Benign Tumors
SETD2 is a tumor suppressor gene. Somatic variants have been detected in a variety of cancers, including clear cell renal cell, gastrointestinal, lung, pancreatic, and osteosarcoma [Li et al 2016, Chen et al 2020]. Somatic variants are also described in primary central nervous system tumors [Viaene at al 2018] and leukemia [Skucha et al 2019].
Chapter Notes
Author Notes
Dr John Pappas: nyulangone.org
Revision History
- 22 September 2022 (sw) Revision: epigenetic signature analysis (Establishing the Diagnosis)
- 30 December 2021 (ma) Review posted live
- 4 January 2021 (jp) Original submission
References
Literature Cited
- Chen R, Zhao WQ, Fang C, Yang X, Ji M. Histone methyltransferase SETD2: a potential tumor suppressor in solid cancers. J Cancer. 2020;11:3349-56. [PMC free article: PMC7097956] [PubMed: 32231741]
- Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008;27:406-20. [PMC free article: PMC2168397] [PubMed: 18157086]
- Heyn P, Logan CV, Fluteau A, Challis RC, Auchynnikava T, Martin CA, Marsh JA, Taglini F, Kilanowski F, Parry DA, Cormier-Daire V, Fong CT, Gibson K, Hwa V, Ibáñez L, Robertson SP, Sebastiani G, Rappsilber J, Allshire RC, Reijns MAM, Dauber A, Sproul D, Jackson AP. Gain-of-function DNMT3A mutations cause microcephalic dwarfism and hypermethylation of Polycomb-regulated regions. Nature Genetics. 2019;51:96-105. [PMC free article: PMC6520989] [PubMed: 30478443]
- Levy MA, McConkey H, Kerkhof J, Barat-Houari M, Bargiacchi S, Biamino E, Bralo MP, Cappuccio G, Ciolfi A, Clarke A, DuPont BR, Elting MW, Faivre L, Fee T, Fletcher RS, Cherik F, Foroutan A, Friez MJ, Gervasini C, Haghshenas S, Hilton BA, Jenkins Z, Kaur S, Lewis S, Louie RJ, Maitz S, Milani D, Morgan AT, Oegema R, Østergaard E, Pallares NR, Piccione M, Pizzi S, Plomp AS, Poulton C, Reilly J, Relator R, Rius R, Robertson S, Rooney K, Rousseau J, Santen GWE, Santos-Simarro F, Schijns J, Squeo GM, St John M, Thauvin-Robinet C, Traficante G, van der Sluijs PJ, Vergano SA, Vos N, Walden KK, Azmanov D, Balci T, Banka S, Gecz J, Henneman P, Lee JA, Mannens MMAM, Roscioli T, Siu V, Amor DJ, Baynam G, Bend EG, Boycott K, Brunetti-Pierri N, Campeau PM, Christodoulou J, Dyment D, Esber N, Fahrner JA, Fleming MD, Genevieve D, Kerrnohan KD, McNeill A, Menke LA, Merla G, Prontera P, Rockman-Greenberg C, Schwartz C, Skinner SA, Stevenson RE, Vitobello A, Tartaglia M, Alders M, Tedder ML, Sadikovic B. Novel diagnostic DNA methylation episignatures expand and refine the epigenetic landscapes of Mendelian disorders. HGG Adv. 2021;3:100075. [PMC free article: PMC8756545] [PubMed: 35047860]
- Li J, Duns G, Westers H, Sijmons R, van den Berg A, Kok K. SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget. 2016;7:50719-34. [PMC free article: PMC5226616] [PubMed: 27191891]
- Lovrecic L, Bertok S, Žerjav Tanšek M. A new case of an extremely rare 3p21.31 interstitial deletion. Mol Syndromol. 2016;7:93-8. [PMC free article: PMC4906427] [PubMed: 27385966]
- Lumish HS, Wynn J, Devinsky O, Chung WK. Brief report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J Autism Dev Disord. 2015;45:3764-70. [PubMed: 26084711]
- Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, Lacombe D, Touraine R, Vidaud M, Pasmant E, Cormier-Daire V. Mutations in SETD2 cause a novel overgrowth condition. J Med Genet. 2014;51:512-7. [PubMed: 24852293]
- Marzin P, Rondeau S, Aldinger KA, Alessandri JL, Isidor B, Heron D, Keren B, Dobyns WB, Cormier-Daire V. SETD2 related overgrowth syndrome: presentation of four new patients and review of the literature. Am J Med Genet C Semin Med Genet. 2019;181:509-18. [PubMed: 31643139]
- McDaniel SL, Strahl BD. Shaping the cellular landscape with Set2/SETD2 methylation. Cell Mol Life Sci. 2017;74:3317-34. [PMC free article: PMC5545052] [PubMed: 28386724]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O'Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012a;338:1619-22. [PMC free article: PMC3528801] [PubMed: 23160955]
- O'Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, Levy R, Ko A, Lee C, Smith JD, Turner EH, Stanaway IB, Vernot B, Malig M, Baker C, Reilly B, Akey JM, Borenstein E, Rieder MJ, Nickerson DA, Bernier R, Shendure J, Eichler EE. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012b;485:246-50. [PMC free article: PMC3350576] [PubMed: 22495309]
- Park IY, Powell RT, Tripathi DN, Dere R, Ho TH, Blasius TL, Chiang YC, Davis IJ, Fahey CC, Hacker KE, Verhey KJ, Bedford MT, Jonasch E, Rathmell WK, Walker CL. Dual chromatin and cytoskeletal remodeling by SETD2. Cell. 2016;166:950-62. [PMC free article: PMC5101839] [PubMed: 27518565]
- Rabin R, Radmanesh A, Glass IA, Dobyns WB, Aldinger KA, Shieh JT, Romoser S, Bombei H, Dowsett L, Trapane P, Bernat JA, Baker J, Mendelsohn NJ, Popp B, Siekmeyer M, Sorge I, Sansbury FH, Watts P, Foulds NC, Burton J, Hoganson G, Hurst JA, Menzies L, Osio D, Kerecuk L, Cobben JM, Jizi K, Jacquemont S, Bélanger SA, Löhner K, Veenstra-Knol HE, Lemmink HH, Keller-Ramey J, Wentzensen IM, Punj S, McWalter K, Lenberg J, Ellsworth KA, Radtke K, Akbarian S, Pappas J. Genotype-phenotype correlation at codon 1740 of SETD2. Am J Med Genet A. 2020;182:2037-48. [PubMed: 32710489]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126-33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Skucha A, Ebner J, Grebien F. Roles of SETD2 in leukemia-transcription, DNA-damage, and beyond. Int J Mol Sci. 2019;20:1029. [PMC free article: PMC6429614] [PubMed: 30818762]
- Suda K, Fukuoka H, Iguchi G, Kanie K, Fujita Y, Odake Y, Matsumoto R, Bando H, Ito H, Takahashi M, Chihara K, Nagai H, Narumi S, Hasegawa T, Ogawa W, Takahashi Y. A case of Luscan-Lumish syndrome: possible involvement of enhanced GH signaling. J Clin Endocrinol Metab. 2021;106:718-23. [PubMed: 33248444]
- Tlemsani C, Luscan A, Leulliot N, Bieth E, Afenjar A, Baujat G, Doco-Fenzy M, Goldenberg A, Lacombe D, Lambert L, Odent S, Pasche J, Sigaudy S, Buffet A, Violle-Poirsier C, Briand-Suleau A, Laurendeau I, Chin M, Saugier-Veber P, Vidaud D, Cormier-Daire V, Vidaud M, Pasmant E, Burglen L. SETD2 and DNMT3A screen in the Sotos-like syndrome French cohort. J Med Genet. 2016;53:743-51. [PubMed: 27317772]
- van Rij MC, Hollink IHIM, Terhal PA, Kant SG, Ruivenkamp C, van Haeringen A, Kievit JA, van Belzen MJ. Two novel cases expanding the phenotype of SETD2-related overgrowth syndrome. Am J Med Genet A. 2018;176:1212-15. [PubMed: 29681085]
- Viaene AN, Santi M, Rosenbaum J, Li MM, Surrey LF, Nasrallah MP. SETD2 mutations in primary central nervous system tumors. Acta Neuropathol Commun. 2018;6:123. [PMC free article: PMC6231273] [PubMed: 30419952]
- Weinberg DN, Papillon-Cavanagh S, Chen H, Yue Y, Chen X, Rajagopalan KN, Horth C, McGuire JT, Xu X, Nikbakht H, Lemiesz AE, Marchione DM, Marunde MR, Meiners MJ, Cheek MA, Keogh MC, Bareke E, Djedid A, Harutyunyan AS, Jabado N, Garcia BA, Li H, Allis CD, Majewski J, Lu C. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature. 2019;573:281-6. [PMC free article: PMC6742567] [PubMed: 31485078]
- Xu Q, Xiang Y, Wang Q, Wang L, Brind'Amour J, Bogutz AB, Zhang Y, Zhang B, Yu G, Xia W, Du Z, Huang C, Ma J, Zheng H, Li Y, Liu C, Walker CL, Jonasch E, Lefebvre L, Wu M, Lorincz MC, Li W, Li L, Xie W. SETD2 regulates the maternal epigenome, genomic imprinting and embryonic development. Nat Genet. 2019;51:844-56. [PubMed: 31040401]
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7-18. [PubMed: 21116306]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: December 30, 2021; Last Revision: September 22, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Pappas J, Rabin R. SETD2 Neurodevelopmental Disorders. 2021 Dec 30 [Updated 2022 Sep 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.



