

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
SNP FAQ Archive [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005-.
This publication is provided for historical reference only and the information may be out of date.

How dbSNP Determines SNP Positions
Annotation of SNPs to Genes
What is coordinate remapping, and how is it used to annotate SNPs to mRNA and proteins?
Annotation of SNPs to mRNA
What is coordinate remapping, and how is it used to annotate SNPs to mRNA and proteins?
Annotation of SNPs at the Amino Acid Level
Annotation of SNPs in Protein/Structure Models
Is it true that any single refSNP (rs) ID maps to only one structure ID at most?
Annotation of SNPs to Splice Sites
What are dbSNP’s rules for classifying a SNP as occurring at a splice site?
Neighbor SNPs
Chromosome Position and Contig Position
What is the difference between a SNP’s contig position and a SNP's chromosome position?
Locate Disappearing RefSNP (rs) Numbers
Merging RefSNP Clusters
RefSNP (rs) not Mapping to Assembly
Why was rs2228570 withdrawn form dbSNP? I can’t find it using Entrez SNP.
RefSNP (rs) Clusters Not Merging as Expected
rs2430561 and rs61923114 refer to the same SNP. Why did they not cluster together?
Why is it that sometimes rs clusters do not merge as expected (e.g., rs1136410 and rs17853760)?
Definition of loctype values
Where can I find a definition for the locType values “range-ins”, “range-del” and “range-exact”?
Forward vs. Reverse strand Orientation
Individual refSNP Orientation Problems
Sequence Notation for Variations
Does dbSNP have a file that contains SNPs in IUPAC code for the entire human genome?
Can you recommend literature that describes standard nomenclature for sequence variations?
Position Notation for Variations
The variation position notation for rs1922237 is: “12382526^12382527”. What does this notation mean?
1-based vs. 0-based (zero based) coordinate policy
Mapping Discrepancies
Why is rs6010717 mapped to an intron in one reference sequence, and mapped to an exon in another?
Why does rs1926736 map to the Celera assembly, but not to the NCBI reference assembly?
Multiple Mapping Positions for a Single SNP
How do I tell if a SNP is uniquely mapped to the genome or if it appears in multiple places?
How do I tell if a SNP is uniquely mapped to the genome or if it appears in multiple places?
Multiple Locations for Specific refSNP Numbers
Why do we find rs386014 displayed in both HLA-A’s and HCG4P6’s batch report?
Mapping Weight
Your descriptions of mapweight in the FTP README for Chromosome Reports and for SNPMapInfo in the data dictionary are contradictory. Which is correct?
The definitions of map weight for chromosome reports and database tables are indeed different:
Chromosome Reports Database Tables Mapweight 1 = Unmapped Mapweight 1 = SNP aligns exactly at one locus Mapweight 2 = Mapped to single position in genome Mapweight 2 = SNP aligns at two locus on same chromosome Mapweight 3 = Mapped to 2 positions on a single chromosome Mapweight 3 = SNP aligns at less than 10 locus Mapweight 4 = Mapped to 3-10 positions in genome (possible paralog hits) Mapweight 5 = Mapped to >10 positions in genome Mapweight 10= SNP aligns at more than 10 locations The mapweight definitions for the database are different for historical reasons, so both definition series are correct: The mapweights defined in the chromosome reports section of the FTP README are true for chromosome reports, and the definitions given for the database tables in the database dictionary are true for all FTP data table files. (07/14/08)
How dbSNP Determines SNP Positions
How does dbSNP determine SNP positions on the genome?
The positions are obtained from mapping the SNPs to the genome. A description of the dbSNP mapping procedure can be found here.
Both chromosome and contig positions are provided, as are the mRNA and protein positions if available. Please see this example of the contig and chromosome positions at the CTG line in a flatfile report.
Your documentation mentions that you localize SNPs using megablast. Do you have details on this method that you could share with me?
Please view this online document:
Annotation of SNPs to Genes
What is coordinate remapping, and how is it used to annotate SNPs to mRNA and proteins?
Coordinate remapping involves projecting a SNP from its mapped contig position to its corresponding mRNA position using the same "best contig and mRNA alignment" that was used during genome annotation pipeline (gpipe). By using shared alignment components with gpipe, this ensures that the mRNA annotated on the genome has SNP annotation that is correct and consistent with all other annotated genomic features.
The diagram below depicts the alignment model used for SNP coordinate remapping from the genomic sequence to mRNA and proteins, with the assignment of SNP functional context as described in the dbSNP Handbook. The “Near Gene” region includes the mRNA region of the gene as well as arbitrary regions of 2K nucleotides upstream and 0.5K nucleotides down stream to allow for potential regulatory regions.
(03/19/09)
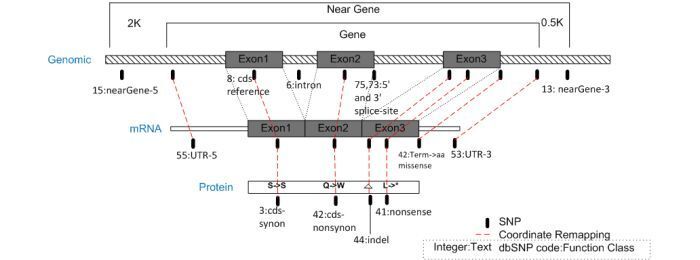
Your documentation states that a SNP in a gene region if it falls 2kb upstream and 500 bp downstream of the gene, while JSNP assigns an association if a gene is 2.5kb in both directions.
As far as I know, dbSNP has used the 2K up, 0.5K down since the beginning of NCBI’s genome annotation pipelines.
I know of no standard for defining the extent of a gene relative to current transcription start sites and termination codons. I think PharmGKB uses 10 K up and 10 K down; while RefSeqGene uses 5K up and 2K down unless a request is received otherwise. (07/16/08)
Do the lists of SNPs in dbSNP that are associated with a particular gene include those SNPs that are near genes, but do not map within the gene itself?
Yes. If a SNP is within 2kb upstream (5' side) or 500 bp downstream (3' side) of the gene, then we associate the SNP with a gene. For a given gene, you can find this information in EntrezSNP, SNP GeneView, or in FTP file reports. (07/23/08)
I'd like to know on what basis intergenic SNPs are assigned to genes in dbSNP. Is it based on a certain distance that the SNP is from the gene?
Intergenic SNPs are assigned to genes in dbSNP based on distance.
They are assigned to a gene if they are within 0.5kb 3' to a gene, or within 2kb 5' to a gene.
A good way to see if a particular SNP is associated with a gene is to look at the SnpFunctionCode for the SNP of interest. To do this, first look in the shared_data directory for your organism on the dbSNP ftp site and download SnpFunctionCode.bcp.gz, which defines the codes used for function class. Then go to the organism_data directory(this link takes you to the directory for human) for your organism of interest, and download SNPContigLocusId.bcp.gz , which contains the snp_id and the functional class code for eachSNP. You can find a description of the columns in the SNPContigLocusId table online.
Looking at the codes defined in SnpFunctionCode.bcp.gz , you’ll see that if the function code is 13 for the SNP, then the functional class is “NearGene–3”, which means that the SNP is within 0.5kb 3' to a gene. If the function code for the SNP is 15, then the functional class is “NearGene–5”, which means that the SNP is within 2kb 5' to a gene. (10/3/07)
Annotation of SNPs to mRNA
What is coordinate remapping, and how is it used to annotate SNPs to mRNA and proteins?
Coordinate remapping involves projecting a SNP from its mapped contig position to its corresponding mRNA position using the same "best contig and mRNA alignment" that was used during genome annotation pipeline (gpipe). By using shared alignment components with gpipe, this ensures that the mRNA annotated on the genome has SNP annotation that is correct and consistent with all other annotated genomic features.
The diagram below depicts the alignment model used for SNP coordinate remapping from the genomic sequence to mRNA and proteins, with the assignment of SNP functional context as described in the dbSNP Handbook. The “Near Gene” region includes the mRNA region of the gene as well as arbitrary regions of 2K nucleotides upstream and 0.5K nucleotides down stream to allow for potential regulatory regions.
(03/19/09)
Would it be possible for dbSNP to determine mRNA position using the “A” in the start codon(ATG) instead of the 1st base of the mRNA?
I’m sorry, but dbSNP must continue starting from the mRNA position, since:
- 1.
We have to be consistent within dbSNP and NCBI
- 2.
Things can get complicated when there are mRNAs that contain multiple start codons, or when there are mRNAs without CDS annotation.
In the future, we hope to change the Geneview page to include the first base of mRNA coordinates as the default and also provide an option to translate into coordinates starting from ATG. We will also consider having this as an available conversion, if demand is high enough.
Until the new default to the Geneview page is implemented, you'll have to continue to do what you're probably already doing:
Current SNP's mRNA position – Codon start position = mRNA position from
ATG. (01/02/08)
Annotation of SNPs at the Amino Acid Level
How are SNP positions on the amino acid level calculated? Are they based on primary translation product or on a processed mature product?
The position of a SNP on amino acid level is calculated from the start codon("ATG"), where the start codon has amino acid position 1.
The number is based on the primary translation product. (05/06/08)
Annotation of SNPs in Protein/Structure Models
Is it true that any single refSNP (rs) ID maps to only one structure ID at most?
A single rs id can map to multiple gene/protein models and neighboring isoforms. (9/13/07)
Annotation of SNPs to Splice Sites
What are dbSNP’s rules for classifying a SNP as occurring at a splice site?
A SNP will be classified as “splice-site” if:
The SNP’s position is one or two bases before the start of an exon.
OR
If the SNP is located one or two bases following the end of an exon.(9/23/08)
Neighbor SNPs
Can a SNP’s structure neighbor protein and the protein that actually includes the SNP ever be the same?
A SNP’s structure neighbors are determined by blast analysis, so it’s possible for the queried protein to match itself if its structure is in the Protein Databank (PDB). (09/10/07)
Mapping to Unplaced Contigs
Can you explain the difference between a refSNP that is mapped to a chromosome but is “unplaced”, versus a refSNP in Chr_Un? What is Chr_Un?
"Placed" contigs are contigs with clear starting and ending positions on a chromosome.
A contig is called "unplaced" if it meets one of the descriptions below:
NT_113953 is an example of an "Unplaced contig".
Other examples of “unplaced’ contigs include the following:
"chr_Un" is a file name for a file in which we place SNPs (with the exception of weight 1 and weight 2 SNPs) that map to "unplaced" contigs.
Please see the FAQ that explains mapping weight.
The reason that weight 1 and weight 2 SNPs are not placed in "chr_Un" is that when a SNP maps uniquely to a placed contig, but also maps to an "unplaced contig", we ignore the "unplaced contig" placement when we assign mapping weight. So it is as if the SNP only maps uniquely to the "placed" contig. (07/14/08)
Why do I sometimes see reference map positions of "unplaced" for SNPs, and what does being "unplaced" mean?
"Unplaced" is an attribute of contigs that are part of the genome assembly but have not yet been assigned a place in a chromosome (usually because there is not enough data to show where these contigs are supposed to go). Therefore, when SNPs map to “unplaced” contigs, dbSNP is unable to assign these SNPs a chromosome position.
Please note that SNP map weight is not affected by mapping to an “unplaced” contig.(10/25/06)
Chromosome Position and Contig Position
What is the difference between a SNP’s contig position and a SNP's chromosome position?
A SNP’s contig position is defined as the position of the SNP on the contig when counting from the first base (base position = 1).
A SNP’s chromosome position is the contig position plus the contig starting position on the chromosome.
If a contig has an "unknown" chromosome position, it cannot be placed on a chromosome. If this "unplaced" contig contains SNPs, these SNPs will have only a contig position — its chromosome position will be "unplaced”. (12/06/05)
Do chromosome sequences consist of several contigs?
Yes. (12/06/05)
Ensembl shows rs3210531 on chromosome 11 strand +1, however, NCBI shows it on chromosome 6 strand -1. Why?
We conduct two different mapping procedures for each SNP. First one places the SNP on contigs, and according to our database, rs3210531 hits chromosome 11 across all assemblies:
snp_id pos chr assembly 3210531 46406925 11 reference 3210531 46598418 11 Celera 3210531 46157042 11 HuRef An additional hit to chr6 was rejected due to our "absolute hit" strategy. This strategy states that if the mapping process finds a 100% identity hit, then the process will reject hits with lower scores as irrelevant.
Mapping to underlying sequences is a separate process and it produced the following hits for rs3210531:
_snp_idoffset accession accession_ver aln_quality 3210531 366 U14972 1 1 3210531 423 BC001955 1 1 3210531 423 BC005012 1 1 3210531 369 BC001032 2 1 3210531 367 BC070235 1 1 3210531 379 BC071946 1 1 3210531 456 BC073799 1 1 3210531 418 NM_001014 3 1 We are working now on a synchronization mechanism for two these processes. (09/29/08)
Locate Disappearing RefSNP (rs) Numbers
Merging RefSNP Clusters
A publication gives rs2857713 a SNP, but when I looked for it in dbSNP, it took me some time to find that it changed to rs2229094. Can’t you inform users when refSNP numbers have changed?
dbSNP does provide notice when refSNPs have merged. Currently, there are three different entry points in dbSNP that will lead you to the partner numbers of a merge:
- 1.
You can now retrieve a list of merged rs numbers from Entrez SNP. Just type “mergedrs” (without the quotation marks) in the text box at the top of the page and click the “go” button. Each entry in the returned list will include the old rs numbers that has merged, and the new rs number it has merged into (with a link to the refSNPpage for the new rs number). You can limit the output to merged rs numbers within a certain species by clicking on the “Limits” tab and then selecting the organism you wish from the organism selection box.
- 2.
If you enter an old rs number (in this case rs2857713) into the “Search for IDs” search on the dbSNP home page, the response page will state that the SNP has been merged, and will provide the new rs number (in this case rs2229094) and a link to the refSNP page for that new rs number.
- 3.
The RsMergeArch table houses the merged SNPs, and is available on the dbSNP ftp site. A full description of the table can be found in the dbSNP Data Dictionary, and the column definitions are located in the dbSNP_main_table.sql.gz, which can be found in the shared
_schema directory of the dbSNP FTP site. (05/15/08:11/03/08)
I heard that RS numbers are not stable. For example, rs17216163 is now rs717620, and rs17231380 is now rs5186. I assume you don't ever reuse the "retired" numbers? Why did you make some numbers obsolete?
The examples you cite are instances where multiple rs numbers were assigned at the same genomic location, and the higher rs number was merged into a lower rs number (this is the dbSNP merge rule for rs numbers). Such a merge can happen when submissions differ in the length and quality of flanking sequence. We only merge rs numbers that have an identical set of mappings to the genome and have the same type of alleles (e.g. both must be the same variation type and share one allele in common). We would not merge a SNP and an indel (insertion/deletion) into a single rs number (different variation classes) since they represent to different types of mutational "events".
The location of the rs number remains valid and we never reuse rs numbers.
We have discussed the issue of supporting query by merged rs numbers more robustly in dbSNP, Entrez and our web based services. That way a retired rs number can be found easily and used as a proxy for the current "live" number. Please note that merging is only used to reduce redundancy in the catalog of rs numbers so each position has a unique identifier.
In the first example you cite, prior to their merge, both rs17216163 and rs717620 would have been the "address" for the same nucleotide. Now only rs717620 is used in annotation, and rs17216163 is retained in our merge history tables. With extended annotation, users would be able to query by the full set of retired rs numbers.
Currently, there are three different entry points in dbSNP that will lead you to the partner numbers of a merge:
- 1.
You can now retrieve a list of merged rs numbers from Entrez SNP. Just type “mergedrs” (without the quotation marks) in the text box at the top of the page and click the “go” button. Each entry in the returned list will include the old rs numbers that has merged, and the new rs number it has merged into (with a link to the refSNP page for the new rs number). You can limit the output to merged rs numbers within a certain species by clicking on the “Limits” tab and then selecting the organism you wish from the organism selection box.
- 2.
If you enter a merged old rs number into the “Search for IDs” search on the dbSNP home page, the response page will state that the SNP has been merged, and will provide the new rs number and a link to the refSNP page for that new rs number.
- 3.
The RsMergeArch table houses the merged SNPs, and is available on the dbSNP ftp site. A full description of the table can be found in the dbSNP Data Dictionary, and the column definitions are located in the dbSNP_main_table.sql.gz, which can be found in the shared
_schema directory of the dbSNP FTP site. (11/07:05/08:11/08)
How do I query dbSNP so that it will return a flat or xml file containing the new RefSNP (rs) ID number into which a previously valid rs recently merged?
You can get the rs merge history of all rs numbers from your organism’s (human in this case) RsMergeArch table located in on the dbSNP ftp site.
The following example shows that rs4344934 has been merged to rs1107123:
gzcat RsMergeArch.bcp.gz | grep 4344934
4344934 1107123 123 1 2004-09-24 18:49:00 2004-10-10
11:55:00 1107123 1Also, you can now retrieve a list of merged rs numbers from Entrez SNP. Just type “mergedrs” (without the quotation marks) in the text box at the top of the page and click the “go” button. Each entry in the returned list will include the old rs numbers that has merged, and the new rs number it has merged into (with a link to the refSNP page for the new rs number). You can limit the output to merged rs numbers within a certain species by clicking on the “Limits” tab and then selecting the organism you wish from the organism selection box. (5/25/05:11/03/08)
Some RefSNP (rs) numbers in dbSNP are merged into one rs number. Where does dbSNP provide the merge history?
You can now retrieve a list of merged rs numbers from Entrez SNP. Just type “mergedrs” (without the quotation marks) in the text box at the top of the page and click the “go” button. Each entry in the returned list will include the old rs numbers that has merged, and the new rs number it has merged into (with a link to the refSNP page for the new rs number). You can limit the output to merged rs numbers within a certain species by clicking on the “Limits” tab and then selecting the organism you wish from the organism selection box.
You can also use the RsMergeArch table located in the dbSNP FTP site. (11/8/05:11/03/08)
Why are SNPs submitted by HGBase that mapped to the genome assembly in previous builds of dbSNP no longer mapped? For example, rs1800324 previously mapped to chromosome X, but is no longer.
It is true that rs1800324, which mapped to the genome assembly in previous releases of dbSNP, no longer maps. The reason is that the internal matching heuristics for NCBI’s MegaBLAST program, which is used for mapping SNPs, has been changing, and this has resulted in mapping changes.
Because of the new matching heuristics, refSNPs with very short flanking sequences can be dropped from the map. rs1800324, submitted by HGBASE, has very short flanking sequences (25 bp on each side), and this may have contributed to it being dropped from the map in b125. If other HGBase submissions also have very short flanking sequences, they may also have been dropped for the same reason. (2/3/06)
RefSNP (rs) not Mapping to Assembly
Why was rs2228570 withdrawn form dbSNP? I can’t find it using Entrez SNP.
rs2228570 has not been withdrawn from dbSNP; however, it does not map to the genome assemblies, which may be due to the fact that this SNP was derived from cDNA. Since rs2228570 does not map to the genome assemblies, you won’t see it in Entrez SNP. I can tell you that rs2228570 is located in NM_000376. Future releases of Entrez SNP will include all SNPs —those that map to genome assemblies, and those that don’t. (2/16/07)
RefSNP (rs) Clusters Not Merging as Expected
rs2430561 and rs61923114 refer to the same SNP. Why did they not cluster together?
Our clustering algorithm requires the mapped chromosome positions of two SNPs to be the identical for them to cluster together. In this case, the two SNPs mapped to two different locations on chromosome 12 — 66838789 and 66838787:
rs61923114
CTG | assembly=reference | chr=12 | chr-pos=66838789 | NT_029419.11 |ctg-start=30695828 | ctg-end=30695828 | loctype=2 | orient=+rs2430561
CTG | assembly=reference | chr=12 | chr-pos=66838787 | NT_029419.11 |ctg-start=30695826 | ctg-end=30695826 | loctype=2 | orient=+Our mapping algorithm provides a "best" placement on the chromosome, but for SNPs like these that have low complexity flanking sequence containing multiple repeats, precise mapping is difficult. We constantly work to improve the mapping algorithm so as to provide better mapping and clustering to fix these problems.(06/10/09)
Some SNPs for A2M seem redundant: rs3832852 and rs1799759 look like the same SNP, as do rs3832850 and rs35904656. rs5796338 and rs3080599 also look identical.
We believe that rs3832852 and rs1799759 in A2M are two separate refSNPs for the following reasons:
- 1.
rs1799759 has variation as -/ACCAT, and rs3832852 has the variation as -/CCATA. If you put the variation in flanking context, the two different chromosome sequences are:
rs1799759 (-/ACCAT)
C-----AG
CACCATAG
rs3832852 (-/CCATA)
CA-----G
CACCATAG
The deleted sequences above for the two refSNPs are shifted one base, so they remain separate SNPs. Currently in dbSNP, we do not have validation (freq or genotype) information for either of these two SNPs. If you have any validation information for either of these SNPs, please contact vog.hin.mln.ibcn@bus-pns and submit your data to dbSNP.- 2.
rs3832850 and rs35904656 are 5 bases apart in mapping, and are therefore distinct SNPs.
- 3.
Similarly, rs5796338 and rs3080599 are 16 bases apart in mapping, and are therefore distinct SNPs. (10/5/07)
I noticed in B125, rs1776148 and rs17856209 represent the same SNP. Why haven’t these two rs clusters been merged?
Merging was not performed in build 125 due to our processing schedule.
In our next build ( B126), we plan to merge those rs numbers that have:
- 1.
The same mapping position on the reference assembly
- 2.
The same SNP allele class (examples of SNP allele classes include: trueSNP, indel, STR, etc.
- 3.
The same mapping loc_type.
rs1776148 and rs17856209 are both in the trueSNP allele class, have the same mapping reference position, and the same loc_type, so these two rs clusters will be merged in the next build. (12/15/05)
Both rs6419492 and rs4601571 seem to describe the same SNP, yet both rs numbers exist separately in dbSNP. Am I missing something?
Yes, these two refSNPs should probably be merged. In general, we cluster submissions that co-locate on the genome, but our heuristics sometimes exclude particular cases. We prefer to err on the side of caution; better to leave a few co-located SNPs unclustered than cluster submissions which do not belong together. In this case, I can see from the submitter comments that the two refSNPs are from the same genomic location. Thanks for bringing this to our attention. (3/13/05)
Why is it that sometimes rs clusters do not merge as expected (e.g., rs1136410 and rs17853760)?
Due to processing timing constraints in b125, some rs numbers that map to the same positions were not merged, but will be merged in the next build. In future builds, SNPs that have different variation classes (and possibly different variation lengths) will not be merged even if they map to the same contig positions. (2/9/06)
Shouldn't the number of new refSNPs be smaller than the number of new submitted SNPs? If so, why are there so many new human rs clusters in B126?
A large number of submitted SNPs did not get clustered in the build 125 release, and were assigned refSNP numbers for them in build 126. There were 2.3 million newly assigned refSNP numbers in build 126, most of which (600,000) are just new clusters of existing submitted SNPs that were submitted in build125.
In the ideal dbSNP world, all SNPs submitted during the time span of a build would be clustered for the release of that build. But in the real dbSNP world, we load submitted SNPs on a daily basis, and by the time all the submitted SNPs are mapped, clustered, and the frequency information for them computed, we end up with many new submitted SNPs that have not been assigned refSNP numbers. We are working to improve the build pipeline, and hope to reduce the time lag between new submitted SNPs loading, and refSNP number assignment.(5/24/06)
Definition of loctype values
Where can I find a definition for the locType values “range-ins”, “range-del” and “range-exact”?
If you go to dbSNP’s online “in-depth explanation of loctypes” you will find that:
range-ins = range insertion = loctype4
range-del = range deletion= loctype6
range-exact= loctype5 (TrueSNP loctype 2 is a special case of this).
These correspond to definitions found in the LocTypeCode file, located in the shared_data directory of the dbSNP FTP site.(02/29/08)
Reassigning loctype
Human build 126 has about 17,000 SNPs mapped to the reference assembly whose submitted class is "MNP”; 15,000 of these SNPs are assigned to the “range” loc_type. Do you plan to reassign these SNPs to the “range substitution” locType?
The current process uses a single IUPac code to represent the variation site, so the SNP mapping program thinks the variation is always one base long. The IUPac code for an MNP is “N”, which is why the loc_type is set to “range” when it should have been set to "range substitution". It may be possible to get the SNP BLAST program to consider the exact variation string and assign the correct loctype. The SNPdev group will discuss this and one of us will get back to you. (10/6/06)
Forward vs. Reverse strand Orientation
Define the term “orientation” as used in dbSNP.
Submissions to our database have arbitrary orientation relative to each other. If multiple submissions refer to the same SNP, they may cluster together in reverse orientation, so we also track the orientation of each submission relative to the exemplar ss. Please bear in mind that submitters to dbSNP are only required to provide some flanking sequence around the SNP for context. The SNPdev team does the positioning using BLAST and the resulting alignments. (3/13/05)
How do you assign strand orientation to a refSNP (rs) in the MapLoc element (from the Primary Sequence element), and how is the "orient" attribute determined?
The "orient" value is determined by mapping SNP flanking sequences to a contig sequence using BLAST.
Please note: a refSNP(rs) flanking sequence will never change orientation even if submitted SNPs in the opposite orientation are assigned to the refSNP (rs) cluster. Also, during a build, refSNP flanking sequences are BLASTed against contigs. If an rs maps to different contigs in each of two different builds, then the strand (or "orient" attribute) of the two different contigs to which the rs maps are in opposite orientation.
Here is an XML example of the above:
<PrimarySequence>
<PrimarySequence_dbSnpBuild>126</PrimarySequence_dbSnpBuild>
<PrimarySequence_gi>8077580</PrimarySequence_gi>
<PrimarySequence_source value="blastmb"/>
<PrimarySequence_accession>AC027456</PrimarySequence_accession>
<PrimarySequence_mapLoc>
<MapLoc>
<MapLoc_asnFrom>189600</MapLoc_asnFrom> <MapLoc_asnTo>189600</MapLoc_asnTo>
<MapLoc_locType value="exact"/>
<MapLoc_alnQuality>0.999994</MapLoc_alnQuality>
<MapLoc_orient value="forward"/>
<MapLoc_leftFlankNeighborPos>628</MapLoc_leftFlankNeighborPos>
<MapLoc_rightFlankNeighborPos>630</MapLoc_rightFlankNeighborPos>
<MapLoc_leftContigNeighborPos>189599</MapLoc_leftContigNeighborPos>
<MapLoc_rightContigNeighborPos>189601</MapLoc_rightContigNeighborPos>
<MapLoc_numberOfMismatches>1</MapLoc_numberOfMismatches>
<MapLoc_numberOfDeletions>0</MapLoc_numberOfDeletions>
<MapLoc_numberOfInsertions>0</MapLoc_numberOfInsertions>
</MapLoc>
</PrimarySequence_mapLoc>
</PrimarySequence>(8/17/06)
Individual refSNP Orientation Problems
dbSNP shows the ancestral allele for rs459552 as T, and that T is the most common allele for all populations. So why do the APC literature and colorectal cancer risk data show the A allele as the most common allele?
rs459552 has an A/T variation, with the T allele being the most common. Your finding that the A allele is the most common allele in the APC gene is also correct, however, since this SNP is located on the negative strand of the gene sequence. You can see this by looking at the refSNP page for rs459552. Please note that the mRNA(NM_000038) maps to the positive strand of the contig, indicating this SNP maps to the negative strand of the gene sequence.
In general, when dbSNP receives a SNP and its flanking sequence from the submitter, we map them as submitted (the submitter may submit the SNP on the positive or negative strand) to the various genome builds as the builds become available. It is therefore normal to see a SNP map to the negative strand of a gene.(8/28/06)
rs4897909 and rs4788229 are 4 base pairs apart, but if I superimpose the flanking sequences of both, the “K” variation of rs4897909 corresponds to an “A” base on the flanks of rs4788229.
The fact that rs4897909's G/T(K) aligns with rs4788229 base "A" in the same orientation does seem curious. These two rs numbers are both from the same computational SNP discovery program (SSAHA — you will find references to this program by reviewing the publications associated with these SNPs), and there are no other submitted SNP clusters that map to the same position. Also, there is no population frequency or individual genotype validation information available for these two SNPs.
Based on above information, I’m guessing that the "K" erroneously aligns to "A" in the same orientation for the following reasons:
First, the NCBI build has progressed to 36, whereas these SNPs were discovered on build 31. It might be that the build 31 contig upon which the SNP comparison was based contains errors. An example supporting this supposition: rs4897909 was based on a single submitted SNP (ss) from SSAHA: WI_SSAHASNP|NT_025920.10_372470, but searching NCBI for NT_025920 shows that this contig has been removed from the current genome build.
Second, dbSNP sets the SNP validation status based on frequency/genotype information or multiple submissions from non-computational methods. Roughly half of the SNPs in dbSNP are validated. At the present time, these two SNPs are not validated, and therefore can be considered suspect. (9/5/06)
I think "alleles" and "db SNP allele" may be switched in rs28944222, where dbSNP shows A/G; S, P; and in rs28944221 where dbSNP shows T/C; N, and D.
Both of these rs numbers mapped to the reverse strand of the contig, while the mRNA mapped to the forward strand:
========================> Contig [Forward]
-------------> mRNA [Forward]
<------ SNP [Reverse]
You must therefore use the complementing nucleotides of the SNP alleles in order to get the correct codon, which will in turn, code for the correct amino acid:
T/C is the complement of A/G and codes for S, P
A/G is the complement of T/C and codes for N, D (1/5/06)
Is it possible for the alleles and the orientation of a refSNP flanking sequence to change either between builds, or upon a new build of dbSNP (e.g. could aacaaaggct [A/C] acggaaggag change to ctccttccgt[G/T]agcctttgtt)?
The case you describe should never happen. (9/22/06)
Sequence Notation for Variations
Why do I sometimes see reference map positions of "unplaced" for SNPs, and what does being "unplaced" mean?
"Unplaced" is an attribute of contigs that are part of the genome assembly but have not yet been assigned a place in a chromosome (usually because there is not enough data to show where these contigs are supposed to go). Therefore, when SNPs map to “unplaced” contigs, dbSNP is unable to assign these SNPs a chromosome position.
Please note that SNP map weight is not affected by mapping to an “unplaced” contig.(10/25/06)
What does TER[*] for a non-synonymous coding change mean?
TER[*] means that the variation changed the codon to a termination codon (Ter or *), which causes premature termination of the protein. (10/25/06)
Does dbSNP have a file that contains SNPs in IUPAC code for the entire human genome?
Sorry, there isn't a file with the variations encoded in IUPAC code for the entire human genome. (1/10/05)
Can you recommend literature that describes standard nomenclature for sequence variations?
Recommendations for description of sequence variations can be obtained from the Human Genome Variation Society. (5/18/05)
Position Notation for Variations
The variation position notation for rs1922237 is: “12382526^12382527”. What does this notation mean?
In this case, the contig has a deletion at the SNP position and actually has a new "allele" (a deletion) that was not reported by the submitters to dbSNP.
When a contig has a deletion at a SNP position, it does not necessarily mean that the SNP is a DIP (Deletion Insertion Polymorphisim), since dbSNP does not always have an exhaustive list of variation alleles for each SNP.
The SNP to contig positions in SNP builds so far have three loc_type(s), each of which has a specific notation:
The variation position notation for rs10600037 is: “DIP(+) seq 12489885..12489886”. What does this notation mean?
If there are multiple bases at a SNP position on the contig, then ".." is used to show the location of the variation. In this example, rs10600037 has the deletion insertion polymorphisim (DIP)” —/TG, where the two bases "TG" align at the SNP variation position: 12489885..12489886 on the contig.
The SNP to contig positions in SNP builds so far have three loc_type(s), each of which has a specific notation:
between, where “asn-to = asn-from+1”, and is written as "asn-from^asn-to”. This loc_type does not depend on the SNP variation class, whether it is a true SNP or a DIP. (9/19/05)
The variation position notation for rs10532925 is: “DIP(+) seq 86843313”, but this notation does not have the “^”or “..” I’ve seen in the variation position notation for other refSNPs. What does this notation mean?
When there is one base at the SNP position on the contig, then just the base position is listed. rs10532925 has the deletion insertion polymorphisim (DIP)”—/AAA, but the contig allele is "A". So this is a case of the contig showing a new allele for the SNP.
The SNP to contig positions in SNP builds so far have three loc_type(s), each of which has a specific notation:
between, where “asn-to = asn-from+1”, and is written as "asn-from^asn-to”. This loc_type does not depend on the SNP variation class, whether it is a true SNP or a DIP. (9/19/05)
The variation position notation for rs4148752 is: “DIP(-) seq 12376835^12376836”. What does this notation mean?
rs4148752 has the deletion insertion polymorphisim: “—/AAAG”, where the contig has the deletion [thus, “DIP(-)”]
The SNP to contig positions in SNP builds so far have three loc_type(s), each of which has a specific notation:
between, where “asn-to = asn-from+1”, and is written as "asn-from^asn-to”. This loc_type does not depend on the SNP variation class, whether it is a true SNP or a DIP. (9/19/05)
1-based vs. 0-based (zero based) coordinate policy
The XML dump for build 126 has a -1 coordinate error that has propagated to all records. Is this change intentional?
In order to meet NCBI guidelines, dbSNP changed the sequence coordinate storage and representation in the XML, ASN.1, .bcp, and the Genotype/ genotype_by_gene files from 1-based to 0-based starting with dbSNP build 125.
ASN.1_flat files, Chromosome Reports, and the web page reports remain 1- based. (04/18/08)
Mapping Discrepancies
Why is rs6010717 mapped to an intron in one reference sequence, and mapped to an exon in another?
Although rs6010717 maps to the same reference contig in a unique position, this SNP is an intron in NM_000913, and an exon in NM_001007125 due to the splice variants listed in different gene models. (10/20/06)
Clicking on SNPs located in the “Graphic display” for the reference mRNA model of rs2534719, shows most of the SNPs to be introns, yet the graphic shows them all as coding. Why?
The problem appears to be that the results of the two mapping pipelines for the SNPs didn't agree. One pipeline maps the SNPs to the genome (results are shown in the Reference Cluster Report), while the other pipeline maps the SNPs to the mRNA (results are shown in the mRNA graphic display). Although such a case is rare, it appears that some of the genomic SNPs (ie. rs2534719) map at the exon/intron boundary, and can therefore be classified as intron or exon depending on whether they map to the genome or the mRNA. (5/26/05)
We have mouse SNPs with Celera IDs, but when we BLAST these sequences, we cannot find any corresponding rs numbers in the gene in question. How do we find these SNPs and how do we report them?
The mapping of SNPs to a gene is completed by BLAST analysis; the mapping is therefore computed rather than experimental. As a result, some submitted SNPs (ss) that have low complexity sequence may therefore map to locations on the genome other than the location at which you are looking.
SNPs are also mapped to multiple assemblies (i.e. reference and Celera), and since these assemblies are different, a SNP may map to different genes within the different assemblies. For example, rs13186575 is mapped to “HSPD1” on the Celera assembly and to “CDH12 on the reference assembly.
Some SNPs have discordance between their computed positions and their experimentally observed positions. We are planning to develop online tools that will allow users to provide annotations and corrections when such discordance arises. (3/3/06)
Why does rs1926736 map to the Celera assembly, but not to the NCBI reference assembly?
rs1926736 is uniquely mapped to both the reference and the Celera assemblies, as can be seen on the refSNP page for rs1926736. However, this SNP was annotated to a gene only on the Celera assembly. This seems to be an annotation error since MapViewer shows rs1926736 located in the NCBI reference assembly on contig NT_077569, and it maps to gene MRC1. rs1926736 will be annotated on the reference assembly and in gene region by the release of B126.(1/30/06)
Why do web queries of rs939820, rs10205833, and rs7597158 return sequences that do not match the exemplar sequences for these SNPs found using a database query?
When you refer to the sequences as ”not matching”, I assume that you are referring the fact that they don’t match at the point of variation, since in rs939820, for example, the two flanking sequences are the same with the exception of the point of variation.
On the RefSNP (rs) page, we show the rs FASTA using the IUPAC code for variations, while on the submitted SNP (ss) page, we show the ss fasta using the submitted observed sequence. In most cases, all member ss of an rs cluster have the same allelic states in the same orientation, so the rs variation matches the ss exemplar variation. There are cases, however, where the rs variation does not match the ss exemplar variation. For example, if an ss exemplar has an A/G variation, and another ss from the cluster in the same orientation has an A/T variation, then the rs allele list will read A/G/T since it includes all member ss alleles. If you viewed the rs allele list converted into IUPAC code (remember the refSNP page shows the flanking sequence in IUPAC), it would show a D representing A or G or T.
Most of the submitted SNPs have the same variations in the rs clusters you mentioned, but one or two of the ss in each cluster have an extra allele. All of the ss in the rs939820 cluster have an A/G variation, with the exception of one ss that has an -/A/G variation. All the ss in the rs10205833 cluster have a C/G variation, with the exception of one ss that has a C/G/T variation. Most of the ss in the rs7597158 cluster have an A/G variation, while the ss exemplar has a -/G variation. In these cases, the refSNP page variation list includes all allelic states: for rs939820, instead of an R, it is N; For rs10205833, instead of an S, it is B that represents for C or G or T; for rs7597158, since the ss exemplar has a -/G variation, while most of the other ss in this cluster have an A/G variation, the refSNP FASTA shows an N at the variation point.
I will update dbSNP’s variation representation for "mixed variation" clusters to include all the allele lists from all the submitted SNPs.(8/18/06)
rs4897909 and rs4788229 are 4 base pairs apart, but if I superimpose the flanking sequences of both, the “K” variation of rs4897909 corresponds to an “A” base on the flanks of rs4788229.
The fact that rs4897909's G/T(K) aligns with rs4788229 base "A" in the same orientation does seem curious. These two rs numbers are both from the same computational SNP discovery program (SSAHA — you will find references to this program by reviewing the publications associated with these SNPs), and there are no other submitted SNP clusters that map to the same position. Also, there is no population frequency or individual genotype validation information available for these two SNPs.
Based on above information, I’m guessing that the "K" erroneously aligns to "A" in the same orientation for the following reasons:
First, the NCBI build has progressed to 36, whereas these SNPs were discovered on build 31. It might be that the build 31 contig upon which the SNP comparison was based contains errors. An example supporting this supposition: rs4897909 was based on a single submitted SNP (ss) from SSAHA: WI_SSAHASNP|NT_025920.10_372470, but searching NCBI for NT_025920 shows that this contig has been removed from the current genome build.
Second, dbSNP sets the SNP validation status based on frequency/genotype information or multiple submissions from non-computational methods. Roughly half of the SNPs in dbSNP are validated. At the present time, these two SNPs are not validated, and therefore can be considered suspect. (9/5/06)
Is it possible for the alleles and the orientation of a refSNP flanking sequence to change either between builds, or upon a new build of dbSNP (e.g. could aacaaaggct [A/C] acggaaggag change to ctccttccgt[G/T]agcctttgtt)?
The case you describe should never happen. (9/22/06)
Multiple Mapping Positions for a Single SNP
Why does the chromosome report show multiple chromosome positions for a given rs number? rs41534544 appears to map to 6 different positions on chromosome 7.
There are a number of reasons why you will find SNPs hitting multiple times to a single chromosome. Let's look at the SNP you used as an example -- rs41534544.
If you look at the integrated maps section of rs41534544's refSNP page
You will see that this particular refSNP maps to the reference assembly, the Celera assembly, and the Venter Diploid assembly (HuRef).
SNPs can map to multiple places within a single chromosome if:
Think of the flanking sequence of a SNP as a linguistic phrase. The longer the phrase is, the more unique it is within the language you are speaking. So the longer a submitted SNP's flanking sequence is, the more likely it is that it will map uniquely to an assembly. So, since not all SNPs are really long, it is quite normal to have multiple hits for single SNP. During the dbSNP mapping process, however, we apply a special filtering algorithm to remove "false positives" (i.e. extra hits which almost every SNP has), which is why we have so many unique hits in our database. Without this algorithm we would have many SNPs that align at more than ten locations (mapping weight=10).
Please take a look at the Database Dictionary description for SNPMAPInfo, which contains the map weight definitions(scroll down to the column "weight"), and the Database Dictionary description for SNPContigLoc, which provides basic instructions for Retrieving a refSNP (rs) number with a "unique" map hit using a direct SQL query.
You may also wish to look at other FAQs in this (Multiple Mapping Positions for a Single SNP) section for more information. (01/31/08)
If a refSNP (rs) ID number is supposed to be unique in position, how can a rs ID locate to multiple chromosomes?
Some refSNPs (rs) can map to multiple chromosomes, if the SNP in question is located in a highly repetitive or duplicated region in our reference genome, which causes the SNP to be mapped to several chromosomes. If you provide us with the particular rs number in question, we will check the mapping positions for you.(5/1/06)
How do I tell if a SNP is uniquely mapped to the genome or if it appears in multiple places?
The NCBI genome assembly now contains sequence from more than one haplotype. At the moment, we assemble one complete reference haplotype and two small alternative haplotypes that span the HLA region. SNPMapInfo.weight is a table that is designed to provide a measure of multiplicity in a single human haplotype, where a weight >1 implies that the SNP hits at more than one locus.
In dbSNP build 119, less than 10% of the refSNPs assigned to human chromosome 7 have single sequence positions—a much lower percentage than for any other chromosome. Why is this?
There is an alternative assembly for chromosome 7. It was submitted from The Hospital for Sick Children in Toronto and has provided a secondary scaffold of chromosome 7 for SNP annotation since (at least) NCBI assembly 34.1. If you are using the dbSNP database tables to mine the data directly, then you need to use the ContigInfo table and restrict you query with ContigInfo.contig_label='reference'.
We count hits to multiple loci using SNPMapInfo.weight (distinguished from total number of hits across alternative haplotypes and alternative assemblies at the same locus). By this measure, our hits to chromosome 7 are over 90% unique (see below). The numbering system for physical positions on the two assemblies is similar, but distinct. Therefore, it is very important to restrict your queries to 'reference' (or 'HSC_TCAG', if you want to study the alternative assembly) to avoid misleading positional information.
Here's a query giving the distinct conti_label fields:
select distinct group_term, contig_label FROM ContigInfo
group_term contig_label
alt_assembly_1 HSC_TCAG
alt_haplotype_1 DR51
ref_haplotype reference
ref_par PARHere is the distribution of SNPMapInfo.weight on CHROMOSOME 7:
select m.weight,count (m.snp_id)
from SNPContigLoc l, SNPMapInfo m
where m.snp_id=l.snp_id AND contig_chr = '7'
group by m.weight
weight
1 788170
2 24824
3 28170
10 3853
Since b125 SNPs are mapped to both the NCBI reference assembly and to the Celera assembly, if an rs number is located on both assemblies, will they in the same position? Which assembly should we use?
The NCBI reference and the Celera assembles will differ depending on the region, but for any one region, one assembly may be more accurate than the other. Please note that the Celera assembly is not updated with new build cycles, and is therefore essentially static. The NCBI reference assembly is updated periodically. (1/12/06)
How do I tell if a SNP is uniquely mapped to the genome or if it appears in multiple places?
The NCBI genome assembly now contains sequence from more than one haplotype. At the moment, we assemble one complete reference haplotype and two small alternative haplotypes that span the HLA region. SNPMapInfo.weight is a table that is designed to provide a measure of multiplicity in a single human haplotype, where a weight >1 implies that the SNP hits at more than one locus.
Multiple Locations for Specific refSNP Numbers
Why does the chromosome report show multiple chromosome positions for a given rs number? rs41534544 appears to map to 6 different positions on chromosome 7.
There are a number of reasons why you will find SNPs hitting multiple times to a single chromosome. Let's look at the SNP you used as an example -- rs41534544.
If you look at the integrated maps section of rs41534544's refSNP page
You will see that this particular refSNP maps to the reference assembly, the Celera assembly, and the Venter Diploid assembly (HuRef).
SNPs can map to multiple places within a single chromosome if:
Think of the flanking sequence of a SNP as a linguistic phrase. The longer the phrase is, the more unique it is within the language you are speaking. So the longer a submitted SNP's flanking sequence is, the more likely it is that it will map uniquely to an assembly. So, since not all SNPs are really long, it is quite normal to have multiple hits for single SNP. During the dbSNP mapping process, however, we apply a special filtering algorithm to remove "false positives" (i.e. extra hits which almost every SNP has), which is why we have so many unique hits in our database. Without this algorithm we would have many SNPs that align at more than ten locations (mapping weight=10).
Please take a look at the Database Dictionary description for SNPMAPInfo, which contains the map weight definitions(scroll down to the column "weight"), and the Database Dictionary description for SNPContigLoc, which provides basic instructions for Retrieving a refSNP (rs) number with a "unique" map hit using a direct SQL query.
You may also wish to look at other FAQs in this (Multiple Mapping Positions for a Single SNP) section for more information. (01/31/08)
Why does rs3823342 have two different chromosome positions?
Starting with b125, the SNPs in dbSNP will be mapped to both the NCBI reference assembly and the Celera assembly. Because rs3823342 is mapped to both the NCBI reference assembly and the Celera assembly, the two different chromosome positions you found are for the two different assemblies. You can see this clearly in the Integrated Maps section of the rs3823342 report. (1/3/06)
Why does rs3128991 have two different local loci?
Starting with b125, the SNPs in dbSNP will be mapped to both the NCBI reference assembly and the Celera assembly. Because rs3128991is mapped to both the NCBI reference assembly and the Celera assembly, the two different local loci you found are for the two different assemblies. You can see this clearly in the Gene view section of the rs3128991 report. (1/3/06)
Why do we find rs386014 displayed in both HLA-A’s and HCG4P6’s batch report?
rs386014 is annotated to HLA-A on the Celera assembly using the gene model (contig---> mRNA---> gene). The same SNP is associated with HCG4P6 because the variation has been mapped within 2 kb of an mRNA transcript for the HLA complex group 4 pseudogene 6.(1/3/06)
dbSNP shows rs3658691 to "map exactly once on NCBI mouse chromosome", yet shows four mapping locations. How do I determine the most likely location for this SNP?
rs3658691 indeed "maps exactly once on NCBI mouse chromosome". "NCBI mouse" in this case, refers to the reference strain ("ref_strain"). But rs3658691 also maps to two locations on the Celera_Mmu16 assembly, as well as one location on alternative assembly called "129_substrain".
Look the column labeled "Group term" and "Group label" in the "Integrated Maps" section of the refSNP page to see the various contig assemblies to which an is mapped. (10/12/05)
I was searching for SNPs on chromosome 6 only, but if I click on rs4209360 in my search results, my search results show it on chromosome 16, too.
Some SNPs have low complexity flanking sequences, and may map by BLAST analysis to multiple chromosomes on the same assembly. (1/30/06)
- Mapping Weight
- How dbSNP Determines SNP Positions
- Chromosome Position and Contig Position
- Locate Disappearing RefSNP (rs) Numbers
- RefSNP (rs) Clusters Not Merging as Expected
- Definition of loctype values
- Reassigning loctype
- Forward vs. Reverse strand Orientation
- Sequence Notation for Variations
- Position Notation for Variations
- Mapping Discrepancies
- The dbSNP Mapping Process - SNP FAQ ArchiveThe dbSNP Mapping Process - SNP FAQ Archive
Your browsing activity is empty.
Activity recording is turned off.
See more...