

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union; Shore CK, Worku TL, Smith CW, et al., editors. Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities. Washington (DC): National Academies Press (US); 2024 Oct 30.

Regulatory Processes for Rare Disease Drugs in the United States and European Union: Flexibilities and Collaborative Opportunities.
Show details[The European Medicines Agency]. . . facilitates research into new medicines and encourages development, thereby translating progress in medical science into medicines with real health benefits for patients. In particular, it promotes the development of medicines for children and drugs to tackle rare diseases.
The European Medicines Agency (EMA) is a decentralized agency of the European Union (EU) that is responsible for evaluating the safety and efficacy of human and veterinary drugs in Europe. As stated on its public website, the agency also serves to protect and promote human health (EMA, n.d.-o). Unlike the U.S. Food and Drug Administration (FDA), EMA does not have the authority to approve medications. Instead, the agency issues guidance and makes authorization recommendations on medical products that the European Commission ultimately approves for marketing in the European Union (EMA, n.d.-o). Day-to-day operations at EMA are carried out by staff who rely on a network of experts from across Europe and collaboration with national competent authorities of EU member states to pool resources and coordinate work to regulate medicines for use in humans (EMA, n.d.-ad) (collectively called the EU medicines regulatory network, which is a World Health Organization–listed authority like FDA).
EMA has regulatory policies in place to support and incentivize drug development for rare diseases and conditions, including an orphan designation program, mechanisms for expedited review, and opportunities for sponsors and people with lived rare disease experience to engage with the agency. These programs may be used separately or in combination; each has its own eligibility criteria and benefits. This chapter examines the regulatory processes, authorities, and mechanisms used by the agency to approve drug products that are relevant to rare diseases. Where available, data are provided on the potential impact of these activities. The chapter is divided into sections on the drug review and approval, standards of evidence, orphan medicine designation, expedited regulatory programs, stakeholder engagement, rare disease programs, and transparency.
DRUG REVIEW AND APPROVAL
In the EU, EMA is responsible for assessing the benefits and risks of medicines. The 27 member states of the EU each have their own regulatory authority for licensing drugs, but EMA provides a centralized procedure that allows a sponsor to apply to one authority, undergo one scientific evaluation, and receive one marketing authorization (EMA, n.d.-s). Once the Committee for Medicinal Products for Human Use (CHMP), which is convened by EMA and made up of experts selected from the 27 member states, issues a positive opinion on a marketing authorization application, the European Commission (EC) makes a final legally binding decision on whether a medicine can be marketed in the EU. This authorization is legally binding and valid in all 27 member states, as well as the other states of the European Economic Area (Iceland, Norway, and Liechtenstein) (EMA, n.d.-e, n.d.-s).
This centralized procedure through EMA is mandatory for certain categories of products, including orphan medicines, advanced therapies (e.g., tissue-engineered medicines, cell- and gene-therapy), medicines derived from biotechnology processes, and medicines with new active substances to treat human immunodeficiency virus (HIV), cancer, diabetes, neurodegenerative diseases, auto-immune and other immune dysfunctions, and viral diseases (EMA, n.d.-e).1
The CHMP is responsible for assessing drug marketing authorization applications and making recommendations regarding those applications to the European Commission (EMA, n.d.-k). The CHMP works with other EMA committees, including the “Committee for Advanced Therapies, which leads the assessment of advanced therapy medicines (gene therapy, tissue engineering and cell-based medicines); the Pharmacovigilance and Risk Assessment Committee for aspects related to the medicine’s safety and risk management; the Paediatric Committee (PDCO) for aspects related to the medicine’s use in children; and the Committee for Orphan Medicinal Products (COMP) for orphan-designated medicines” (EMA, n.d.-r). During an assessment of a marketing authorization application, the CHMP is supported by a team of assessors from membership state agencies with relevant expertise (e.g., pharmaceutical, clinical, statistical), and the EMA secretariat provides technical, scientific, and administrative support (EMA, n.d.-r). See Figure 3-1 for an illustrative overview of the EMA process and the committees involved in each stage.
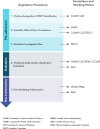
FIGURE 3-1
EMA committees in human medicines regulatory process. SOURCE: Adapted from European Medicines Agency: Presentation—Centralised procedure at the European Medicines Agency, https://www.ema.europa.eu/system/files/documents/presentation/wc500201043_en.pdf (more...)
The EMA secretariate is required to publish a European public assessment report (EPAR) for all medicines that have been granted or refused marketing authorization2 (European Union, 2004). The EPAR includes information about the drug product, including how the marketing authorization submission was assessed, reasons for the committee’s conclusions, and a public-friendly overview (see Table 3-1).
TABLE 3-1
Components of European Public Assessment Reports.
Standards of Evidence
EMA’s decision about whether to recommend a drug for marketing is based on whether its benefits outweigh its risks. This benefit–risk assessment can be complex due to the large amount of data involved and the inherent uncertainty around whether the available evidence is sufficient to assess benefits and risks (EMA, n.d.-f). For products aimed at rare disease, there are two types of “benefit” assessed. First, COMP may give the product orphan designation if the product offers a “significant benefit” compared with other treatments; this means that it offers an advantage in terms of efficacy, safety, or mode of use (EMA, n.d.-u; Thirstrup, 2023). Next, CHMP evaluates the application for marketing approval; the product can be approved if the benefits outweigh the risks (EMA, n.d.-k). The benefit–risk assessment framework allows EMA review to account for factors like small patient populations and limited data within the context of the specific rare disease.
EMA has not issued guidance on drug development for rare diseases and conditions. However, EMA has issued several disease-specific guidelines, many of which concern rare diseases, and held a workshop in 2015 on the demonstration of significant benefit of orphan medicines (EMA, 2016). In addition, there are EMA guidelines and reflection papers on a number of topics that are relevant to the collection of data on rare disease drug development. The details of these guidelines are discussed further in Chapter 4. Table 3-2 includes some guidelines:
TABLE 3-2
EMA Guidelines on Collection of Data.
Inclusion of Pediatric Populations
In EMA, the paediatric investigation plan (PIP) serves to ensure that all needed data to support a marketing authorization for children are collected. PIPs are required to be submitted when the Phase 2 dose is selected at the end of Phase 1 (Ungstrup and Vanags, 2023). All medicines seeking marketing authorization have to include a PIP unless the treatment is exempt due to referral or waiver (EMA, n.d.-h). Typically, waivers are provided to treatments that are likely to be ineffective or unsafe in children, intended for adult-only conditions, or unlikely to provide significant benefit over current treatment available to children (EMA, n.d.-h). The PIP is reviewed and agreed upon by the drug sponsor and EMA’s PDCO (EMA, n.d.-v). In early 2023, EMA launched a stepwise PIP pilot program which is designed to allow greater flexibility for sponsors that are developing innovative treatments. The stepwise PIP will allow sponsors to continue with development with a partial PIP in place rather than waiting for more data to support a full PIP (Al-Faruque, 2023). In qualitative interviews, sponsors indicated that the PIP can be restrictive to drug development and expect the stepwise program to ease some of the issues (see Appendix E for full methodology and results).
ORPHAN MEDICINE DESIGNATION
In 1999, the European Parliament adopted Regulation (EC) 141/2000, which established the COMP, laid out the process for the designation of orphan medicines, and identified the incentives available to designated products. An “orphan medicine” is defined as one that is intended to treat a life-threatening or chronically debilitating condition for which there is no satisfactory method of diagnosis or treatment available, and which affects not more than 5 in 10,000 people in the European Union community. Alternatively, if it is intended to treat a condition that affects more than 5 in 10,000 people, it can still be eligible for orphan designation if the market is unlikely to generate sufficient return on the investment without incentives (European Commission, n.d.).
To receive orphan designation, a drug usually must be targeted at a disease or condition for which there is no treatment available; if a treatment is available, a drug may still be eligible if it provides “significant benefit” to those affected by the condition. Significant benefit is defined as either a “clinically relevant advantage” or “a major contribution to patient care.” A clinically relevant advantage means that compared with previously approved treatments, the product offers either improved efficacy or improved safety and there is a reasonable probability that the patient will actually experience this benefit3 (Thirstrup, 2023). A product that represents a “major contribution to patient care” is one with improved availability or ease of use. For example, a product that does not require refrigeration would be an improvement over one that does require refrigeration, or a product that can be taken by pill would be an improvement over one that requires an injection. As noted in a presentation to the committee, the determination of “major contribution to patient care” can be more difficult than the determination of a “clinically relevant advantage” because it depends in part on what patients consider to be important (Thirstrup, 2023). A “significant benefit” cannot be claimed based on an “alternative mode of action per se, an increase in supply or availability due to a shortage of existing products, or higher pharmaceutical quality” (Thirstrup, 2023).
The condition at which an orphan drug is targeted must be clearly distinct from other conditions; differences in severity or stages do not make a condition distinct from others. “Condition” is defined under EC Guideline (ENTR/6283/00) as “any deviation(s) from the normal structure or function of the body, as manifested by a characteristic set of signs and symptoms (typically a recognized distinct disease or a syndrome)” (European Commission, 2014b). If the targeted condition is a subtype of a more common condition, there must be justification for restricting the medicine to the subgroup of patients. The patients must have “distinct and unique evaluable characteristics,” and such characteristics must be essential for the action of the medicine such that the medicine would not be effective in the larger group of patients with the common condition (European Commission, 2014a). According to Steffan Thirstrup, chief medical officer of EMA, in open session with the committee, differences in biomarkers are currently not accepted as evidence of a distinct condition (Thirstrup, 2023). The determination of whether a condition is considered distinct enough to qualify for orphan designation may change over time as the understanding of disease progresses and new therapies are developed (Thirstrup, 2023).
Process
Orphan medicine designation is a separate process from marketing authorization, with the orphan designation coming before marketing authorization. Sponsors submit a request for orphan medicine designation to the COMP, which examines applications for orphan designation from EMA (EMA, n.d.-d). During the 90-day evaluation process, the COMP reviews the application to assess whether the drug product meets the criteria for orphan medicine designation. The COMP examines whether the condition is life-threatening or chronically debilitating, determines the prevalence of the condition, assesses the medical plausibility that the product will treat the condition, and compares the proposed treatment with existing treatments, if any (EMA, n.d.-u). Typically, products are designated as orphan early in the drug development process, although designation may happen as late as the marketing authorization application (see Figure 3-2). The COMP will adopt a positive opinion or provide a list of questions and invite the sponsor to provide an oral explanation at the next COMP meeting (EMA, n.d.-d). There is an appeals process if the COMP opinion is negative. Following a COMP positive opinion, EMA sends the opinion to the European Commission, which is responsible for issuing a decision within 30 days of receipt (EMA, n.d.-d).
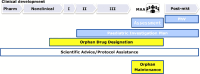
FIGURE 3-2
Orphan product designation and maintenance along drug life cycle. NOTES: I = Phase 1 clinical trials; II = Phase 2 clinical trials; III = Phase 3 clinical trials; MAA = marketing authorization application; Pharm = pharmacology studies; PhV = pharmacovigilance; (more...)
Once a product is designated as orphan, it undergoes the same quality, safety, and efficacy assessment as any other medical product. The CHMP is responsible for assessing the product for authorization, while the COMP is responsible for determining whether an orphan product can maintain its orphan designation (EMA, n.d.-k, n.d.-r). A product could lose its orphan designation if, for example, another product received marketing authorization first and the second product cannot demonstrate a significant benefit over the already-authorized product (EMA, n.d.-c; Thirstrup, 2023).
Benefits
Products that receive orphan designation can access a number of incentives (EMA, n.d.-t). First, sponsors can request protocol assistance from EMA at a reduced fee; this allows sponsors to get answers to questions about what types of studies are necessary to demonstrate the quality, benefits, risks, and significant benefit of the drug. Second, a product with orphan designation is mandated to use the centralized marketing approval process conducted by EMA. Third, products maintaining orphan designation at the time of approval receive 10 years of market exclusivity; this is extended to 12 years for products with an approved pediatric investigation plan. Fourth, sponsors applying for orphan designation pay reduced fees for regulatory activities, including marketing authorization application fees, fees for inspections before authorization, and fees for applications for post-approval changes. In addition, sponsors may be eligible for incentives available through individual EU member states. For companies classified as small and medium-sized enterprises (SMEs) that are developing a product with orphan designation, there may also be administrative and procedural assistance from EMA’s SME office and fee reduction (EMA, n.d.-t).
Approvals of Drugs with Orphan Designation
In the 20 years that EMA has issued orphan designation, over 2,730 products have received orphan designation, and over 230 of these have been recommended for marketing authorization (see Figure 3-3). As of 2023, 142 orphan designations are still active (Thirstrup, 2023). Of the active orphan designations, 98 received full authorization, 26 are under conditional authorization pending confirmatory data, and 18 were approved under “exceptional circumstances” in which the applicant is unlikely to be able to generate more data (Thirstrup, 2023). The largest number of orphan product designations are for congenital, familial, and genetic disorders, with significant numbers of products for blood and lymphatic systems disorders as well as neoplasms (Thirstrup, 2023). Orphan designations are published on the EMA website, along with minutes and agendas of the scientific committee meetings. Reports are published for both the initial assessment of orphan designation, and the assessment of orphan maintenance (Thirstrup, 2023).
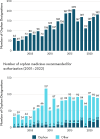
FIGURE 3-3
Designation and authorization of orphan medicines in the EU from 2001 to 2022. NOTE: EU = European Union. SOURCES: Presented to the Committee by Steffan Thirstrup, EMA, on December 4, 2023; adapted from European Medicines Agency: Orphan medicines in the (more...)
As described in Chapter 2, the committee examined the approval rates for orphan and non-orphan medicine applications between 2015 and 2020 (see Appendix D for full methodology). Overall, there was little difference in EMA approval rates for orphan and non-orphan medicine applications (see Figure 3-4) and no discernable differences in approval rates across therapeutic areas (see Figure 3-5).
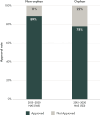
FIGURE 3-4
EMA approval rates for orphan and non-orphan drugs using expedited approval pathways from 2015 to 2020. NOTES: EMA = European Medicines Agency; NAS = new active substance. SOURCE: CIRS Data Analysis, 2024.
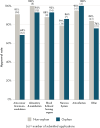
FIGURE 3-5
Approval rates for non-orphan and orphan NAS applications submitted to EMA from 2015 to 2020 per therapeutic area. NOTES: EMA = European Medicines Agency; NAS = new active substance; Other = other therapeutic areas not described in the top 5 therapeutic (more...)
EXPEDITED REGULATORY PROGRAMS
There are four expedited authorization pathways for drugs available through EMA. Each program has its own eligibility criteria, process, and benefits. While these pathways are available for any medicine that meets the criteria, they may be particularly relevant for products with orphan designation because orphan products are more likely to meet the criteria of the programs (e.g., meeting an unmet need, intended to treat a life-threatening disease, extremely rare indication). Expedited pathway programs and orphan medicine designation may be used separately or in combination; medicines targeted at rare, serious diseases—particularly those that have no existing treatment—may be eligible for multiple programs.
Priority Medicines
The Priority Medicines (PRIME) program is targeted at medicines with an unmet need—that is, where no treatment option exists or where a new therapy can offer a major benefit over existing therapies. Applicants must provide data that demonstrate a meaningful improvement of clinical outcomes (e.g., affecting morbidity or mortality of a disease) (EMA, n.d.-y).
Process
Sponsors must apply for PRIME during the early stages of clinical research. It is designed to help sponsors who have preliminary clinical evidence that demonstrates promising potential of the medicine to significantly address an unmet need. Applicants from small businesses and academia, who may have less experience in the regulatory world, can apply for early entry PRIME if they have compelling nonclinical data in a relevant model that shows proof of principle or if first-in-human studies demonstrate the desired effects and safety (EMA, n.d.-y).
Benefits
Applicants selected for the PRIME program receive a number of benefits throughout the regulatory process, including:
- Early appointment of a committee rapporteur
- Meeting with rapporteur and group of experts from EMA
- Appointment of PRIME scientific coordinator
- Iterative scientific advice on development and key issues
- Expedited follow-up scientific advice
- Submission readiness meeting
- Confirmation of potential for accelerated assessment (EMA, n.d.-y)
Applicants who are granted early entry PRIME status receive additional benefits, including an EMA product team, an introductory meeting about regulatory requirements, and total fee exemption for scientific advice for applicants from the European Economic Area (EMA, n.d.-y).
Impact
An EMA analysis of the PRIME program found that since its inception in 2016, PRIME has resulted in reduced overall time to marketing authorization, with PRIME products more likely to be granted accelerated assessment (EMA, 2022c). The analysis found that the benefits of PRIME were most pronounced for more complex products or applications that depend on smaller datasets, such as orphan diseases. EMA analysis found that 56 percent of PRIME products granted eligibility were orphan products (EMA, 2022c).
Accelerated Assessment
Medical products that are expected to be of “major public health interest,” particularly with respect to therapeutic innovation, may be approved for accelerated assessment, which reduces the timeframe for assessment and approval. There is no specific definition of “major public health interest;” each application is reviewed on a case-by-case basis. In general, a product eligible for accelerated assessment is one that involves new methods of therapy or improves on existing methods in order to address unmet needs (EMA, n.d.-a).
Process
Applicants must apply for accelerated assessment at least 2 or 3 months before submitting the application for marketing authorization. EMA recommends that applicants request a pre-submission meeting several months before requesting accelerated assessment in order to discuss their proposal and plan. Applicants who have already received PRIME status may receive confirmation during the clinical development phase that their product is eligible for accelerated assessment (EMA, n.d.-a).
Benefits
Assessment of a marketing authorization application typically takes 210 days (excluding “clock stops” for requests of additional information). Under accelerated assessment, this timeframe is reduced to 150 days (EMA, n.d.-a).
Impact
A study of orphan medicinal product (OMP) approvals in the EU between 2010 and 2022 found that about 24 percent of OMPs were eligible for accelerated assessment, and that use of accelerated assessment for OMPs increased significantly around 2015 (see Figure 3-6). The study found that the use of accelerated assessment is more frequent in OMPs than in nonorphan medicinal products (Bouwman et al., 2024).
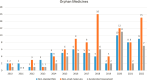
FIGURE 3-6
Evolution of the number of orphan medicinal products that have received a non-standard marketing authorization, are non-small molecules, and have benefited from accelerated assessment. NOTE: MA = marketing authorization. SOURCE: Bouwman et al., 2024. (more...)
Conditional Marketing Authorization
The conditional marketing authorization program is available for products for which the available clinical data are less comprehensive than usual but for which the benefits of providing the public access to the medicine immediately outweigh the risks inherent in approving a product with fewer clinical data. Medicines that are eligible for conditional marketing authorization are those that are intended to treat, prevent, or diagnose seriously debilitating or life-threatening diseases (including orphan medicines), or those that are needed for a public health emergency (e.g., a pandemic) (EMA, n.d.-l). Specifically, products must meet all of the following criteria:
- “the benefit-risk balance of the medicine is positive;
- “it is likely that the applicant will be able to provide comprehensive data post-authorization;
- “the medicine fulfills an unmet medical need;
- “the benefit of the medicine’s immediate availability to patients is greater than the risk inherent in the fact that additional data are still required” (EMA, n.d.-l).
Process
The request for conditional marketing authorization should be submitted along with the notification of intention to submit a marketing authorization application, about 6 to 7 months before the application. The formal request for conditional marketing authorization is submitted with the marketing authorization application, and the CHMP assesses the request and application together (EMA, n.d.-l). EMA encourages applicants to request a pre-submission meeting in order to discuss plans. If a conditional marketing authorization is granted, the sponsor is required to fulfill specific obligations within a defined time (e.g., collecting additional data). If further data indicate that the benefits do not outweigh the risks or if the sponsor fails to comply with its obligations, conditional authorization may be suspended or revoked. Conditional authorization can be converted to standard marketing authorization once obligations are fulfilled and more complete data confirm that the benefits outweigh the risks (EMA, n.d.-l).
Benefits
If a product is granted a conditional marketing authorization, the review timelines could be accelerated if there is sufficient evidence to meet a positive benefit-risk ratio.
Impact
Between 2010 and 2022 of the 192 OMPs that received marketing authorization, 41 (21 percent) were approved via the conditional approval pathway. Over this time period, there has been an increase in the use of conditional approvals, and OMPs have been approved via the conditional pathway more often than non-orphan medicines. Between 2010 and 2022, 6 of the 192 approved OMPs lost authorization; of these 6, 4 were conditional approvals (Bouwman et al., 2024).
Exceptional Circumstances
A marketing authorization applicant that is unable to provide comprehensive data on safety and efficacy may be eligible for authorization under the exceptional circumstances provision4 if one of the following is true:
- the indication for which the product is intended is so rare that the applicant cannot reasonably be expected to provide comprehensive data;
- it is not possible to gather comprehensive information due to the present state of scientific knowledge; or
- generally accepted principles of medical ethics preclude the collection of comprehensive data (Prilla, 2018).
Process
An applicant must submit a request for authorization under exceptional circumstances along with the notification of intention to submit a marketing authorization application. In the formal marketing authorization application, the applicant must provide justification for the inability to collect comprehensive data, a listing of the data that cannot be provided, and proposals for the specific procedures and obligations that will be conducted (e.g., conditions on prescribing the product). Unlike a conditional marketing authorization, the applicant is not required or expected to provide comprehensive data after authorization, and the authorization typically cannot be converted to a full authorization. Authorization is reviewed annually to assess the risk-benefit balance (EMA, n.d.-x).
Benefits
A product that is granted authorization under exceptional circumstances receives authorization with fewer data than are normally required, and the applicant is not expected to provide comprehensive data post-authorization (EMA, n.d.-x).
Impact
Exceptional circumstances is the least common type of approval for orphan products: of the 192 OMPs authorized for marketing between 2010 and 2022, 22 (11.5 percent) were approved under exceptional circumstances (Bouwman et al., 2024). However, orphan products make up the vast majority of approvals under exceptional circumstances; of the products licensed under exceptional circumstances in 2020, 82 percent were designated as orphan products at the time of approval (Marjenberg et al., 2020). Over two-thirds of OMPs approved under exceptional circumstances were biologicals or advanced therapeutic medicinal products (Bouwman et al., 2024).
STAKEHOLDER ENGAGEMENT
Sponsor Engagement
Sponsors will begin to formally work with EMA when submitting for marketing authorization. However, developers are encouraged to reach out to EMA for advice and feedback at any stage of the development of a medical product. The exchange of information between developers and EMA helps to ensure that studies are designed appropriately to generate evidence about safety and efficacy and that no major issues arise during the assessment for marketing authorization. Preparatory meetings are offered early in the process by EMA so that applicants can introduce their proposed plan and receive feedback, identify issues that they may need scientific advice on, ask regulatory questions that are outside the scope of scientific advice, and establish contact with agency staff; these meetings are free-of-charge (EMA, n.d.-z). Applicants shall use EMA’s IRIS platform5 to make requests for scientific advice, share information, and deliver documents. EMA charges a fee for scientific advice; this fee varies by the scope of the advice and fee waivers are available for orphan medicines, SMEs, and products for a public health emergency (EMA, n.d.-z). Individuals from small companies who participated in semi-structured interviews reported some issues with engaging EMA due to the decentralized process (see Appendix E for full methodology and results). They cited difficulties in working with individual countries rather than EMA directly.
Expert Engagement
EMA relies on external experts for a number of tasks. They may serve on committees or steering groups, may provide scientific expertise, or may perform compliance inspections on behalf of EMA. These experts are made available by the national competent authorities of the European Economic Area and must sign a declaration of interests each year. The list of experts is publicly available (EMA, n.d.-n). EMA launched a pilot program in 2020 in which experts are paid by their home institutions but hosted by EMA. The goal of the Collaborating Expert Programme is to provide a mechanism for EMA and external researchers to collaborate on important research questions that address regulatory decision-making (EMA, n.d.-j). Because the Collaborating Expert Programme is still in a pilot phase, the committee cannot comment on its usefulness for rare disease drug development.
Patient Engagement
Organizations, patients, and caregivers interact with EMA in a variety of ways all along the regulatory pathway (EMA, n.d.-q) (see Figure 3-7), a practice that is underpinned by EMA’s broader engagement framework for engaging patients and consumers throughout a medical products lifecycle (EMA, 2022a). Depending on the activity, patients may interact as representatives of their community, as representatives of an organization, or as individual experts. Specific opportunities for engagement include serving on EMA’s management board,6 scientific committees, and initiatives, such as the Accelerating Clinical Trials in the EU initiative (EMA, n.d.-b), which aims to further develop innovative clinical research in the European Union; attending consultations and workshops, assisting with providing advice on science and protocols, and involvement in the Patients’ and Consumers’ Working Party (PCWP) (EMA, n.d.-q). The PCWP was established in 2006 to provide a platform for the exchange of information and discussion of issues between EMA and patients. The group consists of representatives from patient and consumer organizations, representatives of EMA scientific committees, and observers. Organizations currently represented include the European Organisation for Rare Diseases, the Thalassemia International Federation, and the World Duchenne Organization among others (EMA, n.d.-w).
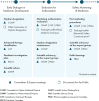
FIGURE 3-7
Patient involvement along the medicines lifecycle at EMA. SOURCE: Adapted from European Medicines Agency: Getting involved, https://www.ema.europa.eu/en/partners-networks/patients-and-consumers/getting-involved (accessed March 15, 2024).
Organizations and individuals can apply to work with EMA if they meet certain requirements. Among other criteria, organizations must be registered in the EU, have a clear mission and objectives, be representative of patients or consumers throughout the EU, and be transparent. Individual patients may also register to have an opportunity to use their real-life experiences to inform EMA’s work. EMA maintains a database7 of patients and calls upon them to provide their expertise in various EMA groups and in reviewing EMA documents prepared for the public (EMA, 2022a).
From January 2021 to May 2022, EMA ran a pilot program that explored how patients and organizations could be involved in the early stages of evaluation for orphan product applications. The pilot program included 37 products and involved identifying applications with orphan status; contacting organizations and inviting them to share information about aspects likely to be useful for evaluation (e.g., quality of life, unmet needs); and sharing this information with rapporteurs and product leads. Rapporteurs and product leads assessed whether the information provided added value, whether it was useful for assessing the application, and whether it should be included in the assessment report. An EMA assessment of the pilot program found that patient input highlighted new and valuable information and contributed to interim assessment reports in the marketing authorization process. This pilot program is being expanded to include health care professionals (EMA, 2022b).
RARE DISEASE INITIATIVES
EMA does not currently have programs specific to rare disease drug development. However, one of EMA’s stated goals is to encourage and facilitate the use of innovative methods in the development of medicines (EMA, n.d.-ac). To this end, EMA has several initiatives that support the development of innovative methods by fostering collaboration with academia and across the regulatory network. In addition to PRIME (described above), two of these initiatives are particularly relevant to rare diseases: the Innovation Task Force (ITF) and the EU Innovation Network (EU-IN).
Innovation Task Force
ITF8 is a multidisciplinary group with the task of ensuring coordination across EMA and providing a forum for early dialogue with applicants about innovative aspects of research and development (EMA, n.d.-aa). ITF plays a number of roles, including:
- Establishing early dialogue with applicants, in particular smaller or less-experienced applicants, in order to identify scientific, legal, and regulatory issues that may arise with emerging technologies, and to identify the need for specialized expertise during the development process;
- Exploring the regulatory and scientific implications of emerging therapies and technologies, in particular with respect to EMA’s scientific, legal, and regulatory requirements;
- Working with committees and other bodies to provide advice relating to research and development, for example, when there are uncertainties about whether a product fits the definition of a medicinal product;
- Increasing awareness of emerging therapies and technologies at EMA (EMA, 2014).
ITF holds briefing meetings with developers of innovative medicines, technologies, and methods. These meetings allow for an informal exchange of information in order to ensure that developers are well-informed about the regulatory process and requirements, and that EMA is prepared to assess emerging developments in innovative medicine. ITF is also responsible for overseeing the regulatory acceptance of “new approach methodologies” that are aimed at replacing animals in research (EMA, n.d.-aa).
EU Innovation Network
EU-IN9 is a group created in 2015 in order to strengthen the collaboration between EMA and regulatory authorities in EU member states, specifically with respect to emerging therapies and associated technologies. The group’s aim is to improve regulatory support at both the national and European levels, in order to make investing in innovative medicines more appealing. EU-IN provides training and regulatory support to developers, facilitates collaboration with the European medicines regulatory network, and works to anticipate how emerging therapies may require additional regulatory support. The group identifies emerging trends in science and technology that are relevant to research and development and publishes reports that explore these topics from a regulatory perspective. EU-IN collaborates with the heads of medicines agencies on a pilot project that supports the repurposing of medicines. The goal of this project is to help academics and nonprofit organizations gather or generate evidence on an established medicine for a new indication, with the aim of obtaining marketing authorization for the new indication (EMA, n.d.-m).
TRANSPARENCY
Under EU law and its own regulations, transparency is an important feature of EMA’s operations. EMA is required by law10 to publish an EPAR for each medicine that it approves or denies a marketing authorization. The EPAR is made up of several documents including a “public friendly” overview; information about the marketing authorization holder; details about the product, labeling, and indications; and the history of EMA’s assessment (e.g., orphan designation assessment, procedural steps taken) (EMA, n.d.-p). The EPAR also includes information on uncertainties about benefits and risks of the product (EMA, 2009). The EPAR is published after the EC issues a decision on an application as well as whenever product information is updated. Prior to the EC decision and EPAR publication, the relevant EMA committee (CHMP or the Committee for Veterinary Medicinal Products) publishes a “summary of opinion” following their adoption of the scientific opinion. Both the summary of opinion and the EPAR are available on the EMA website, and some components are published in multiple languages (EMA, n.d.-p).
A CHMP report in 2007 made recommendations about how CHMP could improve its methodology and could increase the transparency, consistency, and communication of its benefit–risk assessment (EMA, 2007). In 2009, EMA partnered with experts in decision theory on a 3-year project aimed at identifying decision-making models that could be used to make the agency’s work more consistent, transparent, and easier to audit (EMA, n.d.-f). In a 2012 commissioned report, EMA made a recommendation that the agency employ a two-level approach: first, a qualitative analysis using PrOACT-URL (problem formulation, objectives, alternatives, consequences, trade-offs, uncertainties, risk attitude, and linked decisions) along with an MCDA (multiple criteria decision analysis) model for quantitative assessment of more complex situations (EMA, 2012).
Starting in 2016, EMA has published clinical data submitted by sponsors to support marketing applications for human medicines (EMA, n.d.-i). EU law (Article 81 of No 536/2014) mandates that EMA make clinical trial data publicly available while also protecting personal data and commercially confidential information. In accordance with this law, EMA launched the Clinical Trial Information System (CTIS) in January 2022. At the time of the CTIS launch, transparency rules allowed sponsors to apply redactions or defer the publication of certain documents for a certain period of time depending on the type of clinical trial (EMA, 2015; Zhuleku and Preinfalk, 2023). EMA has revised its regulations on clinical data publication through the adoption of EMA/263067/2023 in October 2023. Major changes to the rules include (See EMA (2023b) for a detailed description):
- Selected data will be published using structured data fields that include information on study design, inclusion and exclusion criteria, primary and secondary endpoints, details on the product, and authorization status. These fields were chosen based on relevance for the public and researchers and cannot be redacted.
- The deferral mechanism is removed entirely for every trial category. Timing of document and data publication depends on a number of factors.
- The number of documents published will be rationalized in order to reduce complexity and workload.
In addition to the EPAR and clinical trial data, EMA makes other information available to improve transparency, including dates, agendas, minutes, and outcomes of its scientific committee meetings; information about staff and experts’ conflicts of interest; information about manufacturing inspections; pediatric investigation plans; and orphan designations (EMA, n.d.-ab).
Transparency, on the part of regulators, can be an important component in building trust in regulatory processes. At the same time, transparency must be balanced with the protection of personal and commercially confidential information. The introduction of clinical trial publications by EMA was accompanied by public debate (before and around 2016) about potential downsides of transparency, including the risks of damaging industry competitiveness, false health scares (due to publication of safety data that could be misinterpreted), and compromised patient privacy (Bonini et al., 2014; O’Donnell, 2016). As far as can be ascertained by the committee, none of these risks have materialized. Information provided by EMA may help inform drug developers about the successes and failures of past programs and studies, but it is not possible to quantify the benefits for the drug development ecosystem.
REFERENCES
- Al-Faruque F. EMA launches stepwise ‘PIP’ pilot. 2023. [March 12, 2024]. https://www
.raps.org /news-andarticles/news-articles /2023/2/ema-launches-stepwise-pip-pilot . - Bonini S, Eichler H-G, Wathion N, Rasi G. Transparency and the European Medicines Agency — Sharing of clinical trial data. New England Journal of Medicine. 2014;371(26):2452–2455. [PubMed: 25539105]
- Bouwman L, Sepodes B, Leufkens H, Torre C. Trends in orphan medicinal products approvals in the European Union between 2010–2022. Orphanet Journal of Rare Diseases. 2024;19(1):91. [PMC free article: PMC10900541] [PubMed: 38413985]
- CIRS (Centre for Innovation in Regulatory Science). Data analysis commissioned by the Committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union. National Academies of Sciences, Engineering, and Medicine; Washington, DC: 2024. Data analysis and summary to help inform the National Academies committee on Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union.
- EMA (European Medicines Agency). Points to consider on application with 1. Meta-analyses; 2. One pivotal study. 2001. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /points-consider-application-1meta-analyses-2one-pivotal-study_en.pdf . - EMA. Guideline on clinical trials in small populations. 2006. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /guideline-clinical-trials-small-populations_en.pdf . - EMA. Report of the CHMP working group on benefit-risk assessment models and methods. 2007. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/regulatory-procedural-guideline /report-chmp-working-group-benefit-risk-assessment-models-and-methods _en.pdf . - EMA. EPAR summaries for the public: A further step for the provision of better information about medicines. 2009. [July 11, 2024]. https://www
.ema.europa .eu/en/news/epar-summaries-public-further-step-provision-better-information-about-medicines . - EMA. Benefit-risk methodology project: Work package 4 report: Benefit-risk tools and processes. 2012. [August 1, 2024]. https://eprints
.lse.ac .uk/64630/1/Wok%20package%204.pdf . - EMA. Mandate of the EMA Innovation Task Force (ITF). 2014. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/other /mandate-european-medicines-agency-innovation-task-force-itf_en.pdf . - EMA. Appendix, on disclosure rules, to the “functional specifications for the EU portal and EU database to be audited - EMA/42176/2014”. 2015. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/other /appendix-disclosure-rules-functional-specifications-eu-portal-and-eudatabase-be-audited_en.pdf . - EMA. Demonstrating significant benefit of orphan medicines: Concepts, methodology and impact on access: Workshop report. 2016. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/report /workshop-report-demonstrating-significant-benefit-orphan-medicines-conceptsmethodology-and-impact-access_en.pdf . - EMA. Guideline on registry-based studies. 2021. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /guideline-registry-based-studies_en .pdf-0 . - EMA. Engagement framework: EMA and patients, consumers and their organizations. 2022a. [August 1, 2024]. https://www
.ema.europa .eu/system/files/documents /other/updated _engagement_framework _-_ema_and_patients _consumers_and_their _organisations_2022-en.pdf . - EMA. Pilot on early dialogue with patient organisations for orphan marketing authorisation applications: Outcome Report. 2022b. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/report /pilot-early-dialogue-patient-organisations-orphan-marketing-authorisation-applications-outcome-report_en.pdf . - EMA. PRIME: Analysis of the first 5 years’ experience. 2022c. [August 1, 2024]. https://www
.ema.europa .eu/system/files/documents /report/2022-03 _prime_5_years_report _updated_2022-04-05-en.pdf . - EMA. Reflection paper on establishing efficacy based on single arm trials submitted as pivotal evidence in a marketing authorisation. 2023a. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /reflection-paper-establishing-efficacy-based-single-arm-trialssubmitted-pivotal-evidence-marketing-authorisation_en.pdf . - EMA. Revised CTIS transparency rules. 2023b. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/other /revised-ctis-transparency-rules_en .pdf . - EMA. Reflection paper on use of real-world data in non-interventional studies to generate real-world evidence. 2024. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/scientific-guideline /reflection-paper-use-real-world-data-non-interventional-studies-generate-real-worldevidence_en.pdf . - EMA. Accelerated assessment. [August 15, 2024]. n.d.-a. https://www
.ema.europa .eu/en/human-regulatory-overview /marketing-authorisation /accelerated-assessment . - EMA. Accelerating Clinical Trials in the EU (ACT EU). [August 1, 2024]. n.d.-b. https://www
.ema.europa .eu/en/human-regulatory-overview /research-development /clinical-trials-human-medicines /accelerating-clinical-trials-eu-act-eu . - EMA. Applying for marketing authorisation: Orphan medicines. [July 10, 2024]. n.d.-c. https://www
.ema.europa .eu/en/human-regulatory-overview /marketing-authorisation /orphan-designationmarketing-authorisation /applying-marketing-authorisation-orphan-medicines . - EMA. Applying for orphan designation. [February 20, 2024]. n.d.-d. https://www
.ema.europa .eu/en/human-regulatory-overview /research-development /orphan-designation-research-development /applyingorphan-designation . - EMA. Authorisation of medicines. [March 18, 2024]. n.d.-e. https://www
.ema.europa .eu/en/about-us/what-wedo /authorisation-medicines . - EMA. Benefit-risk methedology. [March 19, 2024]. n.d.-f. https://www
.ema.europa .eu/en/about-us/what-we-do /regulatory-science-research /benefit-risk-methodology . - EMA. The centralised procedure at the EMA. [August 1, 2024]. n.d.-g. https://www
.ema.europa .eu/system/files/documents /presentation/wc500201043_en.pdf . - EMA. Class waiviers. [August 15, 2024]. n.d.-h. https://www
.ema.europa .eu/en/human-regulatory-overview /research-and-development /paediatric-medicines-research-and-development /paediatricinvestigation-plans /class-waivers . - EMA. Clinical data publication. [July 10, 2024]. n.d.-i. https://www
.ema.europa .eu/en/human-regulatory-overview /marketing-authorisation /clinical-data-publication . - EMA. Collaborating experts. [August 15, 2024]. n.d.-j. https://careers
.ema.europa .eu/content/CollaboratingExpert /?locale=en_GB . - EMA. Committee for Medicinal Products for Human Use (CHMP). [August 15, 2024]. n.d.-k. https://www
.ema.europa .eu/en/committees/committee-medicinal-products-human-use-chmp . - EMA. Conditional marketing authorization. [February 20, 2024]. n.d.-l. https://www
.ema.europa .eu/en/humanregulatory-overview /marketing-authorisation /conditional-marketing-authorisation . - EMA. EU Innovation Network (EU-IN). [July 10, 2024]. n.d.-m. https://www
.ema.europa .eu/en/committees/working-parties-other-groups /eu-innovation-network-eu . - EMA. European experts. [March 15, 2024]. n.d.-n. https://www
.ema.europa .eu/en/about-us/how-we-work /european-medicines-regulatory-network /european-experts . - EMA. European Medicines Agency (EMA). [August 15, 2024]. n.d.-o. https:
//european-union .europa.eu/institutions-law-budget /institutions-and-bodies /search-all-eu-institutions-and-bodies /europeanmedicines-agency-ema_en#:~:text =The %20European%20Medicines %20Agency%20(EMA,European %20Economic%20Area%20(EEA) - EMA. European public assessment reports: Background and context. [March 15, 2024]. n.d.-p. https://www
.ema.europa .eu/en/medicines/what-we-publish-medicines-and-when /european-public-assessment-reports-background-and-context . - EMA. Getting involved. [March 15, 2024]. n.d.-q. https://www
.ema.europa .eu/en/partners-networks /patientsand-consumers /getting-involved . - EMA. How EMA evaluates medicines for human use. [August 15, 2024]. n.d.-r. https://www
.ema.europa .eu/en/about-us/what-we-do /authorisation-medicines /how-ema-evaluates-medicines-human-use . - EMA. Obtaining an EU marketing authorisation, step-by-step. [August 15, 2024]. n.d.-s. https://www
.ema.europa .eu/en/human-regulatory-overview /marketing-authorisation /obtaining-eu-marketingauthorisation-step-step . - EMA. Oprhan incentives. [February 19, 2024]. n.d.-t. https://www
.ema.europa .eu/en/human-regulatory-overview /research-and-development /orphan-designation-research-and-development/orphanincentives . - EMA. Orphan designation: Overview. [June 25, 2024]. n.d.-u. https://www
.ema.europa .eu/en/human-regulatory-overview /orphan-designation-overview . - EMA. Paediatric investigation plans. [August 15, 2024]. n.d.-v. https://www
.ema.europa .eu/en/human-regulatoryoverview /research-development /paediatric-medicines-research-development /paediatricinvestigation-plans . - EMA. Patients’ and Consumers’ Working Party. [August 6, 2024]. n.d.-w. https://www
.ema.europa .eu/en/committees/working-parties-other-groups /comp-working-parties-other-groups /patients-consumersworking-party . - EMA. Pre-authorization guidance. [February 10, 2024]. n.d.-x. https://www
.ema.europa .eu/en/human-regulatoryoverview /marketing-authorisation /pre-authorisation-guidance . - EMA. PRIME: Priority Medicines. [August 15, 2024]. n.d.-y. https://www
.ema.europa .eu/en/human-regulatoryoverview /research-development /prime-priority-medicines . - EMA. Scientific advice and protocol assistance. [March 10, 2024]. n.d.-z. https://www
.ema.europa .eu/en/humanregulatory-overview /research-development /scientific-advice-protocol-assistance . - EMA. Supporting innovation. [July 10, 2024]. n.d.-aa. https://www
.ema.europa .eu/en/human-regulatoryoverview /research-development /supporting-innovation . - EMA. Transparency. [March 5, 2024]. n.d.-ab. https://www
.ema.europa .eu/en/about-us/how-we-work /transparency . - EMA. What we do. [July 10, 2024]. n.d.-ac. https://www
.ema.europa .eu/en/about-us/what-we-do . - EMA. Who we are. [July 9, 2024]. n.d.-ad. https://www
.ema.europa .eu/en/about-us/who-we-are . - European Commission. Official Journal of the European Union. 2014a. [August 1, 2024]. Commission notice on the application of Articles 3, 5 and 7 of Regulation (EC) No 141/2000 on orphan medicinal products. https://eur-lex
.europa .eu/legal-content/EN /TXT/PDF/?uri=OJ:JOC_2016_424_R_0003 . - European Commission. Guideline on the format and content of applications for designation as orphan medicinal products and on the transfer of designations from one sponsor to another. 2014b. [August 1, 2024]. https://health
.ec.europa .eu/system/files /2016-11/2014-03_guideline_rev4_final_0 .pdf . - European Commission. Rare diseases. [August 15, 2024]. n.d. https://health
.ec.europa .eu/european-referencenetworks /rare-diseases_en . - European Union. Regulation (EC) NO 726/2004 of the European Parliament and of the Council. 2004. [August 1, 2024]. https://eur-lex
.europa .eu/LexUriServ/LexUriServ .do?uri=OJ:L:2004 :136:0001:0033:en:PDF . - Marjenberg Z, Teague R, Skeldon G. PMU50 a review of exceptional circumstances marketing authorisations granted by the European Medicines Agency. Value in Health. 2020;23:S611.
- O’Donnell P. Will EMA rules calm the disclosure debate. 2016. [August 1, 2024]. https://www
.appliedclinicaltrialsonline .com /view/will-ema-rules-calm-disclosure-debate . - Prilla S. Legal basis & types of approvals. 2018. [August 1, 2024]. https://www
.ema.europa .eu/en/documents/presentation /presentation-legal-basis-and-types-approvals-s-prilla_en.pdf . - Thirstrup S. Orphan medicines in EU: Processes to Evaluate the Safety and Efficacy of Drugs for Rare Diseases or Conditions in the United States and the European Union (Meeting 2 - Hybrid). 2023.
- Ungstrup E, Vanags D. Aligning global drug development for pediatric populations. 2023. [August 15, 2024]. https://www
.pharmalex .com/thought-leadership /blogs/aligning-global-drug-developmentfor-pediatric-populations/ - Zhuleku E, Preinfalk F. Clinical trial regulation: Balancing transparency requirements and protection of CCI – new Q&A published. 2023. [March 10, 2024]. https://www
.allenovery .com/en-gb/global/blogs /life-sciences-talk /clinical-trial-regulation-balancing-transparencyrequirements-and-protection-of-cci--new-qa-published .
Footnotes
- 1
For more information on the scope of EMA centralised procedure, see https://www
.ema.europa .eu/en/about-us/what-we-do /authorisation-medicines (accessed June 13, 2024). - 2
Regulation (EC) No. 726/2004. Available at https://eur-lex
.europa .eu/LexUriServ/LexUriServ .do?uri=OJ:L:2004 :136:0001:0033:en:PDF (accessed July 5, 2024). - 3
Regulation (EC) No. 847/2000.
- 4
For more information, see https://www
.ema.europa .eu/en/glossary/exceptional-circumstances (accessed July 9, 2024). - 5
For more information, see https://iris
.ema.europa.eu/ (accessed July 9, 2024). - 6
EMA’s Management Board sets the agency’s budget and approves its yearly work plans. For more information, see https://www
.ema.europa .eu/en/about-us/who-we-are /management-board (accessed March 18, 2024). - 7
For more information, see https://fmapps
.ema.europa .eu/stakeholders/signup.php (accessed March 15, 2024). - 8
For more information, see https://www
.ema.europa .eu/en/human-regulatory-overview /research-and-development /innovation-medicines (accessed July 9, 2024). - 9
For more information, see https://www
.hma.eu/about-hma /working-groups /eu-innovation-network-eu-in.html (accessed July 7, 2024). - 10
Reference (EC) No. 726/2004.
- EMA Flexibilities, Authorities, and Mechanisms - Regulatory Processes for Rare D...EMA Flexibilities, Authorities, and Mechanisms - Regulatory Processes for Rare Disease Drugs in the United States and European Union
Your browsing activity is empty.
Activity recording is turned off.
See more...