

Download a gene ortholog data package
Download a gene ortholog dataset for a gene using the datasets command-line tool.
Download a gene ortholog data package
Quick overview
Gene orthologs can be retrieved by gene-id, accession or symbol.
gene-id and accession are unique identifiers. As a consequence, the associated taxon is implied. For example: for the human BRCA1 DNA repair associated gene and its gene orthologs in cat and Florida manatee:
symbol is not a unique identifier (human and cat have the same symbol), so it’s necessary to specify a taxon.
datasetsuses human as default species.
| Species | gene-id | accession | symbol |
|---|---|---|---|
| Human | 672 | NM_007297.4 | BRCA1 |
| Cat | 101081937 | XM_019817934.2 | BRCA1 |
| Florida manatee | 101356605 | XM_023725233.1 | LOC101356605 |
In the examples below, we will use NCBI Datasets command line tool
datasets download and datasets summary commands. In short, datasets summary returns only metadata in JSON format, while datasets download retrieves a gene data package
including both metadata and sequence files.
Simplest example: retrieve one gene ortholog set
All of the following commands will download the same gene ortholog set:
datasets download ortholog gene-id 672
datasets download ortholog symbol brca1
datasets download ortholog symbol brca1 --taxon human
datasets download ortholog accession NM_007297.4
Retrieve multiple gene ortholog sets based on a gene list
datasets can retrieve multiple ortholog sets based on a list of symbols, accessions or gene-ids. Currently, datasets does not separate each ortholog set into its own files. All sets will be saved in a single data package.
For example: if we provide a list of gene-ids (one per line or comma-separated) using the flag --inputfile, datasets will iterate over those and save the results as a single data package.
$ cat genelist.txt
672
4157
3206
$ datasets download ortholog gene-id --inputfile genelist.txt --filename ort.zip
$ unzip ort.zip -d ort
$ tree ort
ort
|-- README.md
`-- ncbi_dataset
`-- data
|-- data_report.jsonl
|-- data_table.tsv
|-- dataset_catalog.json
|-- gene.fna
|-- protein.faa
`-- rna.fna
If we want each ortholog data package to be saved separately, we can use a loop instead:
Command:
cat gene.list | while read GENE; do
datasets download ortholog gene-id "${GENE}" --filename "${GENE}".zip;
done
Result:
Found 306 genes in set
Downloading: 672.zip 9.53MB done
Found 259 genes in set
Downloading: 4157.zip 606kB done
Found 409 genes in set
Downloading: 3206.zip 1.33MB done
In this case, the list of genes must have one gene-id per line.
Filter an ortholog gene set by taxon
datasets offers an option to filter the ortholog set by taxon (any level) using the flag --taxon-filter. For example: you can filter the BRCA1 (gene-id 672) ortholog set to include only members of the otter family Mustelidae:
You can get a list of species in the otter family for which gene orthologs of human BRCA1 have been calculated using datasets summary with jq:
datasets summary ortholog gene-id 672 --taxon-filter mustelidae | jq '.genes.genes[].gene.taxname'
"Mustela putorius furo"
"Enhydra lutris kenyoni"
"Mustela erminea"
"Lontra canadensis"
"Neogale vison"
"Meles meles"
The full BRCA1 ortholog set includes 306 species, while the Mustelidae set has only 6 species.
Alternatively, you can download a data package for these otter family gene orthologs:
datasets download ortholog gene-id 672 --taxon-filter mustelidae --filename mustelidae.zip
Found 6 genes in set
Downloading: mustelidae.zip 260kB done
Retrieve an ortholog set by symbol using the --taxon flag
By default, datasets will assume the taxon to be human (Taxonomy ID: 9606) when requesting an ortholog set by symbol. If we request a symbol for which no human gene is included in the ortholog set, we get an error without the --taxon flag.
For example, when we query by the symbol, syna:
$ datasets summary ortholog symbol syna
The gene symbol that you specified, (syna) is either not a recognized gene symbol or not unique for the specified organism. Please try again using a Gene ID or a unique gene symbol."
Error: No genes found for search term
If we specify mouse (TaxId: 10090) with the flag --taxon, then datasets will return the syna ortholog set:
$ datasets summary ortholog symbol syna --taxon 10090
How to retrieve ortholog metadata
Using datasets summary and jq
You can use the summary option in datasets coupled with jq to retrieve ortholog metadata. For example, let’s say that you want to know which species are included in a certain ortholog set, as well as the gene-ids and gene symbols for each of them.
Command:
datasets summary ortholog symbol brca1 | \
jq -r '.ortholog_set_id as $oid
| .genes.genes[].gene
| [$oid, .taxname, .gene_id, .symbol]
| @csv'
Result (first 10 lines):
672,"Sus scrofa","100049662","BRCA1"
672,"Equus caballus","100051990","BRCA1"
672,"Taeniopygia guttata","100224649","BRCA1"
672,"Oryctolagus cuniculus","100347269","BRCA1"
672,"Callithrix jacchus","100388186","BRCA1"
672,"Pongo abelii","100439533","BRCA1"
672,"Ailuropoda melanoleuca","100480891","BRCA1"
672,"Anolis carolinensis","100553919","brca1"
672,"Nomascus leucogenys","100580360","BRCA1"
672,"Loxodonta africana","100653763","BRCA1"
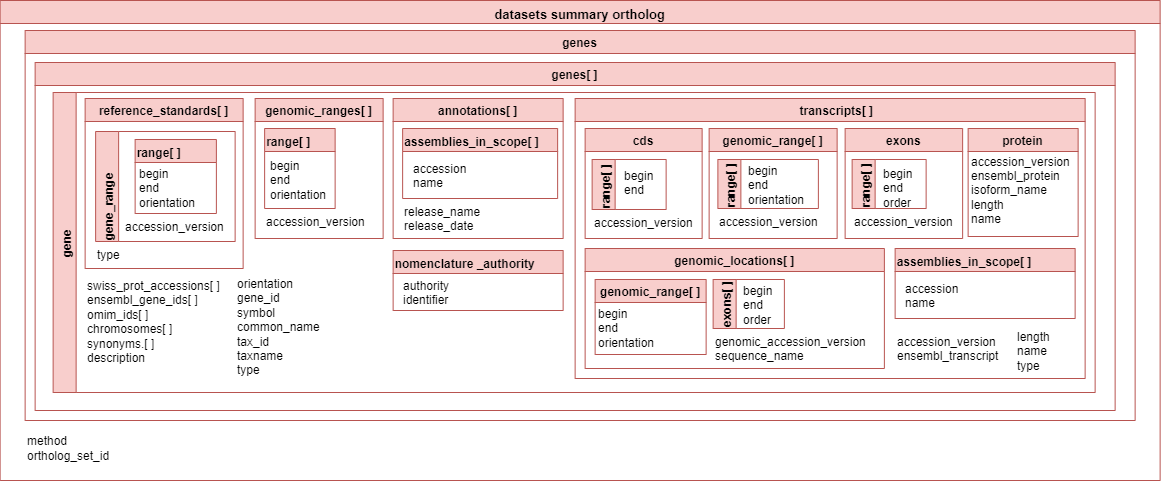
The image below shows how the datasets summary JSON output for orthologs is organized.
Using dataformat
In addition to datasets, we have the NCBI dataformat command line tool
that can be used to extract metadata from the gene data report
included with the data packages.
Download a gene ortholog data package for BRCA1:
datasets download ortholog symbol brca1 --filename brca1.zip
Create a tsv file from the data package using dataformat
dataformat tsv gene --package brca1.zip --fields tax-name,gene-id,symbol > brca1.tsv
head brca1.tsv
Result:
Taxonomic Name NCBI GeneID Symbol
Sus scrofa 100049662 BRCA1
Equus caballus 100051990 BRCA1
Taeniopygia guttata 100224649 BRCA1
Oryctolagus cuniculus 100347269 BRCA1
Callithrix jacchus 100388186 BRCA1
Pongo abelii 100439533 BRCA1
Ailuropoda melanoleuca 100480891 BRCA1
Anolis carolinensis 100553919 brca1
Nomascus leucogenys 100580360 BRCA1
- transcript:
seqtk subseq rna.fna transcript.list > rna_longest.fna - protein:
seqtk subseq protein.faa protein.list > protein_longest.faa