

Abstract
DNA topoisomerase IIα (TOP2A) plays a vital role in replication and cell division by catalytically altering DNA topology. It is a prominent target for anticancer drugs, but clinical efficacy is often compromised due to chemoresistance. In this study, we investigate the role of TOP2A O‐GlcNAcylation in breast cancer cells and patient tumor tissues. Our results demonstrate that elevated TOP2A, especially its O‐GlcNAcylation, promotes breast cancer malignant progression and resistance to adriamycin (Adm). O‐GlcNAcylation at Ser1469 enhances TOP2A chromatin DNA binding and catalytic activity, leading to resistance to Adm in breast cancer cells and xenograft models. Mechanistically, O‐GlcNAcylation‐modulated interactions between TOP2A and cell cycle regulators influence downstream gene expression and contribute to breast cancer drug resistance. These results reveal a previously unrecognized mechanistic role for TOP2A O‐GlcNAcylation in breast cancer chemotherapy resistance and provide support for targeting TOP2A O‐GlcNAcylation in cancer therapy.
Keywords: catalytic activity, chemotherapy resistance, O‐GlcNAcylation, TOP2A
Subject Categories: Cancer; DNA Replication, Recombination & Repair; Post-translational Modifications & Proteolysis
O‐GlcNAcylation at Ser1469 enhances chromatin binding and catalytic activity of TOP2A. O‐GlcNAcylation also enhances the interaction between TOP2A and cell cycle regulators, thereby playing a crucial role in breast cancer progression and Adm resistance.

Introduction
Two isoforms of topoisomerase II (TOP2A and TOP2B) are essential enzymes in regulating the homeostasis of DNA topology and cellular survival during replication, transcription, and cell proliferation (Nitiss, 2009; Pommier et al, 2016). TOP2A is associated with cell mitosis and division by catalyzing cleavage/ligation double‐strand DNA breaks that disentangle the superhelical and interlinked states of genomic DNA (Pommier et al, 2022). Because of its critical role, TOP2A has been identified as a major target for developing antineoplastic chemotherapeutic drugs that hamper its DNA cleavage activity (McClendon & Osheroff, 2007; Chen et al, 2015; Hevener et al, 2018). These drugs are widely used in the clinic for a variety of both hematological and solid malignancies, including breast cancer (Bailly, 2012). Chemotherapy drug (e.g., adriamycin, Adm) stabilize the covalent TOP2A‐DNA cleavage complex and block ligation of the temporary strand breaks, overwhelming cancer cells and causing cell death (Tewey et al, 1984; Nitiss, 2009). However, the development of drug resistance is the most common clinically encountered phenomenon that leads to the failure of breast cancer therapy (Coley, 2008; Li et al, 2008). Exploration of the mechanisms through which TOP2A resistance occurs has huge implications for improving therapeutic efficacy.
The catalytic activity of TOP2A is modulated by several different mechanisms in mammalian cells (Chen et al, 2015; Lotz & Lamour, 2020). Among the multiple folded domains in this evolutionarily conserved enzyme, the C‐terminal domain (CTD) with a highly variable amino acid sequence and low complexity structure is thought to influence TOP2A cellular localization and catalytic activity (Dougherty et al, 2021). Accumulating evidence shows that the TOP2A CTD plays an important role in regulating TOP2A catalytic activity and cancer therapeutic responsiveness (Linka et al, 2007; Kozuki et al, 2017; Hoang et al, 2020). This region contains a variety of posttranslational modifications (PTMs) that influence TOP2A biological functions or stability (Lotz & Lamour, 2020). The hyperphosphorylation of TOP2A was linked to drug response in breast and other cancer cells (Matsumoto et al, 1997; Chikamori et al, 2003; Gmeiner & van Waardenburg, 2021). Introduction of ubiquitin is another mechanism of chemotherapy resistance, as it not only alters TOP2A biochemical activity but also depletes target levels through proteasome degradation (Guturi et al, 2016; Fielding et al, 2018). Recently, we discovered that chromatin complexes are prevalently modified by an O‐linked N‐acetylglucosamine moiety (O‐GlcNAc), which contributes to chemotherapy resistance in cancer cells (Liu et al, 2020). This finding implies that glycosylation may be involved in modulating TOP2A activity.
O‐GlcNAcylation is a monosaccharide modification that occurs on the serine or threonine residues of nuclear and cytoplasmic proteins (Yang & Qian, 2017). Only O‐GlcNAc transferase (OGT) and O‐GlcNAcase (OGA) dynamically and reversibly catalyze this PTM. O‐GlcNAcylation is critical in maintaining chromatin physiological function (Ma et al, 2021). Abnormal chromatin‐associated O‐GlcNAc modification can affect chromatin homeostasis, gene transcription, DNA–protein, and protein–protein interactions in various tumors. Topoisomerase I (TOP1) was previously demonstrated as an O‐GlcNAcylated protein in cell lines and animal models (Noach et al, 2007). O‐GlcNAcylation affects TOP1 DNA relaxation activity and downstream gene expression (Levi et al, 2012). However, it is currently unknown whether TOP2A is O‐GlcNAc‐modified, and the mechanism by which interplay between TOP2A and O‐GlcNAcylation affects breast cancer chemotherapeutic drug resistance remains unclear.
In the current study, we provide evidence that TOP2A is hyper‐O‐GlcNAcylated by OGT at the conserved serine 1469 (S1469) in Adm‐resistant breast cancer cells. We further compared biochemical activity between O‐GlcNAcylated and unmodified TOP2A and revealed that this PTM induces TOP2A catalytic DNA cleavage and decatenation. Meanwhile, O‐GlcNAcylation enhances the interaction between TOP2A and cell cycle regulators, in turn playing a crucial role in breast cancer progression and Adm resistance in vitro and in vivo. Our results provide novel mechanistic insights into TOP2A‐mediated breast cancer malignant progression and provide potential therapeutic opportunities for reversing breast cancer drug resistance by blocking TOP2A O‐GlcNAcylation.
Results
TOP2A and cellular O‐GlcNAcylation are significantly upregulated in breast cancer
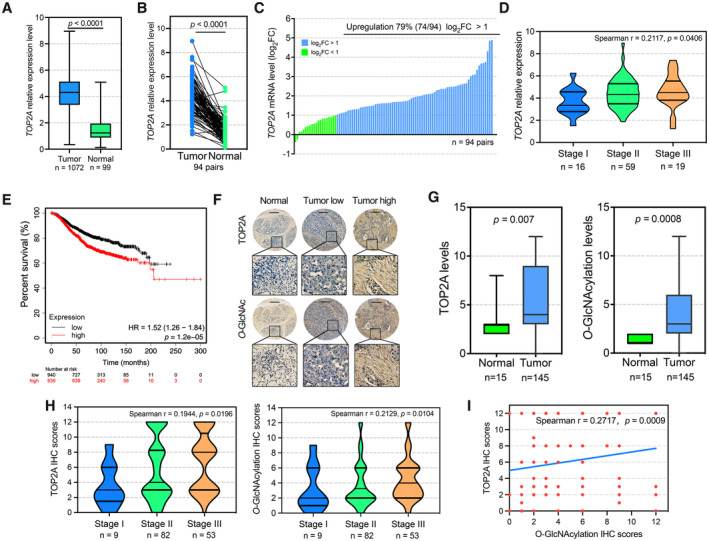
To investigate the clinical relevance of TOP2A in breast cancer, we examined TOP2A transcriptional expression in The Cancer Genome Atlas (TCGA) database (Chang et al, 2013). The results revealed that TOP2A was significantly upregulated in breast cancer tissues (n = 1,072) compared with normal tissues (n = 99, P < 0.0001, Fig 1A). TOP2A mRNA levels in clinical cancer tissue samples were also significantly higher than those in their paired adjacent normal tissues (n = 94, P < 0.0001, Fig 1B and C). Multivariate analysis further showed that high TOP2A mRNA levels were correlated with advanced tumor stage (Fig 1D) and clinical T classification in breast cancer patients (Appendix Table S1). Correspondingly, overall survival in patients with higher TOP2A expression levels was significantly worse compared with patients with lower TOP2A expression (P = 1.2e‐5, Fig 1E). Similar results were found in two other independent breast cancer cohorts (GSE48390 (Huang et al, 2013) and GSE3494 (Miller et al, 2005), Fig EV1), indicating the role of TOP2A in breast cancer malignancy.
Figure 1. Role of TOP2A is linked to cellular O‐GlcNAcylation in breast cancer patients with poor clinical prognosis.
-
AThe mRNA expression of TOP2A in breast tumor tissue (n = 1,072 biological replicates) and normal breast tissue samples (n = 99 biological replicates) according to TCGA database. The box plots show the medians (black lines), 25th and 75th percentiles (boundaries), and minimum/maximum values (whiskers). Unpaired t‐test was used for statistical comparison. The P‐value is indicated.
-
B, CTOP2A mRNA levels were compared between 94 pairs (biological replicates) of breast tumor tissues and their paired adjacent normal tissues from TCGA database. Wilcoxon matched‐pair signed‐rank test was used. The blue bar represents log2FC is > 1. The green bar represents that log2FC is < 1. Paired t‐test was used for statistical comparison. The P‐value is indicated. FC, fold change (tumor/normal).
-
DSpearman correlation coefficient of TOP2A mRNA expression among different progression stages from 94 pairs (biological replicates) of TCGA breast tumor tissues. The violin plots show the 25th, 50th, and 75th percentiles. The Spearman correlation P‐value is indicated.
-
EIn TCGA breast cancer tissues, the samples with higher TOP2A mRNA levels had shorter overall survival times than those with lower TOP2A mRNA levels (n = 1879 biological replicates). Patients with TOP2A expression greater than the median are indicated by the red line, and patients with TOP2A expression below the median are indicated by the black line. HR, hazard ratio. Statistical analysis was performed by the log‐rank test. The P‐values are indicated.
-
FRepresentative IHC staining of TOP2A and cellular O‐GlcNAcylation on a tissue microarray containing 145 breast tumor and 15 adjacent samples. Histological scoring was based on the positive percentages and intensity of stained cells. The micrograph scale bar represents 250 μm.
-
GTOP2A and cellular O‐GlcNAcylation expression levels were compared between breast tumor tissues (n = 145 biological replicates) and normal adjacent tissues (n = 15 biological replicates). The box plots show the medians (black lines), 25th and 75th percentiles (boundaries), and minimum/maximum values (whiskers). Unpaired t‐test was used for statistical comparison. The P‐value is indicated.
-
HSpearman correlation coefficient of TOP2A and cellular O‐GlcNAc expression among different progression stages from tissue microarray. The violin plots show the 25th, 50th, and 75th percentiles. One sample (Stage 0) was excluded. The Spearman correlation P‐value is indicated.
-
ICorrection analysis of TOP2A and O‐GlcNAcylation expression levels in 145 breast tumor tissues. Spearman correlation test was used. The P‐value is indicated.
Figure EV1. mRNA levels of TOP2A positively correlate with overall survival in human breast cancer tissues.
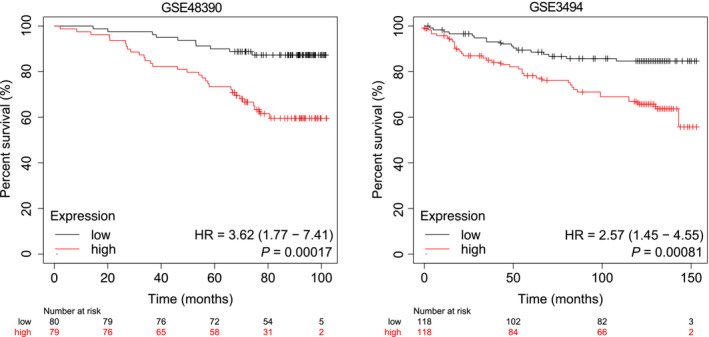
Datasets used for the survival analysis were obtained from the indicated GEO datasets (GSE48390 and GSE3494). Patients with TOP2A mRNA expression greater than the median are indicated by the red line, and patients with TOP2A mRNA expression below the median are indicated by the black line. HR, hazard ratio. Statistical analysis was performed by the log‐rank test. The P‐values are indicated (n = 159 biological replicates for GSE48390, n = 236 biological replicates for GSE3494).
Our previous study revealed that cellular protein O‐GlcNAcylation is an important regulator of cell death thresholds to chemotherapeutic drugs in cancer cells (Liu et al, 2018, 2020). To obtain a comprehensive understanding of the relationship between TOP2A and cellular O‐GlcNAcylation, tissue microarrays of human breast cancer were evaluated by immunohistochemistry (IHC) analysis (Appendix Fig S1). Representative images indicated higher expression of TOP2A and cellular O‐GlcNAcylation in breast cancer tissues compared with adjacent normal tissues (Fig 1F). Our results also showed that TOP2A and cellular O‐GlcNAcylation levels increased correspondingly with the aggressiveness of the disease (Fig 1G and H). The clinical and pathologic features are presented in Appendix Tables S2 and S3. In addition, a positive correlation was found between TOP2A and cellular O‐GlcNAcylation in breast cancer tissues (Spearman correlation coefficient r = 0.2717, P = 0.0009, Fig 1I), indicating that the interplay between TO2A and O‐GlcNAcylation might be an important point in breast cancer malignancy. In summary, these data validated that increased TOP2A and cellular O‐GlcNAcylation were associated with poor clinical outcomes in breast cancer.
TOP2A is hyper‐ O‐GlcNAcylated in chemotherapy‐resistant breast cancer cells
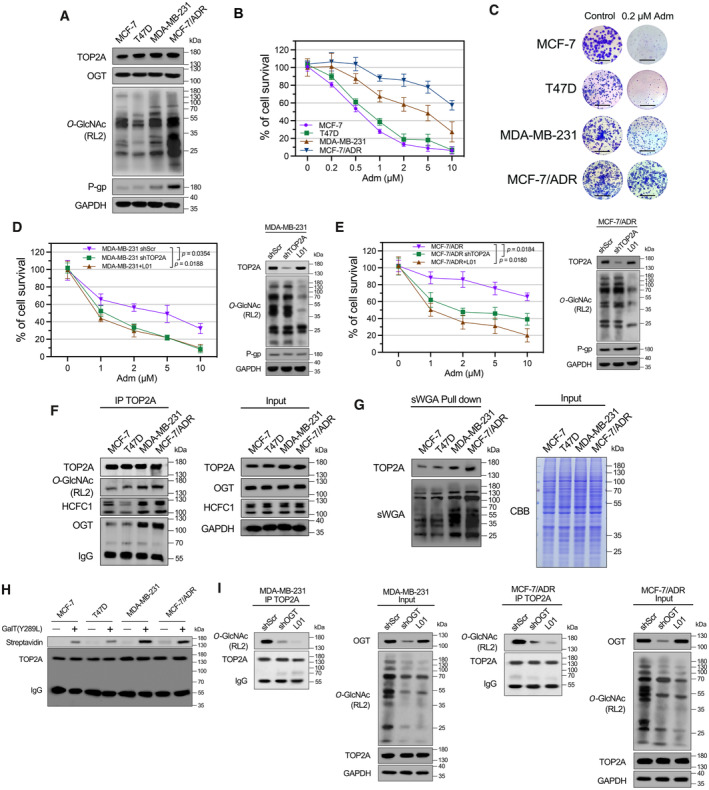
Next, we revealed that TOP2A expression was not much different in adriamycin (Adm)‐resistant breast cancer cell lines (MDA‐MB‐231, MCF‐7/ADR) compared with Adm‐sensitive breast cancer cells (MCF‐7, T47D, Fig 2A–C), indicating the likelihood of other regulatory mechanisms involving TOP2A‐related drug resistance. An increase in cellular O‐GlcNAcylation was found in Adm‐resistant cells with elevated cell survival and colony formation frequency under Adm treatment. However, no clear correlation was identified between OGT levels and cellular response to Adm. In Adm‐resistant MDA‐MB‐231 and MCF‐7/ADR cells, inhibition of O‐GlcNAcylation with L01 (a cell‐permeable OGT inhibitor developed in our previous research; Liu et al, 2017) or TOP2A shRNA‐transfection (shTOP2A) did not affect cell viability alone, while co‐treatment with L01/shTOP2A and Adm significantly reduced the cell viability (Fig 2D and E; Appendix Fig S2). Regardless of the O‐GlcNAcylation levels, the expression levels of multidrug resistance‐related ABC transporter P‐glycoprotein (P‐gp) remained unchanged in MDA‐MB‐231 and MCF‐7/ADR cells. These results suggested that an impact on both TOP2A and O‐GlcNAcylation protects breast cancer cells from Adm‐induced cell death in a P‐gp‐independent manner.
Figure 2. Hyper‐O‐GlcNAcylation of TOP2A increases drug resistance in breast cancer cells.
-
AExpression of TOP2A, OGT, P‐gp, and cellular O‐GlcNAcylation in Adm‐sensitive and Adm‐resistant breast cancer cell lines was analyzed by Western blot analysis.
-
BBreast cancer cells were treated with increasing doses of Adm alone for 48 h, and the cell viability was assessed using CCK‐8 assay. n = 3 biological replicates. The data are presented as means ± SD.
-
CColony formation assays were performed in breast cancer cells exposed to 0.2 μM Adm. The micrograph scale bar represents 5 mm.
-
D, EMDA‐MB‐231 or MCF‐7/ADR cells were transfected with TOP2A shRNA (shTOP2A) or treated with 50 μM L01 and then incubated with the indicated doses of Adm for 48 h. Cell viability was assessed with a CCK‐8 assay. Protein expression was analyzed by Western blot. shScr, scrambled shRNA. n = 3 biological replicates. Paired t‐test was used for statistical comparison. P‐value was indicted. The data are presented as means ± SD.
-
FTOP2A co‐IP was performed, and the immunoprecipitated fractions were analyzed by Western blot for the indicated proteins.
-
GsWGA lectin pull‐down was performed, and precipitated fractions were analyzed by Western blot and lectin blot. CBB, Coomassie Brilliant Blue staining.
-
HThe O‐GlcNAc enzymatic labeling system was employed to confirm the O‐GlcNAcylation of endogenous TOP2A in breast cancer cells. TOP2A IP was performed, and immunoprecipitated fractions were incubated with Y289L GalT1. Streptavidin beads were used to capture O‐GlcNAc proteins. An anti‐TOP2A antibody was used for immunoblotting.
-
IMDA‐MB‐231 or MCF‐7/ADR cells were transfected with OGT shRNA (shOGT) or treated with 50 μM L01 for 48 h. TOP2A co‐IP was performed, and the immunoprecipitated fractions were analyzed by Western blot for the indicated proteins.
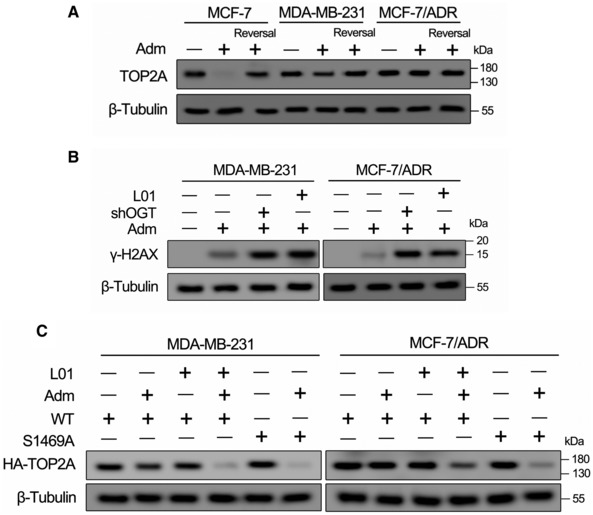
In our previous report, TOP2A was identified as a candidate O‐GlcNAc‐modified protein. Quantitative mass spectrometry showed that the chromatin association of TOP2A was increased (fold change > 2, P < 0.05) in Adm‐resistant MCF‐7/ADR cells compared with parental MCF‐7 cells (Appendix Fig S3; Liu et al, 2020), suggesting that the O‐GlcNAcylation of TOP2A might participate in chemotherapy resistance. To further verify this result, we confirmed that endogenous TOP2A was O‐GlcNAcylated via multiple methods, including co‐immunoprecipitation, succinylated wheat germ agglutinin lectin (sWGA, lectin specifically recognizes the O‐GlcNAc moiety) pull‐down, and the GalT1(Y289L)‐mediated chemical enzymatic method (Fig 2F–H). Remarkably, Adm‐resistant cells showed higher levels of TOP2A O‐GlcNAcylation and TOP2A‐OGT/HCFC1 (an important OGT partner; Daou et al, 2011) interactions compared with Adm‐sensitive cells. Since one of the primary therapeutic mechanisms of Adm is the stabilization of TOP2‐DNA covalent complexes (TOP2Acc), we further elucidated the relationship between Adm‐induced TOP2Acc and TOP2A O‐GlcNAcylation‐related Adm resistance by TOP2A band depletion assays. As shown in Fig EV2A, Adm‐trapped TOP2A into TOP2Acc in Adm‐sensitive MCF‐7 cells. On the contrary, the amount of Adm‐trapped TOP2Acc was greatly reduced in Adm‐resistant MDA‐MB‐231 and MCF‐7/ADR cells, indicating that the Adm resistance in these cells is at least in part due to reduced TOP2Acc. Moreover, both OGT knockdown and the inhibition of cellular O‐GlcNAcylation with L01 restrained TOP2A modification and enhanced DNA damage marker γ‐H2AX expression in MDA‐MB‐231 and MCF‐7/ADR cells (Figs 2I and EV2B). Collectively, these data demonstrated that TOP2A is a novel O‐GlcNAc‐modified protein that its hyperglycosylation contributes to chemotherapy resistance in breast cancer cells.
Figure EV2. Relationship between TOP2Acc and TOP2A O‐GlcNAcylation‐related Adm resistance.
-
AThe amount of TOP2Acc in Adm‐treated breast cells was measured by a band depletion assay. Indicated cells were treated with Adm (1 μM for MCF‐7 cells, 5 μM for MDA‐MB‐231 and MCF‐7/ADR cells) for 2 h. Cells were lysed either immediately or after reversal of the TOP2Acc (Reversal). Cell lysates were analyzed by Western blot.
-
BMDA‐MB‐231 or MCF‐7/ADR cells were transfected with OGT shRNA (shOGT) or treated with 50 μM L01 for 48 h. The cells were further cultured in the presence or absence of Adm (5 μM) for 2 h. Cell lysates were analyzed by Western blotting using anti‐γ‐H2AX antibody.
-
CBand depletion assays were performed in MDA‐MB‐231 and MCF‐7/ADR cells which stable transfected with HA‐tagged TOP2A‐WT or TOP2A‐S1469A (in the presence or absence of 50 μM L01). Breast cancer cells were treated with Adm (5 μM) for 2 h. Cell lysates were analyzed by Western blot.
Source data are available online for this figure.
OGT promotes TOP2A O‐GlcNAcylation at S1469, which strengthens TOP2A chromatin binding.
The online server YinOYang (v1.2) was used to predict potential TOP2AO‐GlcNAcylation site(s), and the majority were predicted within the CTD (Fig 3A). We then prepared three truncated TOP2A variants (TOP2A‐A to TOP2A‐C, Fig 3B). HEK‐293T cell‐derived OGT and these prokaryotically expressed recombinant His‐tagged TOP2A fragments were employed in an in vitro glycosylation assay. Only TOP2A‐C (CTD) could interact with OGT and be further modified (Fig 3C). Similar result was found in OGT overexpressed eukaryotic system (Appendix Fig S4). EThcD‐MS/MS analysis was performed to precisely map the O‐GlcNAcylation site(s) on TOP2A‐C. The results revealed that O‐GlcNAcylation occurs on S1469 (within TOP2A nuclear localization signal sequence, NLS, amino acids 1,454–1,497 (Kozuki et al, 2017), Fig 3B) in the peptide (RKPSTSDDSDSNFEK) spanning residues 1,466–1,480 (Fig 3D). Multiple‐sequence alignment revealed that the S1469 residue is highly conserved across species (Fig 3E). Next, we generated an HA‐tagged TOP2A construct with S1469 mutated to alanine (TOP2A‐S1469A). In Adm‐resistant MDA‐MB‐231 and MCF‐7/ADR cells stably transfected with wild‐type (TOP2A‐WT) or TOP2A‐S1469A constructs, the O‐GlcNAcylation levels were clearly reduced in mutant compared with wild‐type TOP2A. In the presence of the O‐GlcNAcylation agonist PugNAc (an OGA inhibitor), no visible TOP2A‐S1469A O‐GlcNAcylation increase was observed. In contrast, L01 markedly decreased the level of O‐GlcNAcylation in TOP2A‐WT, supporting the notion that the majority of TOP2A O‐GlcNAcylation occurs on S1469 (Fig 3F).
Figure 3. O‐GlcNAcylation at S1469 enhances TOP2A chromatin binding.
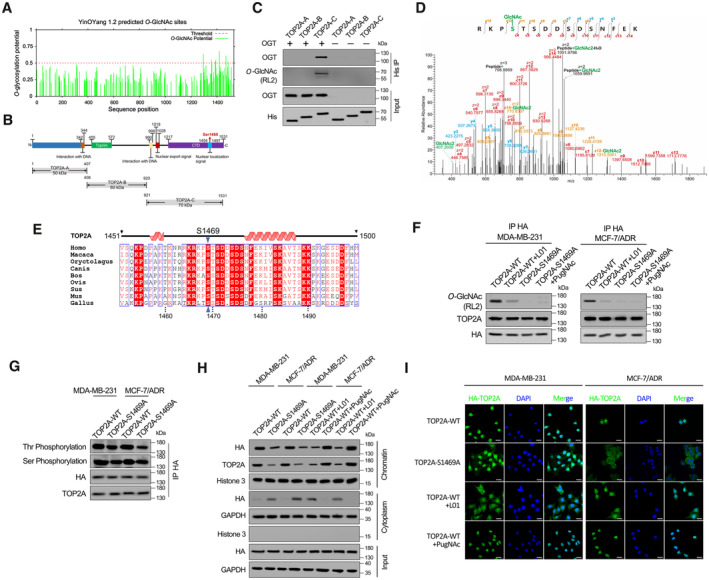
-
AO‐GlcNAc sites of human TOP2A predicted using the YinOYang 1.2. The potential O‐GlcNAcylated Ser/Thr residues and the threshold for O‐GlcNAcylation potential were indicated.
-
BSchematic diagram of key domains in TOP2A. Three truncated variants of TOP2A (TOP2A‐A to C) fused with 6 × His‐tag were generated according to its key domains.
-
CIn vitro glycosylation assay was performed using recombinant His‐tagged truncated variants of TOP2A and purified OGT. The reaction product was detected by co‐immunoprecipitation (co‐IP) and Western blot.
-
DEThcD‐MS/MS spectrum of the TOP2A CTD peptide with the O‐GlcNAcylation site located on S1469. The matched fragment ions are labeled.
-
EAlignment of TOP2A CTD sequence among different species.
-
FMDA‐MB‐231 and MCF‐7/ADR cells which stable transfected with TOP2A‐WT or TOP2A‐S1469A were treated with 50 μM L01 or PugNAc for 48 h. HA‐tag co‐IP was performed, and the immunoprecipitated fractions were analyzed by Western blot for the indicated proteins.
-
GHA‐tag co‐IP was performed in MDA‐MB‐231 and MCF‐7/ADR cells which stable transfected with TOP2A‐WT or TOP2A‐S1469A. Overall threonine (Thr) and serine (Ser) phosphorylation states of TOP2A were analyzed by Western blot.
-
HWestern blots showing the chromatin binding and subcellular localization of TOP2A‐WT or TOP2A‐S1469A with 50 μM L01 or PugNAc treatment in breast cancer cells.
-
IFluorescent staining results showing nuclear localization dependence of TOP2A on O‐GlcNAcylation in MDA‐MB‐231 and MCF‐7/ADR cells. The micrograph scale bar represents 25 μm.
Because O‐GlcNAcylation often competes with phosphorylation in many biological processes (Yang & Qian, 2017), we wondered whether the phosphorylation of TOP2A is affected by different O‐GlcNAcylation states. Experimental results showed that the phosphorylation level (overall serine and threonine) of TOP2A‐S1469A was clearly attenuated compared with that of TOP2A‐WT, suggesting an interplay between O‐GlcNAcylation and phosphorylation of TOP2A (Fig 3G). Because the O‐GlcNAcylation site located in the NLS of this enzyme, we next investigated the role of O‐GlcNAcylation in recruiting TOP2A to chromatin in nucleus. Even though similar expression levels were found for the two recombinant forms of TOP2A, TOP2A‐S1469A showed significantly less chromatin binding capacity and more cytoplasm localization compared with hyper‐O‐GlcNAcylated TOP2A‐WT in MDA‐MB‐231 and MCF‐7/ADR cells, indicating that O‐GlcNAcylation on S1469 affects TOP2A subcellular localization. Furthermore, the suppression of TOP2A‐WT glycosylation by the OGT inhibitor decreased chromatin‐associated TOP2A and elevated cytoplasm localization of this enzyme, whereas the OGA inhibitor PugNAc showed the opposite effect (Fig 3H and I). We also observed that more TOP2A‐S1469A was diffused in the cytosol in MCF‐7/ADR cells than in MDA‐MB‐231 cells. In Fig 3G, threonine phosphorylation level of TOP2A‐S1469A in MCF‐7/ADR cells was lower compared with that of MDA‐MB‐231, indicating the interplay between O‐GlcNAcylation and phosphorylation may also participate in nuclear localization of TOP2A. We next tested whether site‐specific O‐GlcNAcylation reflects the formation of Adm‐induced TOP2Acc. O‐GlcNAcylation site mutation (TOP2A‐S1469A) or L01 treatment increased the amount of TOP2A covalently trapped on DNA compared with wild‐type glycosylated TOP2A‐WT in Adm‐resistant breast cancer cells, suggesting that TOP2A O‐GlcNAcylation impeded the formation of Adm‐induced TOP2Acc and reduced cytotoxicity (Fig EV2C). Together, these results suggest that the abundance of chromatin‐bound TOP2A is modulated by O‐GlcNAcylation at S1469 and this modification can further affect the biological function of this enzyme.
O‐GlcNAcylation of TOP2A promotes catalytic activity
To further assess whether TOP2A CTD O‐GlcNAcylation affects its biochemical function, the endogenous enzyme (purified from MCF‐7/ADR cells) was first shown to retain overall catalytic activity in vitro and to catalyze supercoiled plasmid DNA cleavage in a concentration‐ and time‐dependent manner (Fig EV3). Hyper‐O‐GlcNAcylated TOP2A from Adm‐resistant breast cancer cells displayed greater DNA cleavage efficiency compared with less O‐GlcNAcylated TOP2A from Adm‐sensitive cells (Fig 4A). Following treatment with PugNAc, the DNA cleavage activity of MCF‐7‐extracted TOP2A was enhanced. In contrast, the linear state plasmid was attenuated when TOP2A O‐GlcNAcylation levels were reduced by L01 in MDA‐MB‐231 and MCF‐7/ADR cells. This was further confirmed by using a commercially available recombinant full‐length human TOP2A (Fig 4B) in an in vitro glycosylation assay. Then, we examined the effect of O‐GlcNAcylation at S1469 on TOP2A catalytic activity. HEK‐293T cells stably overexpressing OGT were transfected with HA‐tagged TOP2A‐WT or TOP2A‐S1469A expression plasmids. As shown in Fig 4C, loss of O‐GlcNAcylation led to a noticeable decrease in cleavage activity. Further experiments revealed that the suppression of TOP2A‐WT glycosylation by L01 reduced enzymatic activity, but PugNAc treatment could not increase the catalytic activity of TOP2A‐S1469A. Similar results were obtained using TOP2A‐WT and TOP2A‐S1469A purified from Adm‐resistant breast cancer cells (Fig 4D). These findings suggest that O‐GlcNAcylation is involved in regulating the DNA cleavage activity of TOP2A.
Figure EV3. DNA cleavage assays were performed using the endogenous TOP2A purified from MCF‐7/ADR cells.
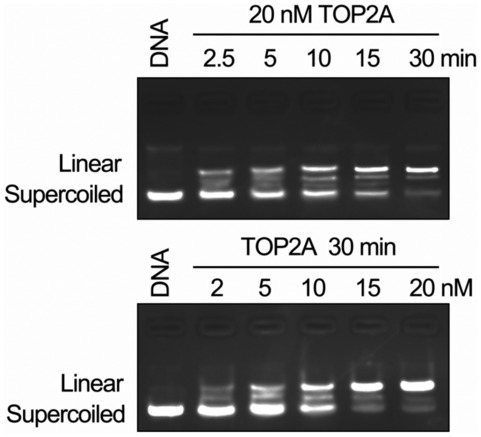
Supercoiled pBR322 plasmid DNA was used as the TOP2A substrate. TOP2A could retain DNA cleavage activity in vitro and catalyzed supercoiled plasmid DNA relaxation in a concentration‐ and time‐dependent manner. The enzyme concentration and reaction time were indicated.Source data are available online for this figure.
Figure 4. O‐GlcNAcylation enhances TOP2A enzymatic activity.
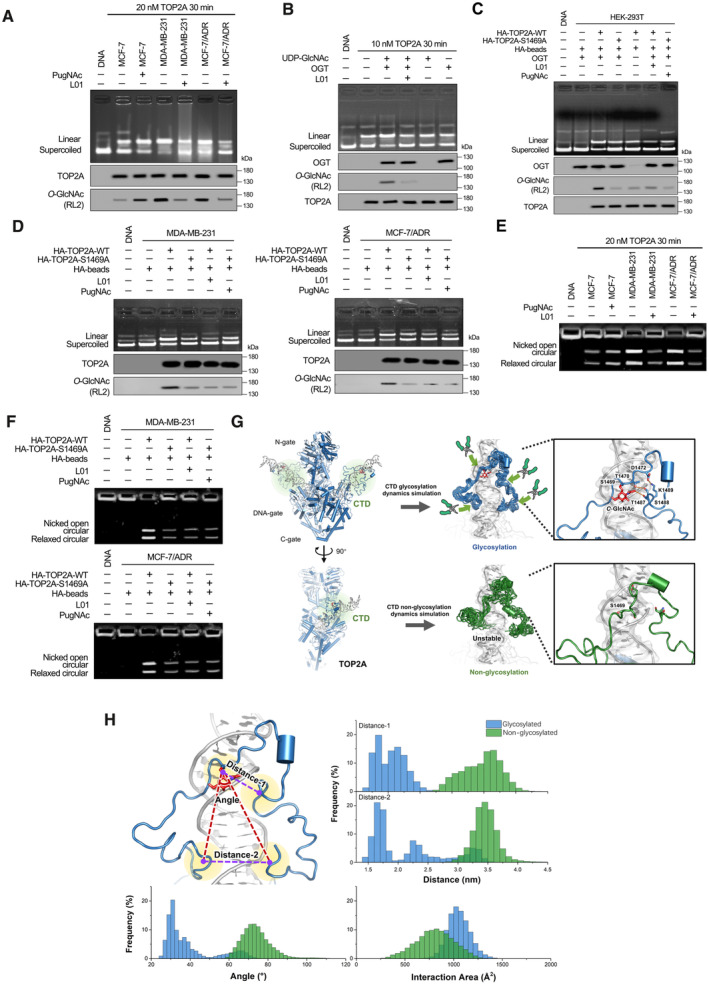
-
ADNA cleavage assays were performed using TOP2A extracted (20 nM, reaction for 30 min) from breast cancer cells with 50 μM L01 or PugNAc treatment (48 h). Supercoiled pBR322 plasmid DNA was used as the TOP2A substrate. TOP2A O‐GlcNAcylation status was detected by IP.
-
B10 nM commercially available recombinant full‐length human TOP2A was O‐GlcNAcylated by an in vitro reaction (with or without 50 μM L01, reaction for 30 min). The reaction product was then used for DNA cleavage assays and co‐IP. The immunoprecipitated fractions were analyzed by Western blot.
-
C, DDNA cleavage assays were performed using TOP2A extracted from OGT stably overexpressed HEK‐293T cells (C) or breast cancer cells (D). These cells were stably transfected with HA‐tagged TOP2A‐WT or TOP2A‐S1469A expression plasmids and treated with 50 μM L01 or PugNAc for 48 h. TOP2A‐WT or TOP2A‐S1469A (20 nM) was immunoprecipitated using anti‐HA magnetic beads from breast cancer cells.
-
EkDNA decatenation assays were measured using TOP2A extracted (20 nM, reaction for 30 min) from breast cancer cells with 50 μM L01 or PugNAc treatment (48 h).
-
FkDNA decatenation assays were performed using TOP2A extracted from TOP2A‐WT or TOP2A‐S1469A overexpressed breast cancer cells with 50 μM L01 or PugNAc treatment (48 h). TOP2A‐WT or TOP2A‐S1469A (20 nM) was immunoprecipitated using anti‐HA magnetic beads from breast cancer cells.
-
GLeft: Two views of the overall model structure of TOP2A (N‐gate, DNA‐gate, C‐gate from Cryo‐EM structure 6ZY8, and CTD structure refined from Alphafold2 modeling structure with MD simulation), with the substrate DNA helix. Middle: The superposition of CTD structures from the cluster results of glycosylated (blue) and nonglycosylated (green) CTD‐DNA systems simulation trajectories. The red stick is the O‐GlcNAc group on S1469. Right: The interaction network around O‐GlcNAc moiety and S1469, act as a pivot point for the tight association with DNA by the N‐ and C‐terminal loops of CTD, which like two arms of a pincher.
-
HThe frequency of some crucial variants from 1 μs MD simulation trajectories for glycosylated (blue) and nonglycosylated (green) CTD‐DNA complex systems, which could describe the difference between these two systems. Distance‐1: center of mass distance between S1469 and 1487‐TSK‐1489. Distance‐2: center of mass distance between 1441‐TKR‐1443 and 1510‐AKS‐1512. Angle: the angle is formed with the center of mass of S1469 as the vertex, and two vectors from the vertex to the center of mass of 1441‐TKR‐1443 and 1510‐AKS‐1512 as the edges. Interaction area: area of the interaction between CTD and DNA.
Because decatenation is another important function of TOP2A catalytic activity (Nitiss, 2009), the ability to unlink catenated DNA circles was measured. Decatenation assays using catenated kinetoplast DNA (kDNA) minicircles showed that hyper‐O‐GlcNAcylated TOP2A appears to decatenate fairly efficiently, in contrast to TOP2A with low O‐GlcNAcylation (Figs 4E and F, and EV4). Moreover, with multiple structural modeling and simulation methods, we found that the O‐GlcNAcylated TOP2A CTD structure could interact with DNA more effectively. The interaction network formed among the O‐GlcNAc moiety and the adjacent residues, for example, T1470, D1472, T1487, S1488, and K1489, provided stable support among the large disordered region of the TOP2A CTD, which is beneficial for the stability and tightness of the interaction between the CTD and DNA (Figs 4G and H, and EV5A–D). Taken together, the present data establish that S1469 O‐GlcNAcylation has a critical role in promoting the catalytic activity of TOP2A.
Figure EV4. kDNA decatenation assays were measured using the endogenous TOP2A purified from MCF‐7/ADR cells.
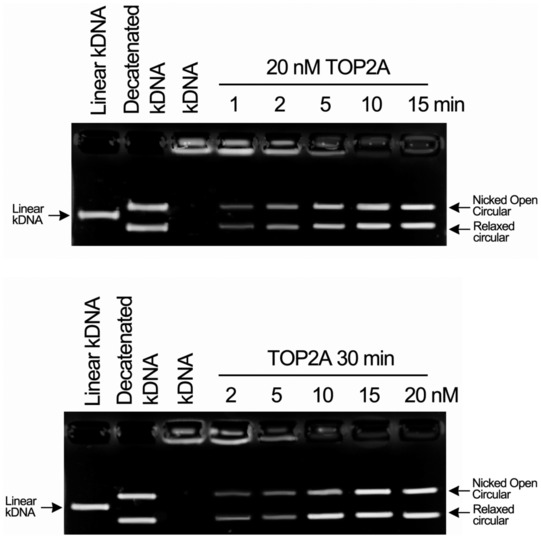
kDNA was used as the TOP2A substrate. TOP2A could retain catalytic activity in vitro and catalyzed kDNA decatenation in a concentration‐ and time‐dependent manner. Linear kDNA was used as the negative control, and decatenated kDNA was used as the positive control. The enzyme concentration and reaction time were indicated.Source data are available online for this figure.
Figure EV5. Molecular simulation of O‐GlcNAcylated TOP2A CTD with DNA.
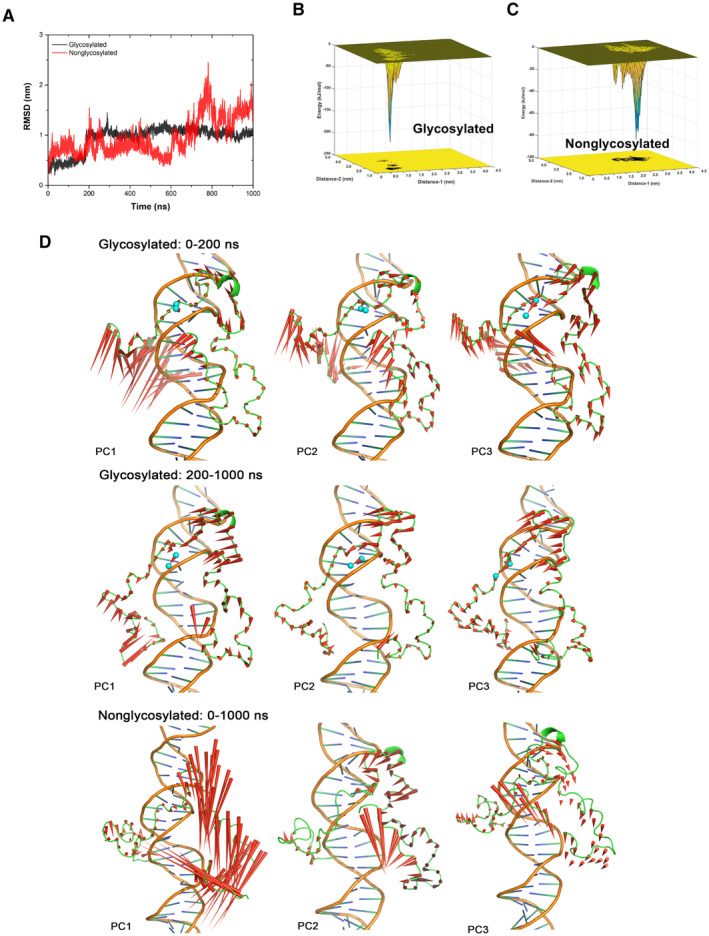
-
AThe curves of root‐mean‐square deviation of CTD Cα atoms along with the time traces from the trajectories of glycosylated and nonglycosylated CTD‐DNA systems. The fluctuation of glycosylated CTD‐DNA system's RMSD was smaller than nonglycosylated system and became steady around 1 nm after 200 ns MD simulation.
-
B, CThe conformational energy surface of glycosylated (B) and nonglycosylated CTD‐DNA systems (C). The energy surface was computed as a function of Distance‐1 (center of mass distance between S1469 and 1487‐TSK‐1489, to indicate the distance between GlcNAc group and adjacent residues) in nm and Distance‐2 (center of mass distance between 1441‐TKR‐1443 and 1510‐AKS‐1512, to indicate the distance between two terminals of CTD) in nm against the overall 1 μs trajectory of each system. Isosurfaces were shown every 1 kJ/mol. There are several small ensemble states and one dominate ensemble state in the energy surface of glycosylated CTD‐DNA system that the deepest energy minimum corresponds to Distance‐1 about 0.8 nm, Distance‐2 about 1.5 nm and energy about −225 kJ/mol. Three consecutive energy minimums in the energy surface of nonglycosylated CTD‐DNA system, as shown in Fig 4G, correspond to Distance‐1 from 1.5 to 2.5 nm, and Distance‐2 around 3.3 nm with energy worse than −90 kJ/mol. This result indicated that glycosylated S1469 enables a tighter conformation ensemble and significantly stabilizes the association between CTD and DNA. Distance‐1: center of mass distance between S1469 and 1487‐TSK‐1489. Distance‐2: center of mass distance between 1441‐TKR‐1443 and 1510‐AKS‐1512.
-
DThe difference in protein motion between the first 200 ns and the rest 800 ns simulation of glycosylated CTD‐DNA system was obvious from the PCA analysis. The protein is quite dynamic, especially the N‐terminal of CTD in the first 200 ns simulation. The interaction network around the glycosylation S1469 (the GlcNAc group shown as cyan sphere) gave the N‐terminal loop (residue 1,435–1,450) enough time to close and enter the major groove of DNA that polar and charged sidechain residues (K1439, 1441‐TKRD‐1444) in this loop could make strong interactions with DNA. The protein dynamics were more stable in the rest 800 ns trajectory. In contrast, the movement in nonglycosylated system was enormous and inhomogeneous in different regions. The residues around S1469 performed strong tendency to move away from DNA, which was unbeneficial for the stable interaction.
Source data are available online for this figure.
O‐GlcNAcylation‐modulated interactions between TOP2A and cell cycle regulators contribute to breast cancer cell drug resistance.
To explore the function of TOP2A O‐GlcNAcylation in Adm resistance, cell proliferation and cell cycle assays were performed. Colony formation assays were performed with MCF‐7 and MCF‐7/ADR cells overexpressing TOP2A‐WT or TOP2A‐S1469A and treated with Adm for 48 h. The colony formation rate was higher in the TOP2A‐WT group and lower in the TOP2A‐S1469A group after Adm treatment, affirming that TOP2A glycosylation promoted Adm resistance in breast cancer cells (Fig 5A). We then investigated the effects of TOP2A O‐GlcNAcylation on cell cycle progression. Of note, the proportion of cells arrested in S phase by Adm treatment was significantly lower in TOP2A‐WT‐overexpressing MCF‐7 and MCF‐7/ADR cells compared with TOP2A‐S1469A‐overexpressing cells (Fig 5B and C), indicating that TOP2A O‐GlcNAcylation at S1469 promotes cell cycle progression.
Figure 5. TOP2A O‐GlcNAcylation accelerates breast cancer cell cycle progression.
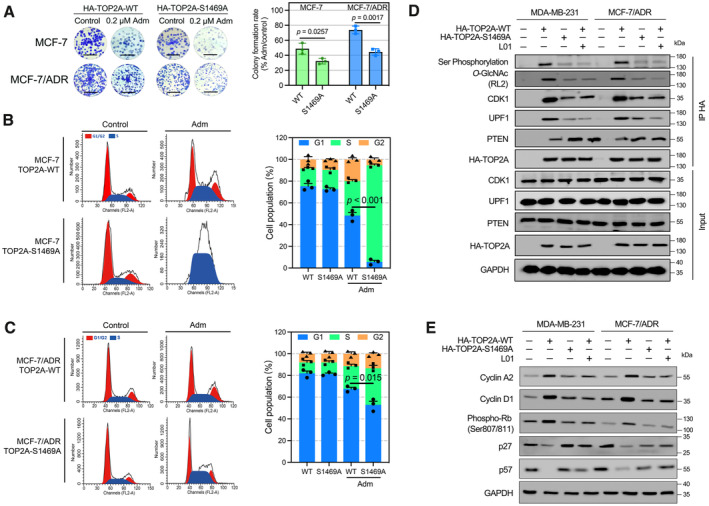
-
AColony formation assays were performed in TOP2A‐WT or TOP2A‐S1469A stably expressed MCF‐7 and MCF‐7/ADR cells. Cells were exposed to 0.2 μM Adm during the assays. The micrograph scale bar represents 5 mm. The colony formation rate was calculated. n = 3 biological replicates. Unpaired t‐test was used for statistical comparison. P‐value was indicted. The data are presented as means ± SD.
-
B, CFlow cytometry was employed to identify the percentage of cells in G1, S, and G2 phases. TOP2A‐WT or TOP2A‐S1469A stably expressed MCF‐7 and MCF‐7/ADR cells were treated with Adm (0.5 μM for MCF‐7 cells, 2 μM for MCF‐7/ADR cells) for 24 h. n = 3 biological replicates. Unpaired t‐test was used for statistical comparison. P‐value was indicted. The data are presented as means ± SD.
-
DBreast cancer cells expressing TOP2A‐WT or TOP2A‐S1469A were immunoprecipitated with anti‐HA magnetic beads, and the interaction between TOP2A and cell cycle regulators was analyzed. Cells were treated with 50 μM L01 for 48 h before immunoprecipitation.
-
ECell cycle‐related protein expression was measured using Western blot in Breast cancer cells expressing TOP2A‐WT or TOP2A‐S1469A. Cells were treated with 50 μM L01 for 48 h before immunoprecipitation.
TOP2A activity can be regulated by interacting with multiple cell cycle regulators (Kang et al, 2015; Jin et al, 2021; Zhu et al, 2021). In the present study, O‐GlcNAcylation site mutation or L01 treatment decreased the interaction of TOP2A with the cell cycle activators CDK1 and UPF1, whereas the TOP2A‐PTEN dimer was increased in Adm‐resistant breast cancer cells (Fig 5D). As a result, hyper TOP2A O‐GlcNAcylation in breast cancer cells resulted in higher levels of the downstream cell cycle‐related proteins cyclin A2, cyclin D1, and phospho‐Rb (S807/811) and lower levels of the cyclin‐dependent inhibitors p27 and p57. O‐GlcNAcylation inhibition had the opposite effect, suggesting a function of TOP2A O‐GlcNAcylation in regulating cell cycle‐related protein expression (Fig 5E). Together, these results demonstrated that O‐GlcNAcylation modulates the interactions between TOP2A and cell cycle regulators and then activates downstream gene expression, resulting in breast cancer cell Adm resistance.
TOP2A O‐GlcNAcylation is critical for mediating resistance to Adm
To test whether O‐GlcNAcylation could impact Adm‐mediated TOP2A inhibition, we examined TOP2A catalytic activity in the presence of Adm. Adm showed no inhibition of TOP2A‐WT‐catalyzed plasmid DNA cleavage or kDNA decatenation in vitro (Fig 6A and B). In contrast, reduction of O‐GlcNAcylation by site mutation increased the ability of Adm to inhibit TOP2A. These data support the idea that O‐GlcNAcylation of TOP2A prevents Adm from inhibiting catalytic activity. Meanwhile, we noticed an additional 20–30% cell survival, which increased with the duration of TOP2A‐WT expression, compared with TOP2A‐S1469A expression in drug‐sensitive MCF‐7 cells (Fig 6C). Similar results were observed in Adm‐resistant MDA‐MB‐231 and MCF‐7/ADR cells (Fig 6D and E). From these results, we confirmed that it is TOP2A O‐GlcNAcylation but not the TOP2A expression itself protects cells from Adm‐induced cell death. Furthermore, five breast cancer patient tumor tissues were examined for TOP2A O‐GlcNAcylation. Even though TOP2A expression was similar, chemotherapy‐resistant or relapsed patient samples (no. 3–5) contained higher levels of O‐GlcNAc‐modified TOP2A and TOP2A‐OGT/HCFC1 interaction compared with sensitive patient samples (no. 1–2, patients' main characteristics are listed in Appendix Table S4). Notably, the interaction between cell cycle activators and TOP2A was increased in resistant samples compared with sensitive samples, and the downstream cell cycle‐related gene expression had corresponding changes (Fig 6F). Taken together, these results suggest that TOP2A activation by O‐GlcNAcylation may be a common mechanism through which cancer cells antagonize chemotherapy‐induced cell death and that this mechanism is at least partially mediated by affecting TOP2A interaction with DNA and other regulators.
Figure 6. O‐GlcNAcylation contributes to Adm resistance in vitro and in vivo .
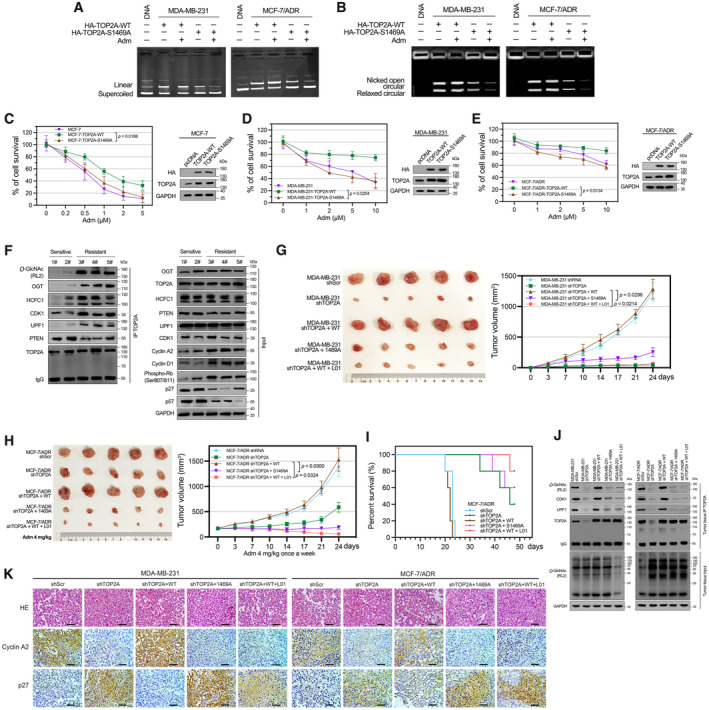
-
A, BO‐GlcNAcylation attenuated Adm‐induced inhibition of TOP2A catalytic activity in DNA cleavage (A) and kDNA decatenation assays (B). TOP2A‐WT or TOP2A‐S1469A (20 nM) was immunoprecipitated using anti‐HA magnetic beads from breast cancer cells. 200 μM Adm was added during the reaction (30 min).
-
C–ETOP2A‐WT or TOP2A‐S1469A overexpressed breast cancer cells were incubated with the indicated doses of Adm for 48 h. Cell viability was assessed with a CCK‐8 assay. Protein expression was analyzed by Western blot. n = 3 biological replicates. Paired t‐test was used for statistical comparison. P‐value was indicted. The data are presented as means ± SD.
-
FTOP2A co‐IP was performed in five breast cancer patient tumor samples (chemotherapy‐sensitive, no. 1‐2; chemotherapy‐resistant or relapsed, no. 3‐5), and the immunoprecipitated fractions were analyzed by Western blot for the indicated proteins.
-
GThe effects of TOP2A O‐GlcNAcylation on tumor xenografts in nude mice. MDA‐MB‐231 cells with stable TOP2A silencing by shRNA (shTOP2A) were transfected with TOP2A‐WT or TOP2A‐S1469A expression plasmids. Cells were injected subcutaneously into the axillae of nude mice (n = 5 biological replicates for each group). 1 mg/kg L01 was administrated by tail vein injection. Volumes of tumors were monitored with caliper twice a week until 24 days. Paired t‐test was used for statistical comparison. P‐value was indicted. The data are presented as means ± SD.
-
HIn vivo antitumor performance of Adm in MCF‐7/ADR bearing nude mice. MCF‐7/ADR cells with stable TOP2A silencing by shRNA (shTOP2A) were transfected with TOP2A‐WT or TOP2A‐S1469A expression plasmids. Cells were injected subcutaneously into the axillae of nude mice (n = 5 biological replicates for each group). 4 mg/kg Adm and 1 mg/kg L01 was administrated. Volumes of tumors were monitored with caliper twice a week until 24 days. Paired t‐test was used for statistical comparison. P‐value was indicted. The data are presented as means ± SD.
-
ISurvival rates of MCF‐7/ADR bearing mice in different treatment groups within 48 d (n = 5 biological replicates).
-
JTOP2A O‐GlcNAcylation and the interaction between TOP2A and cell cycle regulators were measured by co‐IP in tumor tissues. Cellular O‐GlcNAcylation was analyzed by Western blot.
-
KIHC staining of Cyclin A2 and p27 in paraffin sections of MDA‐MB‐231 and MCF‐7/ADR tumors. The micrograph scale bar represents 50 μm.
Blocking TOP2A O‐GlcNAcylation improves the therapeutic efficacy of Adm in a xenograft mouse model.
To evaluate the role of TOP2A O‐GlcNAcylation in regulating the tumorigenic capacity of breast cancer cells and sensitivity to Adm treatment in vivo, breast cancer cells stably transfected with scrambled shRNA (shScr) or TOP2A shRNA (shTOP2A) were further transfected with TOP2A‐WT or TOP2A‐S1469A constructs and subcutaneously injected into female athymic nude mice. Even though knockdown of TOP2A inhibited the tumor proliferation, the mice bearing MDA‐MB‐231 shTOP2A + S1469A cells showed much slower tumor growth than MDA‐MB‐231 shTOP2A + WT‐injected group (Fig 6G). We also observed that L01 treatment significantly decreased tumor volumes for both wild‐type MDA‐MB‐231 (Appendix Fig S5A) and shTOP2A + WT xenografts (Fig 6G), indicating the important role of TOP2A O‐GlcNAcylation but not the expression itself in breast cancer malignancy. Furthermore, a mouse model with MCF‐7/ADR‐derived cells was established. The mice received administration of Adm (4 mg/kg) every 7 days. The average tumor volume was reduced in the presence of TOP2A silencing or TOP2A‐S1469A overexpression, and this reduction was reversed when TOP2A silencing was combined with TOP2A‐WT overexpression (Fig 6H). We also revealed that L01 treatment decreased tumor volumes for MCF‐7/ADR xenografts (Appendix Fig S5B). Notably, the combination of O‐GlcNAc inhibition by L01 and Adm treatment resulted in much more pronounced suppression of tumor growth in mice bearing MCF‐7/ADR shTOP2A + WT cells compared with Adm treatment alone. Kaplan–Meier survival curves showed that mice injected with MCF‐7/ADR shTOP2A, MCF‐7/ADR shTOP2A + S1469A or MCF‐7/ADR shTOP2A + WT + L01 all survived within an experimental period of 30 days (Fig 6I). In contrast, mice that received MCF‐7/ADR shRNA or MCF‐7/ADR shTOP2A + WT cells only had a significantly shorter median survival time (approximately 22 days). In line with the impaired tumor growth, the interaction between TOP2A and cell cycle activators was decreased in TOP2A O‐GlcNAcylation‐inhibited tumor tissues (Fig 6J). Corresponding results were obtained by IHC analysis of Cyclin A2 and p27 in tumor tissues (Fig 6K). These data again demonstrate that O‐GlcNAcylation at S1469 can promote TOP2A activity and further contribute to tumor growth and drug resistance in breast cancer.
Discussion
Although there are numerous complicated mechanisms involved in breast cancer drug resistance (Vasan et al, 2019), including the inactivation of drug targets, the enhancement of drug efflux by overexpressing P‐gp, and the activation of pro‐survival pathways, the precise regulatory processes of drug tolerance in breast cancer remain unclear. In the present study, we uncovered the functional and mechanistic role of TOP2A O‐GlcNAcylation in breast cancer progression and response to the chemotherapeutic drug Adm. We discovered that both of TOP2A and cellular O‐GlcNAcylation expression were increased in breast cancer tissues at the transcriptional and protein levels, and this was accompanied by malignant progression. As a novel PTM on TOP2A (S1469), O‐GlcNAcylation was demonstrated to elevate TOP2A catalytic activity, which is crucial for drug resistance, by intensifying the binding with DNA. These data add a new layer of complexity to the PTM code in modulating TOP2A biochemical functions, which enables increased chemotherapy cell death thresholds in breast cancer.
Our data showed that the TOP2A‐OGT interaction was enhanced in drug‐resistant breast cells/patient samples compared with the drug‐sensitive ones. The underlying molecular mechanism can be explained in two ways. First, our previous research revealed that the synthesis of UDP‐GlcNAc through hexosamine biosynthetic pathway is upregulated in cancer cells with chemoresistance (Liu et al, 2018). This accumulation of the pool of sugar substrate might activate OGT and lead to elevated cellular O‐GlcNAcylation. In the present study, not only TOP2A O‐GlcNAcylation but also global O‐GlcNAcylation levels showed significant increases in Adm‐resistant cells, while OGT expression levels were comparable in Adm‐sensitive and Adm‐resistant breast cancer cells, indicating that the upregulation of UDP‐GlcNAc might be pivotal in regulating OGT activation and the TOP2A‐OGT interaction. In addition, the associations between TOP2A, OGT, and HCFC1 (an auxiliary factor links OGT to chromatin; Daou et al, 2011) were increased in drug‐resistant cells and patient samples. This result suggested that HCFC1 may act as an influential partner to recruit OGT to chromatin‐bound TOP2A. We also revealed that TOP2A O‐GlcNAcylation state regulated the interaction of this enzyme with the cell cycle regulators, suggesting that OGT and HCFC1 might participate in TOP2A‐involved chromatin complex assembly. In this sense, changes in the composition of TOP2A complex could also affect the interaction between OGT and TOP2A.
Because Adm is one of the first‐line chemotherapeutic drugs in oncotherapy, we used this agent to investigate the impact of TOP2A O‐GlcNAcylation on breast cancer resistance. Given one of the primary therapeutic mechanisms of Adm is the stabilization of TOP2Acc (Nitiss, 2009), we provide evidence that O‐GlcNAcylation might affect the conformation of TOP2A‐DNA complexes and antagonize Adm binding and the formation of TOP2Acc. Although O‐GlcNAcylation was found to enhance TOP2A chromatin binding and cleavage in this study, our data suggest that Adm might lose its drug target in resistant cells. Although the deletion of TOP2A O‐GlcNAcylation in MCF‐7/ADR cells did not fully restore the cell's sensitivity to Adm, suggesting that TOP2A glycosylation is not the only factor that can influence the drug resistance of breast cancer cells, our data still demonstrated that deletion of this PTM could reverse TOP2A O‐GlcNAcylation‐triggered Adm resistance in vitro and in vivo. Pharmacological inhibition of OGT was also shown to be as a potential strategy for overcoming Adm resistance in breast cancer‐bearing mice, adding support to the idea that TOP2A O‐GlcNAcylation is a promising therapeutic intervention point for drug resistance.
Although TOP2A is a well‐known nuclear protein, this enzyme has been reported to diffuse in the cytoplasm under particular circumstances (Turley et al, 1997). Previous studies reported that multiple sequences within the CTD contribute to the robust nucleus localization of TOP2A. The NLS sequence in TOP2A is located between amino acids 1,454–1,497 in the CTD and is important for the proper subcellular localization of TOP2A (Mirski et al, 1997; Wessel et al, 1997). Deletion of TOP2A residues 1,174–1,446, which overlap with the CTD, disrupts nuclear localization and mitotic chromatin association despite retaining the major NLS (Adachi et al, 1997; Linka et al, 2007). Published studies also indicated that the CTD is home to multiple PTM sites (Linka et al, 2007; Kozuki et al, 2017; Hoang et al, 2020), which were found to influence TOP2A catalytic activity, protein stability, and subcellular distribution. Based upon these studies, it is reasonable to infer that S1469 O‐GlcNAcylation in NLS could affect the subcellular localization and biochemical functions of this enzyme. The molecular dynamics simulation findings presented here also demonstrated that O‐GlcNAcylation at S1469 induced global landscape changes in the auxiliary DNA‐TOP2A contact region of the CTD and could assist the formation of an enzymatic reaction complex. This may be the mechanism by which O‐GlcNAcylation promotes TOP2A chromatin binding and catalytic activity. Thus, we speculate that the O‐GlcNAcylation and other PTMs of particular residues within the CTD, especially in the NLS of TOP2A, could impact TOP2A subcellular localization and then control its ability to bind chromatin, with a rapid exchange between molecules in the chromatin and cytosolic pools apparently governing TOP2A's biological function. The precise mechanisms underlying this regulation remain to be further identified.
Because O‐GlcNAcylation can influence other nearby PTMs or even modifications in distant sequences (Ma et al, 2022), we also revealed that O‐GlcNAcylation at S1469 enhances the phosphorylation of TOP2A. We cannot rule out the possibility that this change in phosphorylation participates in the O‐GlcNAcylation‐mediated regulation of TOP2A function. Further research is needed to elucidate the interplay between other PTMs and the O‐GlcNAcylation of TOP2A. Nevertheless, our work extends the site‐specific function of this modification in regulating the ability of TOP2A to interact with cell cycle factors, which, in turn, determines downstream cell proliferation‐related gene expression and serves as a potential pro‐survival mechanism. Although it was reported that the expression of cell cycle inhibitor p27 can be affected by OGT (Caldwell et al, 2010). In the present study, we showed that O‐GlcNAcylated TOP2A reduced p27 expression, while site‐specific O‐GlcNAcylation deletion (nonglycosylated TOP2A‐S1469A) led to opposite result. In patient samples, no clear correlation was found between OGT expression levels and drug sensitivity. Drug‐resistant samples with much higher TOP2A O‐GlcNAcylation levels reduced p27 expression compared with that of the sensitive samples. These data suggested that TOP2A O‐GlcNAcylation plays a role in regulating p27 expression.
In summary, the evidence provided here shows that elevated O‐GlcNAcylation impacts TOP2A catalytic activity and breast cancer resistance during chemotherapy. Based on the vital role of TOP2A in cancer progression, we propose that O‐GlcNAcylation is an important regulator of the cancer cell response stimulated with TOP2A‐targeting drugs. Therefore, targeting O‐GlcNAc signaling may be a potential therapeutic approach for conquering breast cancer chemotherapy resistance.
Materials and Methods
Cell culture and reagents
Human breast cancer cell lines (MCF‐7, T47D, and MDA‐MB‐231) and human HEK‐293T cells were purchased from Type Culture Collection Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured at 37°C with 5% (vol/vol) CO2 within 6 months from resuscitation. All the cells were cultured in 90% RPMI‐1640 (Gibco) supplemented with 1% penicillin/streptomycin antibiotics (Gibco) and 10% fetal bovine serum (Gibco). Adriamycin (Adm, Sigma) was added to the cell cultures in stepwise increasing concentrations from 0.1 to 10 μM for about 8 months to develop Adm‐resistant variant, namely MCF‐7/ADR. To maintain the drug resistance, the complete medium supplemented with 1 μM Adm. MCF‐7/ADR cells were maintained in complete medium without Adm for 1 week and cells with > 90% viability before subsequent treatments. Cell lines were periodically authenticated by short tandem repeat profiling. Cell lines were routinely checked for Mycoplasma using Mycoplasma PCR Reagent set (Euroclone). L01 was purchased from BioBioPha Co., Ltd. PugNAc was purchased from Sigma. Other reagents were used as analytic grade or better.
Plasmids, lentiviral production, and transfection
The sequence coding human full‐length (1–1,531) wild‐type (WT) human TOP2A and O‐GlcNAcylation site mutant (Ser1469 → Ala) TOP2A were sequenced and subcloned into lentiviral pLVX‐IRES‐Neo vector containing an N‐terminal HA‐tag. Lentiviral shRNA plasmids for TOP2A (#sc‐36695‐V), OGT (#sc‐40780‐V) were purchased from Santa Cruz Biotechnology. Scrambled shRNA plasmid (#1864) was purchased from Addgene. Lentiviral vectors were co‐transfected with psPAX2 and pMD2.G into HEK‐293T cells with Lipofectamine 3000 (Invitrogen). After 48 h, virus supernatant was collected. Cells were cultured in a 6‐well plate and incubated in virus supernatant for 48 h. Stable transfectants were selected with neomycin or puromycin. The sequence coding Flag‐tagged human full‐length OGT was subcloned into pcDNA3.1 vector. The prokaryotic expression plasmids encoding OGT were a gift from Professor David J. Vocadlo (Simon Fraser University, Canada). The truncated mutants of TOP2A (with 6 × His‐tag in C‐terminal) were subcloned into pET28a(+) prokaryotic expression vector (transformed into E. coli BL21DE3) and pCMV eukaryotic expression vector (transfected into OGT overexpressed HEK293T cells). The primer sequences used in this study are provided in Appendix Table S5.
Western/lectin blot and co‐IP
Total proteins were extracted from cells using western/IP lysis buffer (Beyotime, #P0013) supplemented with protease inhibitor and phosphatase inhibitor cocktail (Roche) at 4°C. Protein concentration of cell lysate was determined by bicinchoninic acid (BCA) protein assay kit (Solarbio, China). Proteins were separated by SDS–PAGE (6–12% gel), followed by Coomassie blue staining or transferred to poly‐vinylidene fluoride membrane (Millipore). The membranes were blocked with 5% nonfat milk solution and hybridized with primary antibody at 4°C overnight. After washing, HRP‐conjugated secondary antibodies were used for visualization. The primary antibodies used were anti‐O‐GlcNAc RL2 (Abcam, #ab93858, 1:1,000), anti‐TOP2A (Proteintech, #24641‐1‐AP, 1:2,000), anti‐OGT (Proteintech, #66823‐1‐Ig, 1:1,000), anti‐HA‐tag (CST, # 3724, 1:1,000), anti‐GAPDH (CST, #5174, 1:1,000), anti‐β‐Tubulin (CST, #2128, 1:1,000), anti‐Histone 3 (CST, #4499, 1:1,000), anti‐HCFC1 (CST, # 69690, 1:1,000), an‐P‐gp (CST, #13978, 1:1,000); γ‐H2AX (CST, #97148, 1:1,000), anti‐CDK1 (Proteintech, #67575‐1‐Ig, 1:2,000), anti‐UPF1 (Proteintech, #66898‐1‐Ig, 1:3,000), anti‐PTEN (Proteintech, #60300‐1‐Ig, 1:1,000), anti‐Cyclin A2 (CST, #67955, 1:1,000), anti‐Cyclin D1 (CST, #55506, 1:1,000), anti‐phospho‐Rb (Ser807/811), (CST, # 8516, 1:1,000), anti‐p27 (CST, #3686, 1:1,000), anti‐phospho‐threonine (CST, #9386, 1:1,000), and anti‐phospho‐serine (Abcam, # ab7851, 1:1,000). Lectin sWGA (Vector Laboratories, #B‐1025S, 1:2,000) was used for lectin blotting. The appropriate secondary antibody used were anti‐mouse IgG‐HRP (CST, # 7076, 1:20,000), anti‐rabbit IgG‐HRP (CST, #7074, 1:20,000), anti‐mouse IgM‐HRP (Abcam, #ab97230, 1:20,000), Streptavidin‐HRP (CST, #3999, 1:50,000).
For immunoprecipitation (IP) and co‐immunoprecipitation (co‐IP), each cell lysate was incubated with anti‐TOP2A (Proteintech, #24641‐1‐AP, 1:500), anti‐His‐tag (Abcam, # ab18184, 1:200) antibodies or anti‐HA‐magnetic beads (Bimake, #B26102) for 2 h at 4°C, added with protein A/G‐magnetic beads (Bimake, #B23201), and rotated at 4°C overnight. Immunoprecipitates were then washed with cold western/IP lysis buffer and then subjected to Western blot analysis. For lectin pull‐down assay, each cell lysate was incubated with sWGA‐bound agarose beads (Vector Laboratories, #AL‐1023S), for 2 h at 4°C. Precipitates were then washed with cold western/IP lysis buffer and then subjected to Western/lectin blot analysis.
Tumor tissues, microarray, and immunohistochemistry
Tissue microarrays consisting of 145 cases of breast cancer tissue and 15 cases adjacent noncarcinoma tissue were analyzed by Bioaitech Co., Ltd. Five breast cancer patient tumor samples (chemotherapy‐sensitive, no. 1–2; chemotherapy‐resistant or relapsed, no. 3–5) were used for co‐IP assay. The study was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). The study and the informed consent obtained from enrolled patients was reviewed and approved by Ethics Committee of the Affiliated Huaian No. 1 People's Hospital of Nanjing Medical University (KY‐2022‐101‐01). Tissue microarrays or paraffin‐embedded tissue sections were stained with primary antibody anti‐O‐GlcNAc RL2 (Abcam, #ab93858, 1:200), anti‐TOP2A (Proteintech, #24641‐1‐AP, 1:200), anti‐Cyclin A2 (CST, #67955, 1:200) or anti‐p27 (CST, # 3686, 1:100), then probed with HRP‐labeled secondary antibody, visualized using diaminobenzidine, stained with hematoxylin, and photographed under microscope. The immunoreactivity was semiquantitatively scored according to the product of categorized percentage of stained cell (0: < 5%, 1, 5–25%, 2: 25–50%, 3: 50–75%, and 4: > 75%) and staining intensity (0: negative, 1: weak, 2: intermediate, and 3: strong), and hence has a range from 0 to 12. An immunoreactivity score below median was considered as low expression, and higher values were considered as high expression. Patients' main characteristics are listed in Appendix Tables S1–S4.
Cell viability and colony formation assays
Cells at a density of ~ 3 × 103 cells/well were seeded in a 96‐well plate and treated with DMSO or Adm with/without L01 or PugNAc for 48 h. The viable cells were determined by Enhanced Cell Counting Kit‐8 (Beyotime, #C0041) according to the manufacturer's instructions. Absorbance was read at 450 nm by microplate reader (Biotek).
For colony formation assay, 1 × 103 cells were seeded and cultured in 6‐well plates for 24 h. Then, the cells were treated with Adm. The medium containing Adm was replaced by the fresh every 3 days. After 15 days, cells were fixed with 4% paraformaldehyde and then stained with crystal violet. The colonies were photographed by a microscope. Colonies with a diameter > 0.5 mm were counted under a microscope.
In vitro glycosylation assay
Recombinant Flag‐tagged OGT purified (Anti‐Flag magnetic beads, Bimake, #B26102) from HEK‐293T cells and His‐tagged truncated variants of TOP2A purified from E. coli were mixed in the reaction buffer (2 mM UDP‐GlcNAc (Sigma), 50 mM Tris–HCl pH 7.5, 12.5 mM MgCl2, 1 mM DTT) in a final volume of 50 μL and then incubated at 37°C for 12 h. The reaction product was detected by co‐IP. A commercially available human recombinant TOP2A was also used in this study (TopoGEN, #TG2000H‐4). For chemoenzymatic labeling of O‐GlcNAcylated TOP2A, Click‐iT™ O‐GlcNAc Enzymatic Labeling System (GalT Y289L) and the Click‐iT™ Glycoprotein detection kit (Biotin alkyne) were used according to the manufacturer's instructions (Invitrogen). Briefly, TOP2A was immunoprecipitated using anti‐TOP2A antibody. Subsequently, the immunoprecipitated fractions were subjected to O‐GlcNAc labeling with GalT1 Y289L using UDP‐GalNAz (labeled with azide), followed by a click reaction with alkyne‐biotin. The labeled proteins were analyzed using Streptavidin‐HRP.
Mass spectrometry
O‐GlcNAcylated TOP2A‐C fragment from the in vitro glycosylation assay was subjected to SDS–PAGE, and the band corresponding to TOP2A‐C was excised from gels. After in‐gel trypsin (Promega) digestion according to a standard protocol, peptides were extracted and analyzed by nano‐LC system coupled to an Orbitrap Fusion Tribrid spectrometer (Thermo Fisher Scientific). The peptides were loaded into an in‐house, 10 cm long analytical column packed with 3 mm C18 resin (Dr. Maisch GmbH, Germany) using mobile phase A (0.1% formic acid). The peptides were then separated by a mobile phase B (ACN/0.1% formic acid) gradient elution with the following three steps: 0–35% for 30 min; 35–80% for 10 min; and 80% for 10 min at a flow rate of 300 nl/min. The mass spectrometer was operated in several modes all in Orbitrap. MS1 spectra were collected in a top‐speed data‐dependent fashion with a dynamic exclusion of the precursor for 20 s after two repeated activation events. Data‐dependent acquisition high‐energy collision dissociation (HCD) spectra were collected using 28 NCE. EThcD was performed in the high‐pressure linear ion trap with an optimized 50 ms reaction time for ETD (2 × 105 reagent AGC) with 25% supplemental collisional energy. The raw data were searched against the Uniprot human database using Proteome Discoverer 2.2 (Thermo Fisher Scientific). Trypsin was selected as the proteolytic enzyme with a maximum allowance of up to two missed cleavages. Peptides were identified with ≥ 95% confidence and filtered at a 1% false discovery rate.
Chromatin complexes isolation
Cells were lysed in hypotonic buffer (10 mM HEPES pH 7.5, 10 mM KCl, 0.1 mM MgCl2, 0.4% Igepal CA‐630 (vol/vol), protease, and phosphatase inhibitor cocktail (Roche)) at 4°C for 20 min. Chromatin was released from the pellets by treatment with lysis buffer (50 mM HEPES pH 7.5, 50 mM NaCl, 0.05% SDS, 2 mM MgCl2, 10% Glycerol, 0.1% Triton X‐100, 10 units of Benzonase Nuclease, protease and phosphatase inhibitor cocktail [Roche]) at 4°C overnight. The supernatant was collected as to obtain the chromatin complexes.
DNA cleavage and decatenation assays
Topoisomerase II cell extracts were prepared by TopII‐NucEx Kit (TopoGEN, #TG1050‐1) according to the manufacturer's instructions. In brief, transfer the cells to an ice‐cold TEMP buffer (10 mM Tris–HCl, pH 7.5, 1 mM EDTA, 4 mM MgCl2, 0.5 mM PMSF) and leave the solution on ice for 10 min. To release nuclei, dounce in a tight‐fitting glass homogenizer, and pellet nuclei by centrifugation. Resuspend nuclear pellet TEP (same as TEMP but lacking MgCl2) and add an equal volume of 1 M NaCl, vortex, and leave on ice for 30 min to extract chromosomal proteins. Centrifuge at 13,000 rpm 20 min and recover the supernatant for topoisomerase II cell extracts. For recombinant TOP2A, TOP2A‐WT or TOP2A‐S1469A was immunoprecipitated using anti‐HA magnetic beads from breast cancer cells and then eluted with 15 μg HA peptide (Sigma) in TBS at 4°C. TOP2A protein was quantified by Bradford protein quantification assay (Bio‐Rad).
DNA cleavage assays were performed using a negatively supercoiled pBR322 plasmid DNA in 20 μL of reaction mixtures (10 mM Tris–HCl, pH 7.9, 100 mM KCl, 0.1 mM EDTA, 5 mM MgCl2, and 2.5% glycerol) at 37°C for 2.5–30 min. Reaction was stopped by adding 2 μL of 10% SDS and 1 μL of 250 mM of EDTA, followed by the addition of 50 μg/ml of proteinase K (Sigma) at 45°C for 30 min. The reaction was then resolved by 1% agarose gel electrophoresis. Double‐stranded DNA cleavage was monitored by the conversion of the negatively supercoiled plasmid DNA to linear molecules.
For decatenation of kinetoplast DNA (kDNA), Topoisomerase II Drug Screening Kit (kDNA based) was employed (TopoGEN, #TG1019). In brief, the reactions contained topoisomerase II cell extracts, kDNA, and 1 mM ATP in 20 μL of reaction buffer were incubated for indicated time at 37°C. 1% agarose gel was run, and the cleavage product was observed.
Band depletion assays
Cells (5 × 105) were treated with 1–5 μM Adm for 2 h. Cells were either lysed immediately or incubated in drug‐free medium for another 30 min at 37°C (to reverse TOP2Acc) before lysis. Then, the samples were incubated at 90°C for 5 min; gross, mechanical fragmentation of the DNA was achieved by passing the sample into a syringe through a 25‐gauge needle. Subsequently, samples were supplemented with MgCl2 (5 mM), and duplicates were incubated with recombinant endonuclease with DNase/RNase activity (benzonase, 0.4 Units/μl, Sigma) for 30 min at room temperature before Western blot using the anti‐TOP2A antibody.
Flow cytometry and immunofluorescence
Cells were seeded in a 6‐well plate and treated with the indicated doses of Adm for 24 h and then fixed in 70% (vol/vol) ethanol overnight at −20°C. After washing with PBS, cells were resuspended with PBS containing RNase A (Sigma) for 30 min at 37°C in the dark and then stained with 5 μL propidium (PI, Sigma) for 30 min at room temperature in the dark. Cells were subsequently analyzed using a flow cytometry (BD).
For immunofluorescence assay, cells on coverslips were fixed in 4% paraformaldehyde for 30 min and permeabilized with 0.1% Triton X‐100. Cells were washed with PBS and blocked for 30 min with 5% goat serum. Then, the cells were incubated overnight with primary antibodies against HA‐tag at 4°C, followed by incubation with goat anti‐rabbit IgG (conjugated with FITC) at room temperature for 2 h. 4′,6‐diamidino‐2‐phenylindole (DAPI) was used for nuclear staining. Images were captured under a confocal microscope (Leica).
TCGA data analysis
Gene expression data and corresponding clinical data for 1,072 patients with breast cancer were obtained from The Cancer Genome Atlas (TCGA) dataset (Chang et al, 2013). The Kaplan–Meier survival curves were generated by “survminer” package of R (Version 3.6.3).
Xenograft mouse model and histopathology
In total, 5–6‐week‐old female nude mice (Changsheng Biotechnology Co., Ltd, Benxi, China) were used for the establishment of breast tumor xenografts. All mice were kept in a sterile and comfortable environment and provided food and water ad libitum. All animal experimental protocols were approved by the ethics committee of the Xuzhou Medical University (202207S098). All mice were randomly divided into different groups. No blinding was performed during animal experiments. Breast cancer cell suspension containing 3 × 106 cells in PBS, followed by injection to the left armpit of the mice (n = 5/group). Mice were administered OGT inhibitor L01 (1 mg/kg) by tail vein injection every other day, for 20 days. Tumor size was monitored with caliper twice a week until 24 days. For in vivo Adm efficacy analysis, the mice were intraperitoneal injected with Adm (4 mg/kg) once a week after the tumor size reached 200 mm3. Mice were administered with OGT inhibitor L01 (1 mg/kg) by tail vein injection every other day, for 22 days. Tumor volume was calculated by formula: volume (mm3) = width2 × length × 0.52. For survival analysis, the mice were treated with Adm; general condition, body weight, and survival of the mice were monitored every 2 days. Mice in each cohort were considered to be dead and euthanized either when the tumor volume increased to 1,000 mm3, weight loss 20%, scruffy coat, hunched appearance or moribund state. The endpoint of the study was stipulated at 48‐day post‐tumor cell inoculation. Mice surviving until the end of the efficacy experiment were euthanized. The tumors were then dissected out, fixed, sectioned, and stained with antibody and hematoxylin.
Molecular simulation
The C‐terminal domain (CTD, residue: 1,435–1,522) of TOP2A was taken from the modeling structure of AlphaFold Protein Structure Database (ID: P11388), and totally 1 μs molecular dynamics (MD) simulation was conducted to refine the modeling structure. The DNA sequence from PDB 6ZY5 was used as the template to build a longer double helix structure (extend the sequence from 17 to 34 bp with a repeat) by web server 3DNA (http://web.x3dna.org/), and a 500 ns MD simulation was taken the relax and refine the modeling structure.
The modeling structures of CTD and DNA double helix were chosen from the largest cluster of the equilibrium period of MD simulation, respectively. The complex structure of CTD‐DNA was calculated with ZDOCK program with S1469 on the interface, and PyRosetta was used for the local refinement of top‐5 results from ZDOCK. Three best scored complexes were taken to carry a 1 μs MD simulation for each. From the cluster and PCA analysis results of the MD trajectories, the most typical complex conformation was taken for further study. The glycosylation structure of CTD‐S1469 was modeled with GlyCAM server. For the glycosylated and nonglycosylated CTD‐DNA complexes, 1 μs MD simulation was conducted in parallel.
All of the MD simulations were performed by Amber20 with Amber14 force field. The structure was solvated in a cubic TIP3P water box with 1 nm distance from the edge, which was neutralized by sodium ions. After four steps of energy minimization, the temperature of the system was gradually heated to 300 K over 100 ps to perform the 10 ns NVT equilibration and 10 ns NPT equilibration subsequently. The production MD simulations at 300 K and 1 atm were carried out with the LINCS algorithm to restrain the hydrogen positions at their equilibrium distances, which allowed the use of an integration time step of 2 fs. Both energies and coordinates were saved every 10 ps for the postproduction analysis of the MD simulations. All MD simulations were performed on a high‐performance computer cluster running the Linux operating system.
Statistical analyses
Statistical differences were determined using the paired/unpaired Student's t‐test comparisons (GraphPad Prism 8.0). Survival curves were plotted according to the Kaplan–Meier method. P‐value < 0.05 was taken as statistically significant. Spearman correlation test was used for correlation analysis. All data were reported as mean ± standard deviation (SD). Detailed n values for each panel in the figures are stated in the corresponding legends.
Author contributions
Yangzhi Liu: Data curation; validation; investigation; visualization; writing – original draft. Kairan Yu: Data curation; formal analysis; validation; investigation; visualization; writing – original draft. Keren Zhang: Resources; formal analysis. Mingshan Niu: Resources. Qiushi Chen: Resources; software; visualization. Yajie Liu: Investigation. Lingyan Wang: Investigation. Nana Zhang: Investigation. Wenli Li: Investigation; writing – original draft. Xiaomin Zhong: Resources. Guohui Li: Software. Sijin Wu: Resources; software; investigation; writing – original draft. Jianing Zhang: Resources; writing – review and editing. Yubo Liu: Resources; data curation; formal analysis; funding acquisition; investigation; visualization; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This study is supported by the National Natural Science Foundation of China (32171282) and the Fundamental Research Funds for the Central Universities (DUT22YG131). We also thank Professor David J. Vocadlo (Simon Fraser University, Canada) for providing plasmids.
EMBO reports (2023) 24: e56458
Contributor Information
Sijin Wu, Email: sijin_wu@foxmail.com.
Jianing Zhang, Email: jnzhang@dlut.edu.cn.
Yubo Liu, Email: liuyubo@dlut.edu.cn.
Data availability
The raw mass spectral data have been deposited to the iProX partner repository (https://www.iprox.cn/) under Project ID: PXD041284. The link access to the raw data is: https://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD041284.
References
- Adachi N, Miyaike M, Kato S, Kanamaru R, Koyama H, Kikuchi A (1997) Cellular distribution of mammalian DNA topoisomerase II is determined by its catalytically dispensable C‐terminal domain. Nucleic Acids Res 25: 3135–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly C (2012) Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem Rev 112: 3611–3640 [DOI] [PubMed] [Google Scholar]
- Caldwell S, Jackson S, Shahriari K, Lynch T, Sethi G, Walker S, Vosseller K, Reginato M (2010) Nutrient sensor O‐GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 29: 2831–2842 [DOI] [PubMed] [Google Scholar]
- Chang K, Creighton CJ, Davis C, Donehower L, Drummond J, Wheeler D, Ally A, Balasundaram M, Birol I, Butterfield YSN et al (2013) The cancer genome atlas Pan‐cancer analysis project. Nat Genet 45: 1113–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Sun Y, Ji P, Kopetz S, Zhang W (2015) Topoisomerase IIα in chromosome instability and personalized cancer therapy. Oncogene 34: 4019–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikamori K, Grabowski DR, Kinter M, Willard BB, Yadav S, Aebersold RH, Bukowski RM, Hickson ID, Andersen AH, Ganapathi R (2003) Phosphorylation of serine 1106 in the catalytic domain of topoisomerase IIα regulates enzymatic activity and drug sensitivity. J Biol Chem 278: 12696–12702 [DOI] [PubMed] [Google Scholar]
- Coley HM (2008) Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat Rev 34: 378–390 [DOI] [PubMed] [Google Scholar]
- Daou S, Mashtalir N, Hammond‐Martel I, Pak H, Yu H, Sui G, Vogel JL, Kristie TM, Affar EB (2011) Crosstalk between O‐GlcNAcylation and proteolytic cleavage regulates the host cell factor‐1 maturation pathway. Proc Natl Acad Sci USA 108: 2747–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty AC, Hawaz MG, Hoang KG, Trac J, Keck JM, Ayes C, Deweese JE (2021) Exploration of the role of the C‐terminal domain of human DNA topoisomerase IIα in catalytic activity. ACS Omega 6: 25892–25903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding AB, Concannon M, Darling S, Rusilowicz‐Jones EV, Sacco JJ, Prior IA, Clague MJ, Urbé S, Coulson JM (2018) The deubiquitylase USP15 regulates topoisomerase II alpha to maintain genome integrity. Oncogene 37: 2326–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmeiner WH, van Waardenburg RC (2021) Targeting DNA topoisomerases: past & future. Cancer Drug Resist 4: 758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guturi KKN, Bohgaki M, Bohgaki T, Srikumar T, Ng D, Kumareswaran R, El Ghamrasni S, Jeon J, Patel P, Eldin MS (2016) RNF168 and USP10 regulate topoisomerase IIα function via opposing effects on its ubiquitylation. Nat Commun 7: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener K, Verstak TA, Lutat KE, Riggsbee DL, Mooney JW (2018) Recent developments in topoisomerase‐targeted cancer chemotherapy. Acta Pharm Sin B 8: 844–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang KG, Menzie RA, Rhoades JH, Fief CA, Deweese JE (2020) Reviewing the modification, interactions, and regulation of the C‐terminal domain of topoisomerase IIα as a Prospect for future therapeutic targeting. EC Pharmacol Toxicol 8: 27–43 [Google Scholar]
- Huang C‐C, Tu S‐H, Lien H‐H, Jeng J‐Y, Huang C‐S, Huang C‐J, Lai L‐C, Chuang EY (2013) Concurrent gene signatures for han chinese breast cancers. PLoS One 8: e76421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Li J, Hu R, Xu B, Huang G, Huang W, Chen B, He J, Cao Y (2021) Cyclin A2/cyclin‐dependent kinase 1‐dependent phosphorylation of Top2a is required for S phase entry during retinal development in zebrafish. J Genet Genomics 48: 63–74 [DOI] [PubMed] [Google Scholar]
- Kang X, Song C, Du X, Zhang C, Liu Y, Liang L, He J, Lamb K, Shen WH, Yin Y (2015) PTEN stabilizes TOP2A and regulates the DNA decatenation. Sci Rep 5: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozuki T, Chikamori K, Surleac MD, Micluta MA, Petrescu AJ, Norris EJ, Elson P, Hoeltge GA, Grabowski DR, Porter AC (2017) Roles of the C‐terminal domains of topoisomerase IIα and topoisomerase IIβ in regulation of the decatenation checkpoint. Nucleic Acids Res 45: 5995–6010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi I, Segev Y, Priel E (2012) Type 1 diabetes affects topoisomerase I activity and GlcNAcylation in rat organs: kidney, liver and pancreas. Glycobiology 22: 704–713 [DOI] [PubMed] [Google Scholar]
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu M‐F, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC (2008) Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 100: 672–679 [DOI] [PubMed] [Google Scholar]
- Linka RM, Porter AC, Volkov A, Mielke C, Boege F, Christensen MO (2007) C‐terminal regions of topoisomerase II α and II β determine isoform‐specific functioning of the enzymes in vivo . Nucleic Acids Res 35: 3810–3822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ren Y, Cao Y, Huang H, Wu Q, Li W, Wu S, Zhang J (2017) Discovery of a low toxicity O‐GlcNAc transferase (OGT) inhibitor by structure‐based virtual screening of natural products. Sci Rep 7: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cao Y, Pan X, Shi M, Wu Q, Huang T, Jiang H, Li W, Zhang J (2018) O‐GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis 9: 485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen Q, Zhang N, Zhang K, Dou T, Cao Y, Liu Y, Li K, Hao X, Xie X (2020) Proteomic profiling and genome‐wide mapping of O‐GlcNAc chromatin‐associated proteins reveal an O‐GlcNAc‐regulated genotoxic stress response. Nat Commun 11: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz C, Lamour V (2020) The interplay between DNA topoisomerase 2α post‐translational modifications and drug resistance. Cancer Drug Resist 3: 149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wu C, Hart GW (2021) Analytical and biochemical perspectives of protein O‐GlcNAcylation. Chem Rev 121: 1513–1581 [DOI] [PubMed] [Google Scholar]
- Ma J, Hou C, Wu C (2022) Demystifying the O‐GlcNAc code: a systems view. Chem Rev 122: 15822–15864 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Takano H, Fojo T (1997) Cellular adaptation to drug exposure: evolution of the drug‐resistant phenotype. Cancer Res 57: 5086–5092 [PubMed] [Google Scholar]
- McClendon AK, Osheroff N (2007) DNA topoisomerase II, genotoxicity, and cancer. Mutat Res 623: 83–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET (2005) An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA 102: 13550–13555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirski SE, Gerlach JH, Cummings HJ, Zirngibl R, Greer PA, Cole SP (1997) Bipartite nuclear localization signals in the C terminus of human topoisomerase IIα. Exp Cell Res 237: 452–455 [DOI] [PubMed] [Google Scholar]
- Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9: 338–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noach N, Segev Y, Levi I, Segal S, Priel E (2007) Modification of topoisomerase I activity by glucose and by O‐GlcNAcylation of the enzyme protein. Glycobiology 17: 1357–1364 [DOI] [PubMed] [Google Scholar]
- Pommier Y, Sun Y, S‐yN H, Nitiss JL (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17: 703–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, Nussenzweig A, Takeda S, Austin C (2022) Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol 23: 407–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewey K, Rowe T, Yang L, Halligan B, Liu L‐F (1984) Adriamycin‐induced DNA damage mediated by mammalian DNA topoisomerase II. Science 226: 466–468 [DOI] [PubMed] [Google Scholar]
- Turley H, Comley M, Houlbrook S, Nozaki N, Kikuchi A, Hickson I, Gatter K, Harris A (1997) The distribution and expression of the two isoforms of DNA topoisomerase II in normal and neoplastic human tissues. Br J Cancer 75: 1340–1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan N, Baselga J, Hyman DM (2019) A view on drug resistance in cancer. Nature 575: 299–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel I, Jensen PB, Falck J, Mirski SE, Cole SP, Sehested M (1997) Loss of amino acids 1490Lys‐ser‐Lys1492 in the COOH‐terminal region of topoisomerase IIα in human small cell lung cancer cells selected for resistance to etoposide results in an extranuclear enzyme localization. Cancer Res 57: 4451–4454 [PubMed] [Google Scholar]
- Yang X, Qian K (2017) Protein O‐GlcNAcylation: emerging mechanisms and functions. Nat Rev Mol Cell Biol 18: 452–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Zhang L, Zhao S, Dai W, Xu Y, Zhang Y, Zheng H, Sheng W, Xu Y (2021) UPF1 promotes chemoresistance to oxaliplatin through regulation of TOP2A activity and maintenance of stemness in colorectal cancer. Cell Death Dis 12: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]