

Abstract
We present two cases of malignant ossifying fibromyxoid tumor (OFMT) which eluded diagnosis due to compelling clinicopathologic mimicry, compounded by similarly elusive underlying molecular drivers. The first is of a clavicle mass in a 69 year-old female, which histologically showed an infiltrative nested and trabeculated proliferation of monomorphic cells giving rise to scattered spicules of immature woven bone. Excepting SATB2 positivity, the lesion showed an inconclusive immunoprofile which along with negative PHF1 FISH led to an initial diagnosis of high-grade osteosarcoma. Next generation sequencing revealed a particularly rare CREBBP::BCORL1 fusion. The second illustrates the peculiar presentation of a dural-based mass in a 52 year-old female who presented with neurologic dyscrasias. Sections showed a sheeted monotonous proliferation of ovoid to spindle cells, but in contrast to Case #1, the tumor contained an exuberance of reticular osteoid and woven bone deposition mimicking malignant osteogenic differentiation. Next generation sequencing showed a novel CREBZF::PHF1 fusion. Both tumors recurred locally less than one year post-operatively. As such we reiterate that careful morphologic examination is axiomatic to any diagnosis in this discipline, but this paradigm must shift to recognize that molecular diagnostics can provide closure where traditional tools have notable limitations.
Keywords: Malignant ossifying fibromyxoid tumor, fusion, PHF1, CREBZF1, BCORL1, BCOR, CREBBP1, Osteosarcoma
Introduction
Musculoskeletal oncologic literature describing ossifying fibromyxoid tumor (OFMT) has evolved over the years to recognize its recurrent molecular signature as well as its clinicopathologic and biologic heterogeneity. An entity of still ambiguous derivation, OFMT’s inherently nonspecific immunoprofile along with its documentation in a variety of sites render it capable of masquerading as other benign as well as frankly malignant processes. Indeed, the diagnostic crux often rests upon molecular testing when all other tools – morphology included - have been exhausted.
The majority of OFMT pursue an innocuous clinical course with complete surgical excision. Local recurrence and distant metastases are observed rarely, a phenomenon usually confined to histologically ‘atypical’ or malignant subsets. In the latter case, morphology rather than molecular identity often predicts adverse behavior, although criteria across these variants are not well-defined - predicated on limited series and follow-up information.
Herein, we present the narrative of two clinicopathologically peculiar cases of malignant OFMT originally misclassified as high-grade osteosarcoma, including one of four reported cases primary to the brain which also harbored a heretofore undescribed CREBZF::PHF1 fusion.1–3 Pitfalls inherent to the differential diagnosis are discussed.
Case #1
A 69 year-old female presented with a right distal clavicle mass, appearing on pre-operative plain films as ill-defined mineralization over a portion of the distal clavicular circumference. Periosteal reaction of the superior cortex was observed, but there was otherwise no evidence of fracture. It was unclear at the time whether the lesion was intra- or extraosseous in etiology. The radiologic differential included osteosarcoma, tumoral calcinosis, or tophaceous pyrophosphate deposition (Figure 1).
Figure 1.
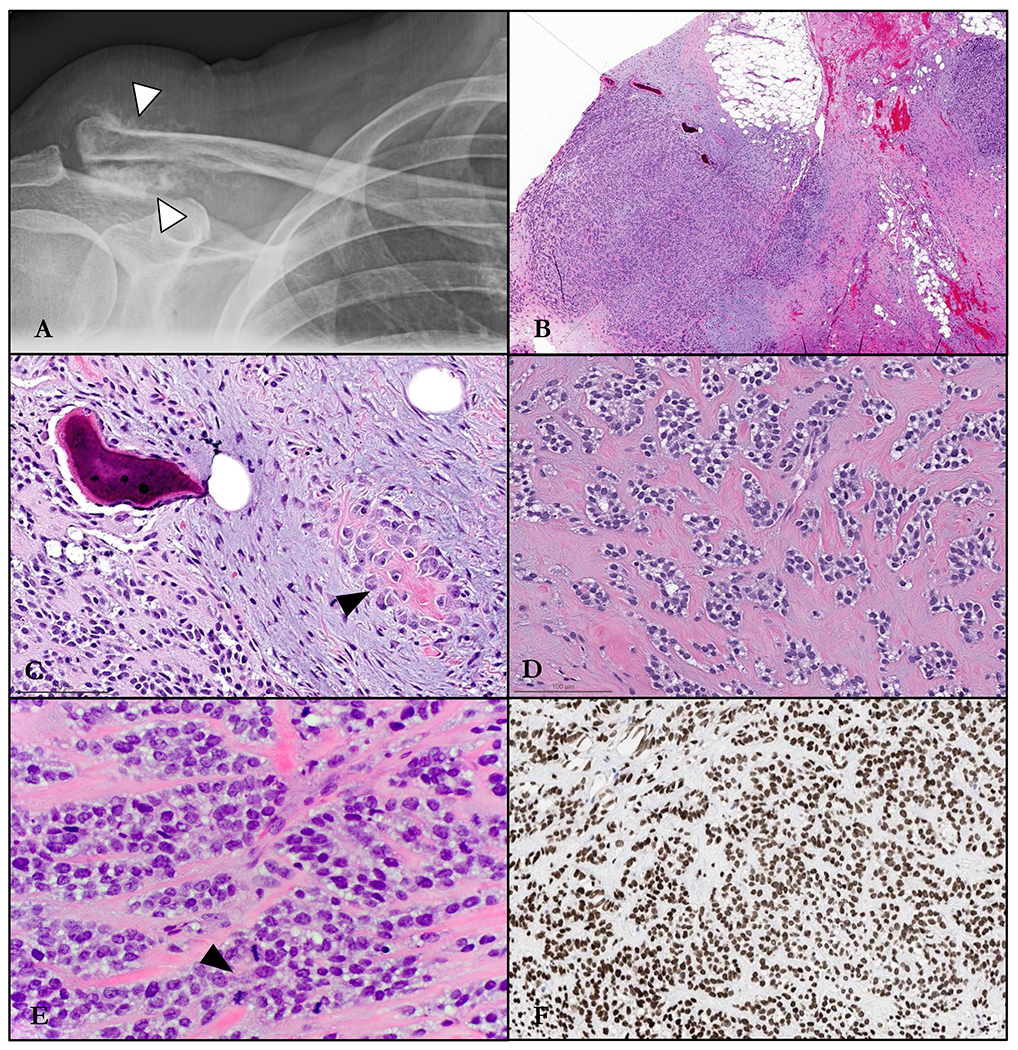
Radiology and histomorphology of Case #1. A. Plain film showing slight lucencywithin and flocculent calcifications surrounding the distal clavicle (arrowhead). B. Destructive infiltration into soft tissue and bone. C. Spicule of immature matrix (arrowhead) elaborated by neoplastic ovoid cells which are embedded in a dense myxoid stroma containing shards of lamellar bone. D. Irregular nests of epithelioid cells mouldedby a fibrocollagenous matrix. E. Nested and corded epithelioid cells with increased mitotic activity (arrowhead). F. Immunohistochemical stain for SATB2, showing strong and unequivocal positivity in the trabeculated tumor cells.
Incisional biopsy showed an infiltrative soft tissue mass with alternating loose fibromyxoid to glassy hyaline stroma which imparted a distinctly trabeculated and nested cellular architecture (Figure 1). The lesional population was comprised by mildly atypical but overall monotonous epithelioid cells with variably abundant pale to clear cytoplasm and round nuclei with homogenous chromatin. A subset of cells showed a more fusiform morphology with amphophilic cytoplasmic processes. Embedded within the tumor were scattered minute foci of both unmineralized osteoid matrix elaborated by neoplastic cells, along with likely pre-existing remodeled lamellar bone with osteoblastic rimming and peripheral deposition of new matrix. Mitotic activity reached up to 10 per 10 high-power fields (all conventional forms), and necrosis was not identified.
Immunohistochemical stains demonstrated the cells to be diffusely and strongly positive for SATB2, with equivocal reactivity for synaptophysin and myosin heavy chain. Epithelial (EMA, AE1/AE3, CAM5.2), myoepithelial (GFAP, SOX10, S100, calponin, p63), melanocytic (HMB45, MelanA), and myogenic/myofibroblastic (SMA, desmin) markers were all negative. FISH studies were negative for rearrangements in EWSR1, NR4A3, and PHF1 genes. Due to the non-specific immunoprofile with only potential evidence of osteoblastic differentiation, the rendered diagnosis was most consistent with high-grade osteosarcoma. To elucidate any possible therapeutic targets, the patient’s oncologist subsequently requested that the tumor undergo comprehensive DNA Next generation sequencing, which revealed an underlying in-frame CREBBP::BCORL1 fusion (Figure 3A). FISH analysis using custom BAC break-apart probes for BCORL1 and CREBBP confirmed rearrangements in both genes.4–6 In the resulting chimeric transcript, exons 1-30 (of 31) of CREBBP (NM_004380.2) were fused to exons 4-12 of BCORL1 (NM_021946.4) [fusion breakpoint chr16:3,781,193::chrX:129,154,960]. The predicted fusion protein contained most of the coding sequence of CREBBP, including the CREB-binding protein domain.
Figure 3.
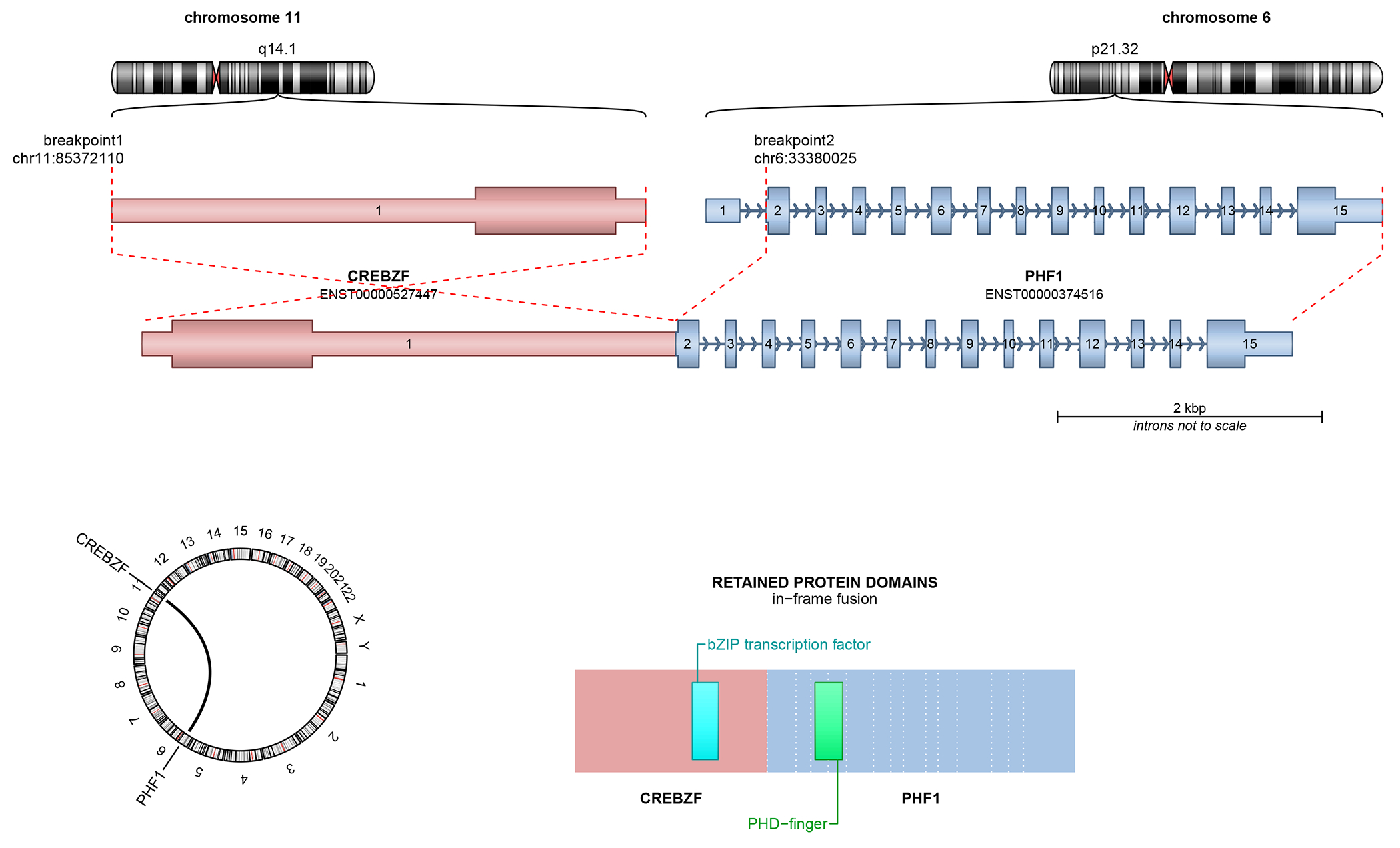
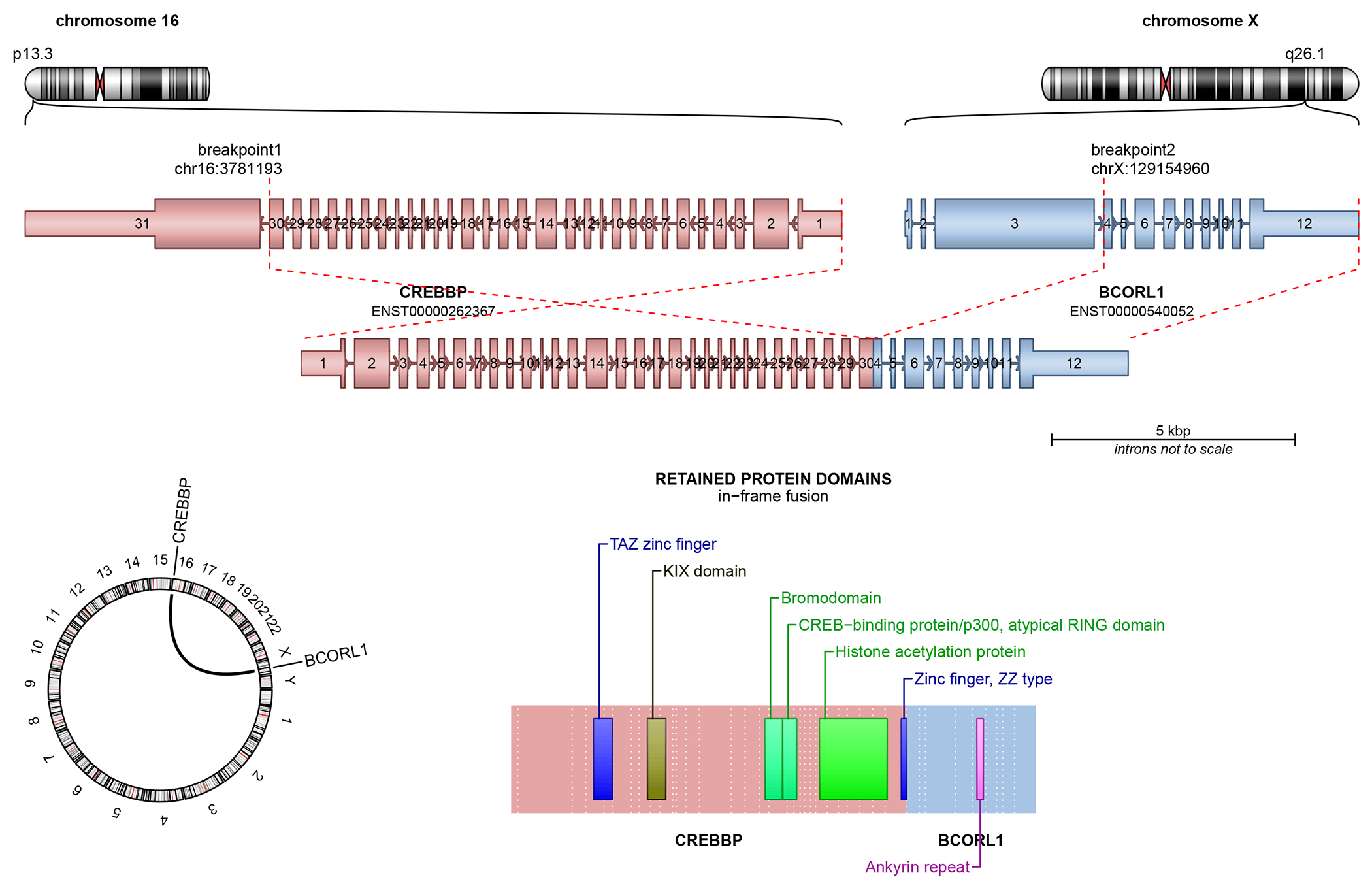
Structural schematics of CREBZF::PHF1 (top) and CREBBP::BCORL1 (bottom) fusions in the malignant ossifying fibromyxoid tumors.
Resection of the mass was performed three months later, showing an 11.2 cm mass focally present at the surgical margins. Mitotic rate was 21 per 10 high-power fields, with approximately 10% overall necrosis. External beam radiation was recommended but declined by the patient. The tumor recurred 3 months later in the acromion, detected initially at a size of 4 cm which expanded rapidly over the course of a month to 9 cm. She refused chemotherapy and immunotherapy, and was subsequently lost to follow-up.
Case #2
A 52 year-old female presented with a sudden-onset history of sensorineural abnormalities over the left upper extremity, which progressed to posturing of the arm, shaking, and ultimately loss of consciousness. In the emergency room, brain MRI demonstrated a 2.6 cm dural-based ring-enhancing lesion – hypointense on T1 and T2 sequences – in the right fronto-parietal region with peripheral vasogenic edema (Figure 2). The lesion appeared entirely contained within the brain parenchyma without dural breach or osseous involvement of the inner skull plate.
Figure 2.
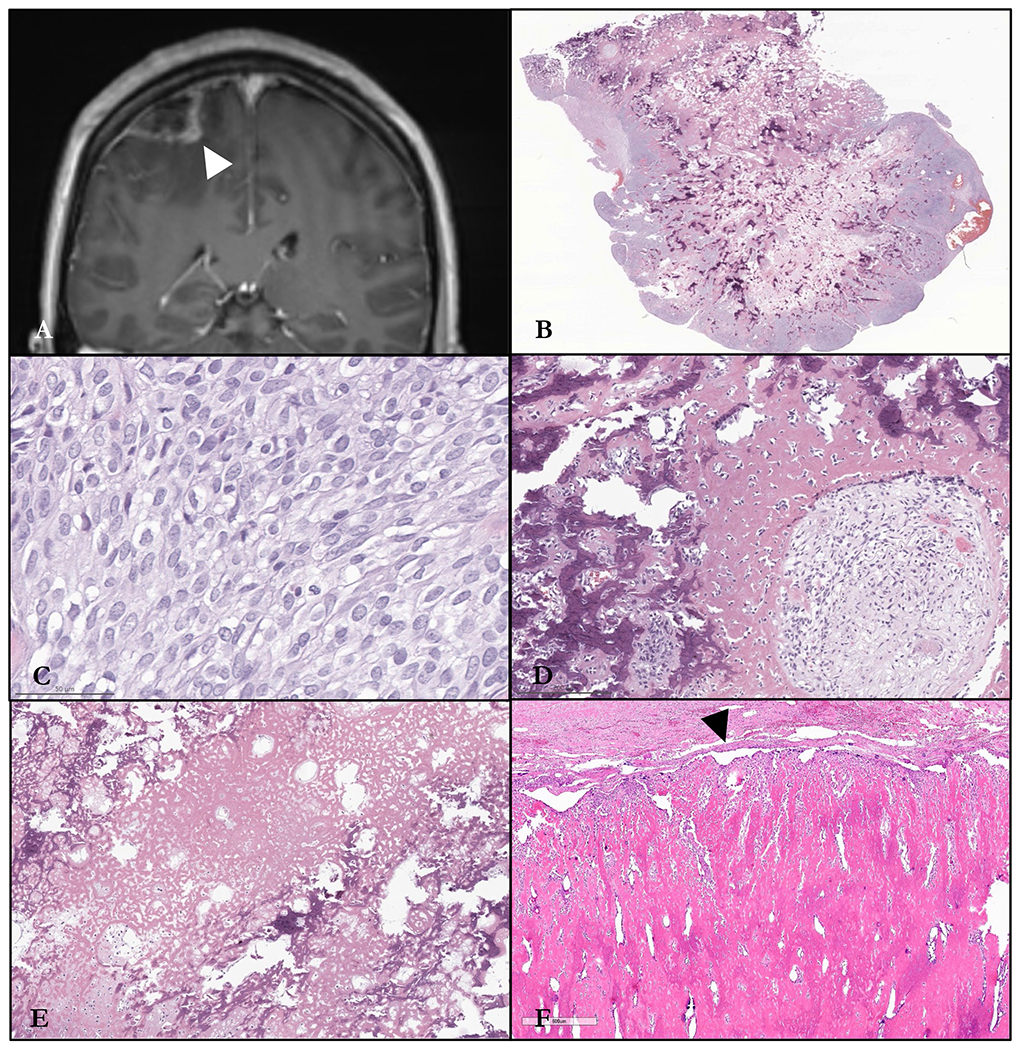
Radiology and histomorphology of Case #2. A. T1 coronal MRI showing a hypointense frontoparietal dural-based ring-enhancing lesion. B. Low-power image illustrating central and peripheral mineralized bony trabeculae. C. Syncytial sheets of monomorphic ovoid to round cells with pale amphophilic cytoplasm. D. Thick anastomotic woven bone with neoplastic cells in myxoid matrix. E. Extensive deposition of reticular osteoid matrix reminiscent of a high-grade osteoblastic osteosarcoma. F. Dural-based recurrence eroding into the skull plate, with extensive deposition of sclerotic bone matrix with a peripheral rind of neoplastic cells (arrowhead).
The patient underwent craniotomy and resection of the mass. The specimen consisted of a portion of tan-grey dura with an adherent calcified mass. Cut surface was focally fleshy but otherwise firm with a gritty consistency. Low-power appearance of the tumor, in comparison to that of Case #1, was remarkable for an internal meshwork of anastomotic and patchily mineralized, coarsely trabecular woven bone (Figure 2), the periphery of which was focally infiltrative into glial tissues. The intervening ovoid to polygonal cells showed moderate atypia without pleomorphism, along with amphophilic cytoplasm, and pale speckled chromatin. They were arranged in syncytial, streaming sheets between and appositional to the bone, but also focally molded into discrete small nests and cords by a dense hyaline matrix. Acellular reticular osteoid was also extensive. Other disparate morphologies were not identified to suggest heterologous osteosarcomatous differentiation from another histologic primary. Mitotic activity was brisk (up to 22 per 10 high-power fields, including atypical forms), and necrosis was focally identified. Immunohistochemically, the tumor cells were strongly positive for vimentin, with weak, patchy reactivity for NKX2.2. They were negative for OSCAR, AE1/AE3, EMA, GFAP, MART1, SOX10, SMA, desmin, ERG, and synaptophysin. Surgical margins were close but free of tumor. The final diagnosis was that of a high-grade extraskeletal osteosarcoma involving the brain.
FoundationOne® sequencing revealed an in-frame CREBZF::PHF1 fusion (Figure 3B), along with a targetable PIK3CA mutation (E545G) and CCND3 amplification. In the chimeric transcript, exon 1 (of 1) of CREBZF (NM_001039618.2) was fused to exons 2-14 of PHF1 (NM_002636.4) [fusion breakpoint chr11:85,368,608::chr6:33,380,025]. In the predicted fusion protein, the entire coding region of CREBZF was fused to the entire coding region of PHF1. The tumor was microsatellite stable with a tumor mutational burden of 3 Muts/Mb. Post-operatively, the patient reported persistent numbness over the lateral aspects of the left side of her body (upper extremity more severe, most pronounced in the fourth and fifth digits), but otherwise was regaining strength and ambulation. She was followed serially with MRI scans. Eleven months after initial resection, imaging revealed a 1.1 cm recurrence along the dura at the anterior aspect of the prior craniotomy. This was resected and interpreted once again as high-grade osteosarcoma (not available for review). Fourteen months after initial presentation, the patient is alive with disease and being considered for radiation therapy.
Discussion
Ossifying fibromyxoid tumor (OFMT) is a soft tissue neoplasm characterized by recurrent PHF1 and rarely, BCOR and BCORL1 genetic rearrangements. 4,7–9 Classically encountered as primary in the superficial soft tissues of the extremities and trunk with a recognized male predilection, OFMT can also infrequently present intraosseously.10 Radiologic features are generally nonspecific and can include an inconsistently high signal on fluid-sensitive sequences due to either a predominance of collagenous or myxoid to fibromyxoid matrix.11 Plain films might demonstrate a sclerotic rind of bone peripheral to an otherwise radiolucent mass. Aggressive features including infiltration and adjacent cortical destruction (when deep and abutting bone) can be observed, and may simulate an osteogenic or Ewing sarcoma.
Histologically, conventional OFMT varies from a well-circumscribed solid mass to a more infiltrative, multinodular lesion. Appreciable on low-power, a histologic hallmark is a shell of peripheral bone which may be of woven or lamellar quality. This osseous tissue (conjectured as metaplastic rather than neoplastic in nature) can be completely absent or so exuberant as to obscure the underlying characteristic stromal elements and cellular architecture. Similar to other translocation-driven tumors, the constituent neoplastic population of OFMT is monomorphic with minimal frank atypia - predominantly ovoid or epithelioid but also spindled in some cases. Cells are arranged in patternless sheets to distinct cords and nests molded by intervening fibrocollagenous to myxoid matrix. OFMT may express S100 or desmin most frequently (more reliably in those with underlying PHF1 rearrangement), along with unpredictable positivity for a host of other lineage- and or tumor-specific mesenchymal markers such as SMA, GFAP, cytokeratins, and even MUC4 and panTRK - rendering an overall nonspecific immunoprofile and definitive diagnosis consequently challenging on purely morphologic grounds.12,13 The continuum from conventional to ‘atypical’ to ‘malignant’ OFMT is rather vaguely defined by a combination of severe atypia, increased cellularity with concomitant stromal rarefication, and a proliferation index of >2 mitoses/50 high-power fields. Other attributes such as necrosis, destructive tissue invasion, and presence of metastasis, are calculated into this subjective gestalt.14–17
Case #1 exemplifies the diagnostic challenges of OFMT at virtually any musculoskeletal site. The clavicle and its peripheral soft tissues are an uncommon location for any mesenchymal tumor, enabling perhaps a wider degree of plausible considerations – not the least of which was osteosarcoma. Complicating interpretation of the different qualities of bone matrix within the specimen was the uncertain location of the tumor – deriving from within the bone with soft tissue extension or vice versa. As such, fragments of both lamellar bone without features of remodeling and overlying matrix deposition, along with scattered islands of new bone formation produced by the cellular population could feasibly be interpreted as either a reparative/reactive, metaplastic, or genuinely neoplastic process. Together with this putatively neoplastic matrix, strong SATB2 positivity (a marker, albeit nonspecific, of osteoblastic differentiation which has not to our knowledge been systematically explored in the context of OFMT), and lack of any other convincing lineage, extraskeletal osteosarcoma became quite a plausible diagnosis of exclusion.
Areas of myxoid stroma with architecturally distinct clusters to trabeculae of ovoid cells, however, are typically not observed in osteosarcomas of any phenotype, and were rather more evocative of a myoepithelial neoplasm,extraskeletal myxoid chondrosarcoma, or BCOR-rearranged sarcoma. While the first two can be positive for S100 (like OFMT), salient ossification is not a characteristic feature of any of these three. Absence of immunohistochemical evidence of myoepithelial differentiation (cytokeratins, EMA, SOX10, GFAP, SMA) and lack of EWSR1 rearrangement effectively excluded a malignant myoepithelial tumor.18 Extraskeletal myxoid chondrosarcoma can also show similar cytologic plasticity and structured architecture within glassy fibromyxoid matrix; they are otherwise characterized by a canonical NR4A3 rearrangement (usually EWSR1::NR4A3 fusion),19 which can be queried with FISH or RNA sequencing. Finally, we include BCOR-altered sarcoma as a potent diagnostic pitfall in most intraosseous malignancies of children and adolescents, displaying a distinct myxoid matrix populated by a variable combination of monomorphic ‘small round cells’ along with short fascicles of spindle cells. Like some OFMT, these should be unequivocally positive for the BCOR immunostain, and likewise for BCOR FISH – which can justifiably misdirect the diagnostic conclusion.20
Indeed, OFMT might have been a strong contender synthesizing the body of evidence, but the negative FISH study for PHF1 rearrangement (the most common gene involved in to 85% of these tumors) unfortunately deterred the diagnosis. Notably, comprehensive sequencing which led to the correct diagnosis and amended report , was requested for therapeutic rather than diagnostic purposes. Although in many settings this modality is financially prohibitive for solely the latter, the case highlights clinicopathologic inconsistencies which are occasionally reconciled only via comprehensive genomic interrogation. In a situation such as this where the diagnosis of OFMT is suspected but thwarted by negative PHF1 FISH, FISH or even immunohistochemistry for BCOR are alternative and less expensive tests that may confirm this less common fusion partner. To complicate matters further, BCOR-rearranged OFMT are distinct in that not only do they pursue a worse clinical trajectory, but also consistently show variant malignant histology without expression of S100 or desmin characteristic of their PHF1-rearranged counterparts.5,6,13,21 In contrast, one of the two previously described cases of CREBBP::BCORL1-fusion OFMT showed conventional ‘benign’ histology with rare S100-positive cells and negative desmin, which involved the same genetic breakpoint as that detected in the current case.6 The other CREBBP::BCORL1-fusion OFMT was considered malignant by virtue of brisk mitotic activity, necrosis, peripheral infiltration, and lymphovascular invasion; it also was focally positive for S100 and negative for desmin.6
Case #2 stands alone as an addition to the OFMT literature considering not only one but two singular features: 1) intracranial epicenter (only a few of which have ever been documented in the repertoire of this already uncommon tumor), and 2) underlying CREBZF::PHF1 fusion, the latter of which has (to our knowledge) never been described in the context of OFMT. While bone tumors are not a first-line consideration when faced with an intracranial lesion, extensive ossification predominantly in the form of lace-like mineralized osteoid resembling that generated by a high-grade conventional osteoblastic osteosarcoma would certainly redirect diagnostic considerations (Figure 2). Perhaps reflective of the novel fusion, this particular matrix quality is not an expected feature of even atypical or malignant OFMT as outlined in the earliest series by Enzinger and Weiss (nor those subsequent). As mentioned, rearrangement of PHF1 is characteristic of OFMT aligned with an assortment of partners including EP400, EPC1, and TFE3.4,8,21,22, Including these, other uncommon gene partners underlying OFMT share the common thread of involvement in histone modification. In contrast, the CREBZF locus encodes for a transcription factor which, among other regulatory roles, is involved in stabilization and transcriptional enhancement of the tumor suppressor TP53. It has only ever r been implicated once in fusion-associated tumorigenesis prior to this case – a bladder urothelial carcinoma with a CD44::CREBZF fusion.23
Admittedly, these two cases showed quite contrasting architectural features, but both demonstrated a critical cytomorphologic commonality: nuclear uniformity, which should intimate the presence of an underlying fusion-driven pathophysiology. Regardless of the subtype, most intra-osseous and extraskeletal osteosarcomas instead harbor complex genomic alterations as nonrecurrent copy number alterations and mutations, which translates to histology as frank sarcoma in the form of pleomorphism, high-grade nuclear atypia, and abnormal mitotic activity. With rare exceptions, osteosarcoma is not characterized by cytologic monotony disproportionate to other features of malignancy including destructive permeation, geographic coagulative necrosis, and neoplastic matrix deposition. It should be noted that all the aforementioned features of malignancy characteristic of osteosarcoma can be observed in ‘atypical’ and malignant OFMT, although none in isolation are necessary or sufficient for this qualification. These subsets have been described only in a few series, and as such our threshold for and understanding of this entity is somewhat tenuous.22, 23
Conclusion
We describe two unique cases and expand the molecular profile of the already enigmatic ossifying fibromyxoid tumor, which were confounded not only by their clinicopathologic peculiarities – both elderly female patients, one with a clavicular and the other with a brain-based tumor and radiology favoring a malignant bone-forming lesion – but also by their documented but uncommon genomic identities. As molecular testing continues to more optimally partition historic mimics, knowledge of histologic overlap exemplified by the above narratives becomes crucial towards initiating these more granular examinations of tumor biology.
Footnotes
Conflicts of Interest: The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article
References:
- 1).Pisapia DJ, Ohara K, Bareja R, et al. Fusions involving BCOR and CREBBP are rare events in infiltrating glioma. Acta Neuropathol Com-mun. 2020;8(1):80. doi: 10.1186/s40478-020-00951-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Yamazaki A, Arai Y, Fukuoka K, et al. Diffusely infiltrating glioma withCREBBP-BCORL1 fusion showing overexpression of not onlyBCORL1 but BCOR: a case report. Brain Tumor Pathol. 2022;39(3):171–178. doi: 10.1007/s10014-022-00435-4 [DOI] [PubMed] [Google Scholar]
- 3).Beyer S, Sebastian NT, Prasad RN, et al. Malignant ossifying fibromyx-oid tumor of the brain treated with post-operative fractionated ste-reotactic radiation therapy: a case report and literature review. SurgNeurol Int. 2021;12:588. doi: 10.25259/SNI_827_2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Suurmeijer AJH, Song W, Sung YS, et al. Novel recurrent PHF1-TFE3fusions in ossifying fibromyxoid tumors. Genes Chromosomes Cancer.2019;58(9):643–649. doi: 10.1002/gcc.22755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Antonescu CR, Sung YS, Chen CL, et al. Novel ZC3H7B-BCOR,MEAF6-PHF1, and EPC1-PHF1 fusions in ossifying fibromyxoidtumors—molecular characterization shows genetic overlap with endo-metrial stromal sarcoma. Genes Chromosomes Cancer. 2014;53(2):183–193. doi: 10.1002/gcc.22132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Kao YC, Sung YS, Zhang L, Chen CL, Huang SC, Antonescu CR.Expanding the molecular signature of ossifying fibromyxoidtumors with two novel gene fusions: CREBBP-BCORL1 andKDM2A-WWTR1. Genes Chromosomes Cancer. 2017;56(1):42–50.doi: 10.1002/gcc.22400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Graham RP, Weiss SW, Sukov WR, et al. PHF1 rearrangements inossifying fibromyxoid tumors of soft parts: a fluorescence in situhybridization study of 41 cases with emphasis on the malignant vari-ant. Am J Surg Pathol. 2013;37(11):1751–1755. doi: 10.1097/PAS.0b013e31829644b4 [DOI] [PubMed] [Google Scholar]
- 8).Gebre-Medhin S, Nord KH, Möller E, et al. Recurrent rearrangementof the PHF1 gene in ossifying fibromyxoid tumors. Am J Pathol. 2012;181(3):1069–1077. doi: 10.1016/j.ajpath.2012.05.030 [DOI] [PubMed] [Google Scholar]
- 9).Srivastava P, Zilla ML, Naous R, et al. Expanding the molecular signa-tures of malignant ossifying fibromyxoid tumours with two novelgene fusions: PHF1::FOXR1 and PHF1::FOXR2. Histopathology. 2023;82(6):946–952. doi: 10.1111/his.14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Sbaraglia M, Bellan E, Gambarotti M, et al. Primary malignant ossify-ing fibromyxoid tumour of the bone. A clinicopathologic andmolecular report of two cases. Pathologica. 2020;112(4):184–190. doi: 10.32074/1591-951X-207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11).Finkelstein D, Foremny G, Singer A, et al. Differential diagnosis of T2hypointense masses in musculoskeletal MRI. Skeletal Radiol. 2021;50(10):1981–1994. doi: 10.1007/s00256-021-03711-0 [DOI] [PubMed] [Google Scholar]
- 12).Graham RP, Dry S, Li X, et al. Ossifying fibromyxoid tumor ofsoft parts: a clinicopathologic, proteomic, and genomic study. Am J Surg Pathol. 2011;35(11):1615–1625. doi: 10.1097/PAS.0b013e3182284a3f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Linos K, Kerr DA, Sumegi J, Bridge JA. Pan-Trk immunoexpression in asuperficial malignant ossifying fibromyxoid tumor with ZC3H7B-BCORfusion: a potential obfuscating factor in the era of targeted therapy. J Cutan Pathol. 2021;48(2):340–342. doi: 10.1111/cup.13915 [DOI] [PubMed] [Google Scholar]
- 14).Enzinger FM, Weiss SW, Liang CY. Ossifying fibromyxoid tumor ofsoft parts. A clinicopathological analysis of 59 cases. Am J Surg Pathol.1989;13(10):817–827. doi: 10.1097/00000478-198910000-00001 [DOI] [PubMed] [Google Scholar]
- 15).Folpe AL, Weiss SW. Ossifying fibromyxoid tumor of soft parts: aclinicopathologic study of 70 cases with emphasis on atypical andmalignant variants. Am J Surg Pathol. 2003;27(4):421–431. doi: 10.1097/00000478-200304000-00001 [DOI] [PubMed] [Google Scholar]
- 16).Dantey K, Schoedel K, Yergiyev O, McGough R, Palekar A, Rao UNM.Ossifying fibromyxoid tumor: a study of 6 cases of atypical and malig-nant variants. Hum Pathol. 2017;60:174–179. doi: 10.1016/j.humpath.2016.10.012 [DOI] [PubMed] [Google Scholar]
- 17).Kilpatrick SE, Ward WG, Mozes M, Miettinen M, Fukunaga M,Fletcher CD. Atypical and malignant variants of ossifying fibromyxoidtumor. Clinicopathologic analysis of six cases. Am J Surg Pathol. 1995;19(9):1039–1046. doi: 10.1097/00000478-199509000-00007 [DOI] [PubMed] [Google Scholar]
- 18).Hornick JL, Fletcher CD. Myoepithelial tumors of soft tissue: aclinicopathologic and immunohistochemical study of 101 cases withevaluation of prognostic parameters. Am J Surg Pathol. 2003;27(9):1183–1196. doi: 10.1097/00000478-200309000-00001 [DOI] [PubMed] [Google Scholar]
- 19).Suurmeijer AJH, Dickson BC, Swanson D, et al. A morphologicand molecular reappraisal of myoepithelial tumors of softtissue, bone, and viscera with EWSR1 and FUS gene rearrangements.Genes Chromosomes Cancer. 2020;59(6):348–356. doi: 10.1002/gcc.22835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Kao YC, Owosho AA, Sung YS, et al. BCOR-CCNB3 fusion positivesarcomas: a clinicopathologic and molecular analysis of 36 cases withcomparison to morphologic spectrum and clinical behavior of otherround cell sarcomas. Am J Surg Pathol. 2018;42(5):604–615. doi: 10.1097/PAS.0000000000000965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Linos K, Kerr DA, Baker M, et al. Superficial malignant ossifying fibro-myxoid tumors harboring the rare and recently described ZC3H7B-BCOR and PHF1-TFE3 fusions. J Cutan Pathol. 2020;47(10):934–945.doi: 10.1111/cup.13728 [DOI] [PubMed] [Google Scholar]
- 22).Schneider N, Fisher C, Thway K. Ossifying fibromyxoid tumor: mor-phology, genetics, and differential diagnosis. Ann Diagn Pathol. 2016;20:52–58. doi: 10.1016/j.anndiagpath.2015.11.002 [DOI] [PubMed] [Google Scholar]
- 23).Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implica-tions in the development and treatment of human cancers. Cell Rep.2018;23(1):227–238.e3. doi: 10.1016/j.celrep.2018.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]