

Abstract
Objective:
Inflammasomes modulate the release of bioactive IL-1β. Excessive IL-1β levels are detected in patients with systemic juvenile idiopathic arthritis (sJIA) and cytokine storm syndrome (CSS) with mutated and unmutated inflammasome components, raising questions on the mechanisms of IL-1β regulation in these disorders.
Methods:
To investigate how the NLRP3 inflammasome is modulated in sJIA, we focused on Tmem178, a negative regulator of calcium levels in macrophages, and measured IL-1β and caspase-1 activation in wild-type (WT) and Tmem178−/− macrophages following calcium chelators, silencing of Stim1, a component of store-operated calcium entry (SOCE), or by expressing a Tmem178 mutant lacking Stim1 binding site. Mitochondrial function in both genotypes was assessed by measuring oxidative respiration, mitochondrial reactive oxygen species (mtROS), and mitochondrial damage. CSS development was analyzed in Perforin−/−/Tmem178−/− mice infected with LCMV in which inflammasome or IL-1β signaling was pharmacologically inhibited. Human TMEM178 and IL1B transcripts were analyzed in datasets of whole blood and peripheral blood monocytes from healthy controls and active sJIA patients.
Results:
TMEM178 levels are reduced in whole blood and monocytes from sJIA patients while IL1B levels are increased. Accordingly, Tmem178−/− macrophages produce elevated IL-1β compared to WT cells. The elevated intracellular calcium levels following SOCE activation in Tmem178−/− macrophages induce mitochondrial damage, release mtROS, and ultimately, promote NLRP3 inflammasome activation. In vivo, inhibition of inflammasome or IL-1β neutralization prolongs Tmem178−/− mouse survival to LCMV-induced CSS.
Conclusion:
Downregulation of TMEM178 levels may represent a marker of disease activity and help identify patients that could benefit from inflammasome targeting.
Keywords: sJIA, NLRP3 inflammasome, IL-1β, Ca2+, Tmem178, Stim1
Graphical Abstract
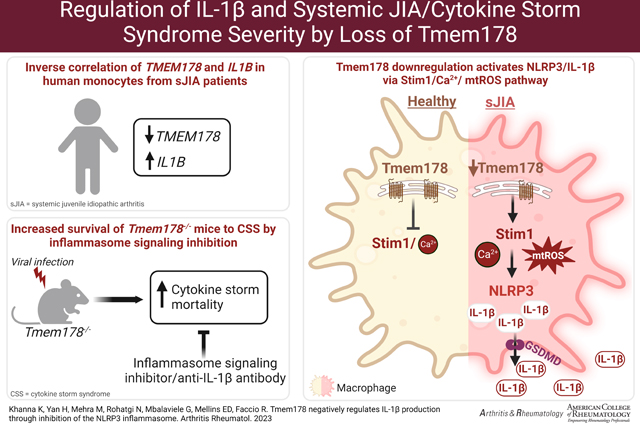
Introduction
Systemic juvenile idiopathic arthritis (sJIA) is a serious clinical complication of systemic inflammatory disorders characterized by arthritis and fever (1) (2). Almost 300,000 children in U.S.A. are currently fighting some form of arthritis, which is more than the number of patients for cystic fibrosis, leukemia and juvenile diabetes combined. About 3–10% of sJIA patients develop a life-threatening complication named macrophage activation syndrome (MAS) or cytokine storm syndrome (CSS), where uncontrolled T cell and macrophage activation result in an exaggerated systemic inflammatory response (3). Although the causes of MAS/CSS are unknown, this complication is also observed in critically ill COVID19 patients (4), cancer patients receiving CAR T-cell immunotherapies (5), and patients with Kawasaki disease (6). Thus, understanding the driver/s of this deadly inflammatory storm can have important therapeutic implications for a variety of conditions.
Studies using animal models as well as samples from sJIA patients developing CSS have shown elevated levels of pro-inflammatory cytokines in particular IL-1β and IL-18 (7) (8) (9). IL-1β and IL-18 are modulated by multimolecular complexes, named inflammasomes, sensing microbe-derived pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (10). Activation of caspases, caspase-1 in particular, by the inflammasomes allows the cleavage of the immature pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into their active forms and their subsequent release (11).
Whole exome sequencing in sJIA patients developing recurrent CSS identified a variant in the gene CASP1, which encodes for caspase-1, causing inflammasome gain-of-function (12). An activating-NLRC4 mutation was further shown to induce autoinflammation with recurrent CSS (13). Similarly, NLRP3 inflammasome overactivation has been observed in CSS patients infected by SARS-CoV-2 (14). However, inflammasome mutations or variants in CASP1 are extremely rare, underscoring the need to understand how inflammasomes are activated during CSS and whether inflammasome-mediated inflammatory responses are drivers of disease progression.
We recently identified Tmem178, a transmembrane protein in the endoplasmic reticulum, as an important negative modulator of inflammatory cytokine production in macrophages through regulation of calcium fluxes (15). Macrophages lacking Tmem178 develop severe inflammatory responses in two mouse models of MAS/CSS, one induced by repeated TLR9 stimulation in response to CpG administration and a second driven by lymphocytic choriomeningitis virus (LCMV) infection in the perforin null background (16). The drivers of disease severity in Tmem178−/− mice remain to be defined.
In this study, we demonstrate that loss of Tmem178 induces NLRP3 inflammasome activation and IL-1β release via increased Ca2+ levels through Stim1, a component of the store operated calcium entry (SOCE), and mitochondrial damage. Administration of an inflammasome signaling inhibitor or IL-1β targeting prolongs the survival of Tmem178−/− mice to LCMV-induced CSS. Importantly, TMEM178 levels are reduced in two sJIA patient datasets of whole blood and monocytes and negatively correlate with IL1B expression. Our data provides novel mechanistic insights into regulation of NLRP3 inflammasome and IL-1β production in sJIA/CSS and suggests a new therapeutic opportunity to treat patients with low TMEM178 levels and high IL-1β with drugs targeting inflammasomes, to potentially prevent development of CSS.
Materials and Methods
Details on materials and procedures for in vitro and in vivo experiments used in the present study are described in Supplementary Materials.
Mice.
WT, Tmem178−/−, Nlrp3−/−, Prf−/−, and correspondent double knockout mice were used for the experiments. Tmem178−/− mice (Strain B6;129S5-Tmem178tm1Lex/Mmucd) were generated by the trans-NIH Knock-Out Mouse Project (KOMP) and purchased from the KOMP Repository at University of California Davis (https://www.komp.org) (stock no. 032664-UCD) and crossed by speed congenic to obtain pure C57/B6 background. C57/B6 WT, Nlrp3−/−and Prf−/− mice were purchased from Jackson Laboratory (Bar Harbor, ME). Tmem178−/− mice were crossed with Nlrp3−/− or Prf−/− mice to generate Tmem178+/−;Nlrp3+/− or Tmem178+/−;Prf+/− ( double heterozygous, F1) mice, which were, in turn, inter-crossed to generate F2 homozygous breeders (Tmem178−/−;Nlrp3−/− or Tmem178−/−;Prf−/− ). F3 mice were used for all experiments. All mice were on C57/BL6 background and housed ad libitum with free access to food and water. Female and male animals were used for all experiments since no gender differences were noted. When different groups were compared, the same numbers of males and/or females used in each group. All experiments were approved by Washington University School of Medicine animal care and use committee.
In vivo animal models.
For LPS-induced inflammation, LPS (5 or 10mg/kg) from Escherichia coli 0111:B4 (Sigma) was injected intraperitoneally into 6–8 week-old male mice. Animals were euthanized after 6 or 16 hours following LPS injection to collect blood, peritoneal fluid, and liver samples for analysis of IL-1β levels. For the CpG-induced CSS model, 50μg CpG 1826 (IDT) was injected intraperitoneally into 6–8 week-old male mice every 2 days. On day 9, the mice were euthanized to collect liver samples to analyze IL-1β levels. For the LCMV-induced CSS model, 2×105 plaque-forming units (PFUs) of LCMV-Armstrong were administered intraperitoneally in 8–12 week-old female Prf−/− or Prf−/−/:Tmem178−/− mice and animals were monitored daily to record survival.
Statistical analysis.
Data is represented as mean +/− SEM using GraphPad Prism 9. Statistical significance was determined by unpaired Student’s t-test or in experiments with multiple comparisons by the two-way ANOVA in conjunction with Sidak’s multiple comparison test, as indicated. For human dataset analysis, significance was determined by Wald test using DESeq2. For survival studies, the significance was determined by log-rank (Mantel-Cox) test. P value <0.05 is set as statistically significant.
Results
Tmem178 deficiency increases IL-1β production and NLRP3 inflammasome activation
To evaluate IL-1β levels in Tmem178−/− mice under inflammatory conditions, we injected a sublethal dose of the TLR4 ligand LPS (5mg/kg) or delivered the TLR9 ligand CpG every other day for 9 days into 6–8 weeks old, sex matched WT and Tmem178−/− animals. A significant increase in IL-1β was detected 16 hours after LPS stimulation in the serum, peritoneal fluid, and liver lysate of Tmem178−/− mice compared to WT (Fig. 1A). IL-1β levels were also increased in liver lysates of Tmem178−/− mice following CpG stimulation, although remained undetectable in the serum and peritoneal fluid (Fig. 1B and data not shown).
Figure 1.

Tmem178 deficiency increases IL-1β production. IL-1β levels in WT or Tmem178−/− mice challenged with (A) 5mg/kg LPS i.p. (16 hours) in peritoneal fluid (n = 7–8/group), liver lysate (n = 8/group) and serum (n = 5/group) or (B) 50μg CpG i.p. (8 days) in liver lysates (n = 5/group). WT and Tmem178−/− BMDMs were primed with 100ng/ml LPS for 4hr followed by nigericin for indicated time/dose and subjected to (C) western blotting to detect pro-IL-1β and mature IL-1β protein levels, (D-E) ELISA to measure IL-1β release in the culture supernatants (n = 3–4/group), (F) pro-caspase-1 and NLRP3 protein analysis by immunoblotting, (G) fluorescence imaging for active caspase-1 (green) and DAPI staining (blue) (Scale=10μm), (H) quantification of cells with active caspase-1 in WT (n = 8) and Tmem178−/− (n = 11) cultures, (I) GSDMD and cleaved GSDMD protein analysis in WT (W) and Tmem178−/− (T) lysates. Data are expressed as mean ± SEM. Immunoblots are representative of three independent experiments and β-actin was used as loading control. Statistical significance is determined by two-tailed t-test (A, B, E, H) or two-way ANOVA for multiple comparisons (D). *P < 0.05 , **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Synthesis and release of IL-1β is a two-step process (10). In the first or priming step, activation of NF-κB induces expression of IL-1β transcripts and pro-IL-1β protein. In the second step, activation of the inflammasome induces the cleavage of pro-IL-1β into the mature active form and its release outside of the cell. Similar to osteoclasts (15), we did not detect differences in the phosphorylation and degradation of IκB-α, a readout for NF-κB activation (20), between WT and Tmem178−/− bone marrow derived macrophages (BMDMs) stimulated with LPS (Supplemental Fig. 1A–C). However, pro-IL-1β transcripts were significantly higher in the Tmem178−/− cell cultures compared with WT, while cytoplasmic pro-IL-1β protein levels were similarly increased by LPS in both genotypes (Supplemental Fig. 1D–E).
NLRP3 polymorphisms are associated with the pathophysiology of sJIA (21), and NLRP3 inflammasome overactivation is considered a trigger for development of CSS after SARS-CoV-2 infection (22). To determine whether Tmem178 deficiency increases IL-1β release by controlling the NLRP3 inflammasome, WT and Tmem178−/− BMDMs were primed with LPS for 4 hours followed by stimulation with the NLRP3 inflammasome activator, nigericin. Intracellular pro-IL-1β levels were reduced at a faster rate in Tmem178−/− cells, with a concomitant increase in IL-1β release compared with WT (Fig. 1C–D, Supplemental Fig. 1F–G). Furthermore, a dose-response to different concentrations of nigericin showed higher IL-1β levels in Tmem178−/− cultures compared to WT (Fig. 1E).
Active caspase-1 cleaves pro-IL-1β into its mature form (23). While both genotypes expressed similar levels of pro-caspase-1 protein at baseline (Fig. 1F, Supplemental Fig. 1I), treatment with LPS and nigericin led to a slightly faster reduction of pro-caspase-1 in Tmem178−/− BMDMs (Fig. 1F). LPS induced NLRP3 expression as previously shown (24). To test the hypothesis that the activated state of the NLRP3 inflammasome is higher in knock-out compared to control cells, we quantified the percentage of cells with activated caspase-1, which binds the FLICA™ FAM-YVAD-FMK probe (25). We observed a higher number of Tmem178−/− cells with active caspase-1 compared with WT (Fig. 1G, H). By contrast, the levels of caspase-8, involved in inflammasome independent IL-1β release, were similar in WT and knock-out cells (not shown). Cleavage of gasdermin-D (GSDMD) is dependent on caspase-1 activation in response to NLRP3 inflammasome activators. We observed higher levels of cleaved GSDMD in the null cells compared to controls (Fig. 1I, Supplemental Fig. 1J–K). Importantly, NLRP3 levels were similarly expressed in both genotypes, suggesting that increased caspase-1 activation was not a reflection of increased NLRP3 levels (Fig. 1F, Supplemental Fig. 1H).
Tmem178 controls NLRP3 inflammasome activation via SOCE
We previously reported that Tmem178 deficiency leads to increased intracellular Ca2+ levels in myeloid cells (18). To determine whether Tmem178 modulates NLRP3 inflammasome via a calcium-dependent pathway, we cultured WT and Tmem178−/− BMDMs in 2mM Ca2+, Ca2+ free media or with the calcium chelator BAPTA. As expected, the percentage of cells with active caspase-1 and IL-1β release were higher in Tmem178−/− cultures in the presence of physiological calcium levels (Supplemental Fig. 2A–D). However, both WT and Tmem178−/− cells had barely detectable active caspase-1 and IL-1β release in Ca2+ free media or in the presence of BAPTA. While this result confirms that Ca2+ plays a key role in NLRP3 inflammasome signaling, it does not explain how Tmem178 deficiency regulates this pathway.
Since Tmem178 binds to Stim1 (18), a critical component of SOCE and modulator of intracellular Ca2+ levels (26) (27), next we wondered whether Tmem178 modulates inflammasome activation through SOCE. We confirmed that Stim1-shRNA decreased Stim1 levels in WT and Tmem178−/− BMDMs while not affecting pro-IL-1β and pro-caspase-1 protein and transcript levels (Fig. 2A, Supplemental Fig. 3A–B). Interestingly, the percentage of caspase-1 positive cells and IL-1β release following LPS and nigericin stimulation were significantly reduced in Stim1-knockdown Tmem178−/− but not WT BMDMs (Fig. 2B–D), indicating that SOCE drives NLRP3 inflammation activation in the context of Tmem178 deficiency.
Figure 2.

Tmem178 controls NLRP3 inflammasome activation via SOCE. WT and Tmem178−/− BMDMs infected with shRNA-control or shRNA-Stim1 were (A) subjected to immunoblotting to measure Stim1, pro-IL-1β and pro-caspase-1. (B-C) Representative images of cells with active caspase-1 (green) following LPS (100ng/ml, 4hr) and nigericin (15μM, 45min) treatments (B). Quantification of the percentage of cells with active caspase-1 (n = 7–8/group) (C) and IL-1β levels in culture supernatants by ELISA (n = 6–7/group) (D). (E-H) WT or Tmem178−/− BMDMs infected with empty vector (EV), HA-tagged full length Tmem178 (Tmem178-WT) or mutant lacking the Stim1 binding site (Tmem178-Mut) were (E) subjected to immunoblotting using anti-HA antibody. (F-H) Representative images of cells with active caspase-1 (green) following 100ng/ml LPS for 4hr, and 15μM nigericin for 45min (Scale=10μm) (F) and correspondent quantification (n = 10–12) (G). IL-1β levels in culture supernatants measured by ELISA (n = 6) (H). Data are presented as mean ± SEM. Nuclei were stained with DAPI (blue). Statistical significance is determined by two-way ANOVA for multiple comparisons. *P < 0.05 ,**P < 0.01 , ***P < 0.001 and ****P < 0.0001.
To better understand how Tmem178 controls NLRP3 inflammasome activation via Stim1, we expressed HA-tagged Tmem178-WT or the Tmem178 mutant lacking the Stim1 binding site (Tmem178-L212W;M216W as described in (18)) in Tmem178−/− BMDMs. First, we confirmed similar protein and mRNA levels of Tmem178 and caspase-1 in the infected cells (Fig. 2E, Supplemental Fig. 3C–D). Next, we observed that expression of Tmem178-WT did not alter inflammasome activation in WT cells but reduced the percentage of caspase-1+ cells and IL-1β release in Tmem178−/− BMDMs (Fig. 2F–H). Importantly, the expression of a Tmem178 mutant lacking the Stim1 binding in Tmem178−/− cells restored active caspase-1 and IL-1β release to empty vector levels (Fig. 2F–H). These results indicate that loss of a direct interaction between Stim1 and Tmem178 is required to enhance NLRP3 activation.
Loss of Tmem178 induces mitochondria dysfunction
Mitochondrial damage is a known intracellular activator of inflammasomes, triggered by calcium overload (28). Because Tmem178−/− BMDMs have increased basal intracellular calcium (Fig 3A–B and (18)), we wondered whether treatment with nigericin could further increase calcium levels following SOCE activation. WT and Tmem178−/− BMDMs plated in calcium free medium with or without nigericin were stimulated with 2mM calcium to activate SOCE. While nigericin per se did not change calcium levels, SOCE activation led to a much higher calcium increase, as measured by the area under the curve (AUC), in nigericin-treated Tmem178−/− BMDMs compared with WT (Fig. 3C–D).
Figure 3.

Loss of Tmem178 induces mitochondria dysfunction. (A-D) Calcium traces and area under curve (AUC) in WT and Tmem178−/− BMDMs in calcium free medium stimulated with nigericin or PBS followed by 2mM calcium. (E) Measurement of oxygen consumption rate (OCR) before and after injections of oligomycin, FCCP and antimycin A in resting and LPS- stimulated (100ng/ml, 4hr) WT and Tmem178−/− BMDMs. (F-G) Quantification of damaged mitochondria (MitoGreenhigh and MitoRedlow) (F; n = 6/group) and mtROS (MitoSOXhigh) (G; n = 6/group) in WT and Tmem178−/− BMDMs stimulated with LPS (100ng/ml, 4hr) and nigericin (15μM, 45 min). (H) Fold changes of MitoSOXhigh WT and Tmem178−/− BMDMs treated with 2-APB (25μM and 50μM for 30 min) after priming with LPS (100ng/ml, 4 hr) and nigericin (15μM, 45 min) (n = 6/group). MitoSOXhigh cells exposed to LPS and nigericin were used as basal controls. Data are presented as mean ± SEM. Statistical significance is determined by two-tailed t-test (B, C, D) or two-way ANOVA for multiple comparisons (E-H). *P < 0.05 and ****P < 0.0001.
To examine the effects of Tmem178 loss on mitochondria fitness, we measured the Oxygen Consumption Rate (OCR). Strikingly, Tmem178−/− BMDMs had reduced OCR in basal conditions and after LPS simulation compared with WT (Fig. 3E). Next, we stained BMDMs with MitoTracker Red that is taken up by negatively charged mitochondria and is used to determine mitochondrial functionality (29). We also used MitoTracker Green, which reacts with cysteine residues on mitochondrial proteins and is used to represent mitochondrial mass (30). At steady state, we did not detect differences between WT and Tmem178−/− BMDMs, with less than 6% of cells having damaged mitochondria (Fig. 3F). LPS increased mitochondria damage in both genotypes, but it was further enhanced in Tmem178−/− BMDMs after nigericin stimulation (Fig. 3F, Supplemental Fig. 4A–B). Next, we measured mtROS, a known activator of the NLRP3 inflammasome (31), by staining the cells with the MitoSOX dye. Consistent with elevated mitochondria damage, mtROS levels were higher in Tmem178−/− cells treated with LPS and nigericin compared with WT (Fig. 3G, Supplemental Fig. 4C–D).
Finally, to determine if increased SOCE activation induces mitochondria dysfunction in Tmem178−/− cells, we treated BMDMs with different doses of the SOCE inhibitor 2- Aminoethoxydiphenyl borate (2-APB), and measured mtROS levels. Strikingly, 2-APB significantly reduced mtROS in Tmem178−/− cells but not WT (Fig. 3H). These findings indicate that loss of Tmem178 activates SOCE and induces mitochondrial damage and mtROS, events that lead to NLRP3 inflammasome activation.
Inhibition of inflammasome signaling ameliorates CSS in Tmem178−/− mice
To further demonstrate the importance of Tmem178 in modulating IL-1β production via the NLRP3 inflammasome in vivo, we generated WT, Tmem178−/−, Nlrp3−/, Tmem178−/−;Nlrp3−/− mice and examined IL-1β circulating levels following 6-hour stimulation with 10mg/kg LPS. Confirming our initial observations, IL-1β levels in the serum were significantly increased in Tmem178−/− mice compared to WT, while barely detectable in Nlrp3−/− and Tmem178−/−;/Nlrp3−/− mice (Fig. 4A). These results indicate that loss of Tmem178 requires the NLRP3 inflammasome for IL-1β production, thus excluding the involvement of other inflammasomes.
Figure 4.

Inhibition of inflammasome signaling ameliorates CSS in Tmem178−/− mice. (A) IL-1β levels in serum measured by ELISA in WT, Tmem178−/−, Nlrp3−/−, and Tmem178−/−;Nlrp3−/− mice following i.p. injection with 10mg/kg LPS or PBS for 6hr prior to sample collection (n = 5–7). (B-C) Percent of survival of LCMV-infected Prf−/− mice or Tmem178−/−;Prf−/− treated with (B) CuET (1mg/Kg i.p.; n = 5/group), starting 2 days prior to LCMV infection and continued every other day for a total of 5 doses. Mice receiving vehicle were used as control (n = 3 Tmem178−/−;Prf−/− and n = 4 Prf−/− mice) or (C) anti-IL-1β (10μg, n = 3/group) starting 4hr after LCMV-infection and continued every other day for 30 days. Data are presented as mean ± SEM. Statistical significance is determined by two-way ANOVA (A), Log-rank (Mantel-Cox) test (B, C). *P < 0.05 ,**P < 0.01 and ****P < 0.0001.
Next, we used a well-established CSS mouse model consisting of Perforin (Prf)−/− mice infected with LCMV, which develop severe macrophage-driven inflammatory responses due to the inability of T cells to clear the virus (32). To inhibit inflammasome activation, mice were administered bis(diethyldithiocarbamate)-copper (CuET), the active metabolite of disulfiram, a compound in current use to inhibit GSDMD, the common effector of inflammasomes, including the NLRP3 inflammasome (33). Animals were administered with 1mg/Kg CuET or vehicle given every alternate day, starting 2 days prior up to 6 days post virus infection. Similar to our previous finding, Prf−/−;Tmem178−/− mice were highly susceptible to LCMV infection, with all animals succumbing within 10–13 days, as compared to Prf−/− which died within 20–22 days. CuET did not improve the overall survival of LCMV-infected Prf−/− animals, while significantly prolonged the life of Prf−/−;Tmem178−/− animals for additional 2 weeks (Fig. 4B). Confirming that CuET can target GSDMD downstream of the NLRP3 inflammasome, IL-1β levels were significantly reduced by CuET in Tmem178−/− BMDM following stimulation with LPS and nigericin (Supplemental Fig. 5). Furthermore, neutralizing IL-1β antibody, administered every other day for the duration of the experiment, also increased the survival of Prf−/−;Tmem178−/− similar to Prf−/− mice (Fig. 4C). Collectively, these results support the contribution of the NLRP3 inflammasome in the development of CSS in the context of Tmem178 deficiency.
TMEM178 negatively correlates with IL1B expression
sJIA patients have elevated levels of IL-1β in circulation. To investigate whether increased IL1B expression correlates with reduced TMEM178 levels, we analyzed two published data set consisting of whole human blood (GSE80060) and human monocytes (GSE147608) from sJIA patients and healthy donors (34, 35). Microarray analysis of whole human blood showed TMEM178A downregulation and IL1B upregulation in the sJIA patient dataset compared to healthy controls (Fig. 5A–B). Similarly, bulk RNA sequencing of human monocytes from sJIA patients showed reduction of TMEM178A and significant upregulation of IL1B, STIM1 and CASP1 compared to healthy donors (Fig. 5C–D). Reduced TMEM178 levels were also detected in the THP1 human monocyte line exposed to plasma from sJIA patients (Fig. 5E). Interestingly, we also observed that human monocytes from a healthy donor exposed to plasma from sJIA patients during disease flare had significantly lower levels of TMEM178 compared to patients at quiescence (Fig. 5F). Regression analysis further showed an inverse relation between Tmem178 mRNA and IL-1β levels in WT BMDMs treated with sJIA plasma from different patients versus healthy controls. A similar trend was observed using a limited number of CD14+ monocytes isolated from sJIA patients (data not shown). Finally, to demonstrate that reduced Tmem178 expression drives IL-1β release, WT and Tmem178−/− BMDMs were incubated with healthy or sJIA plasma and stimulated with LPS and nigericin. As expected, IL-1β release in WT cells was increased by sJIA plasma (Fig. 5G), a condition that reduces Tmem178 expression. Interestingly, IL-1β levels were equally elevated in Tmem178−/− BMDMs exposed to either healthy or sJIA plasma (Fig. 5H), thus demonstrating that loss of Tmem178 drives maximal IL-1β release.
Figure 5.

TMEM178 negatively correlates with IL1B expression. (A-D) Differential gene expression in (A-B) whole human blood from sJIA patients (n=33) versus healthy controls (n = 22) and (C-D) human PBMC from sJIA patients (n = 16) versus healthy controls (n = 11). Volcano plot with genes of interest shown in red; P-valueadjusted(BH)< 0.05 shown in black; P-valueunadjusted< 0.05 in blue; genes with no significant difference in grey (A, C). Normalized gene counts (B, D). (E-F) TMEM178 mRNA expression in (E) THP1 cells exposed to control or sJIA plasma or (F) human blood monocytes treated with plasma from patients at disease flare or quiescence. (G) Regression analysis showing Tmem178 transcripts relative to IL-1β in the supernatant of WT BMDMs treated with sJIA versus healthy plasma. (H) IL-1β ELISA in WT and Tmem178−/− BMDMs cultured in the presence of healthy (n = 6) or sJIA (n = 12) plasma. Data are presented as mean ± SEM. Statistical significance is determined by moderated t-statistics (A-B), Wald test using DESeq2 (C-D), two-tailed t-test (E, F) or two-way ANOVA for multiple comparisons (H). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
Identifying sJIA patients who might develop severe inflammation associated with cytokine storm syndrome and understanding the etiology of CSS are unmet clinical needs. Various diagnostic tools have been developed for identifying CSS in the setting of sJIA, based on clinical (e.g., fever) and laboratory features (e.g., cytopenia) (36). However, none is reliably sensitive and specific. Here, we have identified Tmem178 as a negative regulator of inflammasome activation, whose expression levels in monocytes/macrophages negatively correlate with IL-1β in sJIA. Most importantly, we report that the premature death of Tmem178−/− mice in a LCMV-induced CSS model can be prevented by inflammasome inhibition. Our data supports TMEM178 expression as a marker of sJIA patients at risk of increased disease activity and provide a new therapeutic opportunity to treat patients with low TMEM178 levels and high IL-1β with drugs targeting inflammasomes, and potentially prevent development of CSS.
Elevated IL-1β and IL-18 have been observed in sJIA and CSS (37). While blocking IL-1β alone by canakinumab does not reduce the risk to develop CSS in sJIA patients; anakinra, a recombinant IL-1β receptor antagonist, induces remission with normalization of lab abnormalities and fever to some patients poorly responsive to more traditional therapies (38) (39). Free IL-18 is also highly elevated in CSS/MAS patients. Blockade of IL-18 receptor reduces inflammation in a murine model of CSS (8). Importantly IL-18 inhibition in combination with anakinra successfully improved life-threatening hyperinflammation in a patient with a dominant heterozygous mutation in the NLRC4 inflammasome (13). These clinical funding suggests that aberrant activation of inflammasomes could be driving disease progression in sJIA patients developing CSS.
The causes of elevated IL-1β and IL18 levels in sJIA and CSS are not known. Upregulation of AIM2 and NLRC4 inflammasomes, and elevation in S100 proteins, known for priming the NLRP3 inflammasome, have been detected in children with sJIA (40). Furthermore, a gain-of-function mutation of NLRC4 (13), and a heterozygous missense variant in the CASP1 gene encoding pro-caspase-1 have been reported to induce severe recurrent CSS in patients with sJIA (12). However, activating-mutations in inflammasomes or their components are very rare.
In this study, we show that downregulation of Tmem178 levels in macrophages/monocytes drives IL-1β production via SOCE-dependent NLRP3 inflammasome activation. While increased IL-1β transcripts are also observed in Tmem178−/− cells, NF-kB activation, NLRP3 levels, and cytoplasmic pro-IL-1β levels are similar to WT. This apparent controversy might be dependent on a more efficient cleavage of pro-IL-1β by caspase-1 due to increased inflammasome activation. Indeed, in support of this hypothesis, Tmem178−/− cultures show increased number of caspase-1 positive cells and higher levels of GSDMD, the pore forming protein involved in IL-1β release.
Our findings are in line with a previous gene profiling study in PBMCs from patients with sJIA with early-stage CSS that identified a cluster enriched for genes involved in innate immune responses (41). Our results are clinically relevant as we find reduced TMEM178 while increased IL1B levels in two human data sets from sJIA patients. Furthermore, naïve human monocytes exposed to sJIA plasma from patients with flare disease have significantly lower TMEM178 expression than the quiescent patients, supporting the idea that TMEM178 downregulation could be a marker of increased disease activity. Importantly, our data also demonstrate that Tmem178 deficiency confers a survival disadvantage in CSS that can be reversed by inflammasome/ IL-1β inhibition. Considering that loss of Tmem178 in macrophages upregulates several inflammatory cytokines (16), the improved survival of Tmem178 null mice to CSS following inflammasome inhibition, confirms a key role for IL-1β and possibly IL-18 in disease severity in the context of Tmem178 deficiency. These findings suggest a new therapeutic opportunity for patients with low TMEM178 levels and high IL-1β by targeting inflammasomes, to potentially prevent CSS development.
Several studies have shown that calcium regulates NLRP3 inflammasome activation by increasing the assembly of inflammasome components (42) (43). Both intracellular calcium release from the ER or mitochondria, and extracellular calcium entry via GPCRs and calcium channels can activate the NLRP3 inflammasome (43), (44). These findings support our results using BAPTA, to chelate intracellular calcium, and calcium free medium, to remove the extracellular source of calcium. However, the role SOCE on NLRP3 inflammasome activation is controversial. While a recent publication indicated the involvement of the Stim1 SOCE component in inducing NLRP3 upregulation and cytokine production in lung epithelial cells infected with influenza virus (45), studies using Stim1−/−/Stim2−/− mice indicated that SOCE is dispensable for NLRP3 inflammasome activation in macrophages (46). The novelty of our finding consists in demonstrating a key role of SOCE activation in the context of Tmem178 deficiency to drive NLRP3 inflammasome activation and IL-1β release in macrophages.
Calcium overload has been shown to cause mitochondrial damage (28). Tmem178−/− BMDMs have higher basal calcium levels than WT, and now we find that SOCE activation in the presence of nigericin leads to an even higher increase in calcium. Thus, it is very likely that elevated calcium in Tmem178−/− BMDMs induces mitochondria damage, further confirmed by reduced OCR and increased mtROS. Consistent with this posture, the SOCE inhibitor 2-APB reduces mtROS levels in Tmem178−/− but not WT BMDMs. Mitochondrial damage and mtROS have been implicated in NLRP3 activation (31). Recently, increased cleaved GSDMD and its insertion into the mitochondria has been also related to mtROS generation and IL-1β maturation in THP1 macrophages (47), while loss of GSDMD confers protective effects in a mouse model of CSS (48). We find increased GSDMD cleavage in Tmem178−/− cells and higher susceptibility to CSS. Considering the increased clinical interest in the modulation of inflammasome activation in a variety of diseases, our findings are highly significant as they provide a better mechanistic understanding of the regulation of NLRP3 in sJIA/CSS, via downregulation of Tmem178, increased SOCE and cleavage of GSDMD, and induction of mtROS (Fig. 6).
Figure 6.

Model of Tmem178-mediated regulation of IL-1β levels in sJIA. Downregulation of Tmem178 in sJIA drives the activation of SOCE and increases intracellular calcium levels. Calcium overload leads to mitochondria damage and the release of mtROS, that ultimately activates NLRP3 inflammasome assembly. Increased pro- IL-1β and generation of mature IL-1β by caspase-1 cleavage, together with its secretion through the pore forming protein GSDMD, leads to excessive release of IL-1β in circulation in sJIA pathologies.
In the recent era of COVID-19, numerous research studies showing disease severity in COVID-19 patients suggests the involvement of NLRP3 inflammasome, activation of key inflammatory molecules like active caspase-1, and elevated levels of IL-18 in the sera/tissue or PBMC samples (14). Considering that loss of Tmem178 can drive exuberant NLRP3 inflammasome activation, it would be important to monitor Tmem178 levels in COVID-19 patients to determine the risk of developing CSS. In conclusion, herein we report a novel mechanism of NLRP3 inflammasome/IL-1β regulation via loss of Tmem178 through modulation of SOCE and mitochondria damage, suggesting that TMEM178 levels could be used as a biomarker of disease severity and inflammasomes could be considered a potential therapeutic target for sJIA, CSS or other inflammatory disorders accompanied by loss of TMEM178 expression in monocytes/macrophages.
Supplementary Material
Acknowledgements
This work was supported by the Shriners Hospital Grant 85170 (to R.F.), NIH Grants R01 AR066551, CA235096 and CA270030 (to R.F.), and Siteman Cancer Center (to R.F.), NIH/NIAMS AR076758 and AI161022 grants (to G.M.) and by the NIH P30 Grants AR057235 and P30 AR074992. We thank Dr. Claudia Macaubas from Stanford University for helping with the patient plasma collection.
Footnotes
Conflict of Interest
Authors declare no conflict of interest.
References
- 1.Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12(5):259–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yasin S, Schulert GS. Systemic juvenile idiopathic arthritis and macrophage activation syndrome: update on pathogenesis and treatment. Curr Opin Rheumatol. 2018;30(5):514–20. [DOI] [PubMed] [Google Scholar]
- 3.Bracaglia C, Prencipe G, De Benedetti F. Macrophage Activation Syndrome: different mechanisms leading to a one clinical syndrome. Pediatr Rheumatol Online J. 2017;15(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimabukuro-Vornhagen A, Gödel P, Subklewe M, Stemmler HJ, Schlößer HA, Schlaak M, et al. Cytokine release syndrome. J Immunother Cancer. 2018;6(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma C, Ganigara M, Galeotti C, Burns J, Berganza FM, Hayes DA, et al. Multisystem inflammatory syndrome in children and Kawasaki disease: a critical comparison. Nat Rev Rheumatol. 2021;17(12):731–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toplak N, Blazina Š, Avčin T. The role of IL-1 inhibition in systemic juvenile idiopathic arthritis: current status and future perspectives. Drug Des Devel Ther. 2018;12:1633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girard-Guyonvarc’h C, Palomo J, Martin P, Rodriguez E, Troccaz S, Palmer G, et al. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood. 2018;131(13):1430–41. [DOI] [PubMed] [Google Scholar]
- 9.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. 2018;131(13):1442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407–20. [DOI] [PubMed] [Google Scholar]
- 11.Man SM, Kanneganti TD. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol. 2016;16(1):7–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jørgensen SE, Christiansen M, Høst C, Glerup M, Mahler B, Lausten MM, et al. Systemic juvenile idiopathic arthritis and recurrent macrophage activation syndrome due to a CASP1 variant causing inflammasome hyperactivation. Rheumatology (Oxford). 2020;59(10):3099–105. [DOI] [PubMed] [Google Scholar]
- 13.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet. 2014;46(10):1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vora SM, Lieberman J, Wu H. Inflammasome activation at the crux of severe COVID-19. Nat Rev Immunol. 2021;21(11):694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Decker CE, Yang Z, Rimer R, Park-Min KH, Macaubas C, Mellins ED, et al. Tmem178 acts in a novel negative feedback loop targeting NFATc1 to regulate bone mass. Proc Natl Acad Sci U S A. 2015;112(51):15654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahajan S, Decker CE, Yang Z, Veis D, Mellins ED, Faccio R. Plcγ2/Tmem178 dependent pathway in myeloid cells modulates the pathogenesis of cytokine storm syndrome. J Autoimmun. 2019;100:62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15(8):1477–88. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z, Yan H, Dai W, Jing J, Yang Y, Mahajan S, et al. Tmem178 negatively regulates store-operated calcium entry in myeloid cells via association with STIM1. J Autoimmun. 2019;101:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Benjamini YaH Y, Journal of the Royal statistical society: series B (Methodological) , 57 (1), pp.289–300. Controlling the false discovery rate: a practical and powerful approach to multiple testing.: Journal of the Royal statistical society: series B (Methodological); 1995. p. pp.289–300. [Google Scholar]
- 20.Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu Rev Immunol. 2000;18:621–63. [DOI] [PubMed] [Google Scholar]
- 21.Yang CA, Huang ST, Chiang BL. Association of NLRP3 and CARD8 genetic polymorphisms with juvenile idiopathic arthritis in a Taiwanese population. Scand J Rheumatol. 2014;43(2):146–52. [DOI] [PubMed] [Google Scholar]
- 22.Ratajczak MZ, Kucia M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia. 2020;34(7):1726–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galliher-Beckley AJ, Lan LQ, Aono S, Wang L, Shi J. Caspase-1 activation and mature interleukin-1β release are uncoupled events in monocytes. World J Biol Chem. 2013;4(2):30–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurung P, Li B, Subbarao Malireddi RK, Lamkanfi M, Geiger TL, Kanneganti TD. Chronic TLR Stimulation Controls NLRP3 Inflammasome Activation through IL-10 Mediated Regulation of NLRP3 Expression and Caspase-8 Activation. Sci Rep. 2015;5:14488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.den Hartigh AB, Fink SL. Detection of Inflammasome Activation and Pyroptotic Cell Death in Murine Bone Marrow-derived Macrophages. J Vis Exp. 2018(135). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437(7060):902–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287(4):C817–33. [DOI] [PubMed] [Google Scholar]
- 29.Buckman JF, Hernández H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. J Neurosci Methods. 2001;104(2):165–76. [DOI] [PubMed] [Google Scholar]
- 30.Xiao B, Deng X, Zhou W, Tan EK. Flow Cytometry-Based Assessment of Mitophagy Using MitoTracker. Front Cell Neurosci. 2016;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–5. [DOI] [PubMed] [Google Scholar]
- 32.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–43. [DOI] [PubMed] [Google Scholar]
- 33.Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol. 2021;6(64):eabj3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulert GS, Pickering AV, Do T, Dhakal S, Fall N, Schnell D, et al. Monocyte and bone marrow macrophage transcriptional phenotypes in systemic juvenile idiopathic arthritis reveal TRIM8 as a mediator of IFN-γ hyper-responsiveness and risk for macrophage activation syndrome. Ann Rheum Dis. 2021;80(5):617–25. [DOI] [PubMed] [Google Scholar]
- 35.Brachat AH, Grom AA, Wulffraat N, Brunner HI, Quartier P, Brik R, et al. Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on canakinumab therapy. Arthritis Res Ther. 2017;19(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boom V, Anton J, Lahdenne P, Quartier P, Ravelli A, Wulffraat NM, et al. Evidence-based diagnosis and treatment of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2015;13:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu M. Macrophage activation syndrome in systemic juvenile idiopathic arthritis. Immunol Med. 2021;44(4):237–45. [DOI] [PubMed] [Google Scholar]
- 38.Sönmez HE, Demir S, Bilginer Y, Özen S. Anakinra treatment in macrophage activation syndrome: a single center experience and systemic review of literature. Clin Rheumatol. 2018;37(12):3329–35. [DOI] [PubMed] [Google Scholar]
- 39.Rajasekaran S, Kruse K, Kovey K, Davis AT, Hassan NE, Ndika AN, et al. Therapeutic role of anakinra, an interleukin-1 receptor antagonist, in the management of secondary hemophagocytic lymphohistiocytosis/sepsis/multiple organ dysfunction/macrophage activating syndrome in critically ill children*. Pediatr Crit Care Med. 2014;15(5):401–8. [DOI] [PubMed] [Google Scholar]
- 40.Brown RA, Henderlight M, Do T, Yasin S, Grom AA, DeLay M, et al. Neutrophils From Children With Systemic Juvenile Idiopathic Arthritis Exhibit Persistent Proinflammatory Activation Despite Long-Standing Clinically Inactive Disease. Front Immunol. 2018;9:2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fall N, Barnes M, Thornton S, Luyrink L, Olson J, Ilowite NT, et al. Gene expression profiling of peripheral blood from patients with untreated new-onset systemic juvenile idiopathic arthritis reveals molecular heterogeneity that may predict macrophage activation syndrome. Arthritis Rheum. 2007;56(11):3793–804. [DOI] [PubMed] [Google Scholar]
- 42.Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach-Mansky R, Sacks DB, et al. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature. 2012;492(7427):123–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Murakami T, Ockinger J, Yu J, Byles V, McColl A, Hofer AM, et al. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2012;109(28):11282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossol M, Pierer M, Raulien N, Quandt D, Meusch U, Rothe K, et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun. 2012;3:1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu CC, Miao Y, Chen RL, Zhang YQ, Wu H, Yang SM, et al. STIM1 mediates IAV-induced inflammation of lung epithelial cells by regulating NLRP3 and inflammasome activation via targeting miR-223. Life Sci. 2021;266:118845. [DOI] [PubMed] [Google Scholar]
- 46.Vaeth M, Zee I, Concepcion AR, Maus M, Shaw P, Portal-Celhay C, et al. Ca2+ Signaling but Not Store-Operated Ca2+ Entry Is Required for the Function of Macrophages and Dendritic Cells. J Immunol. 2015;195(3):1202–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, et al. Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep. 2018;25(6):1525–36.e7. [DOI] [PubMed] [Google Scholar]
- 48.Tang S, Yang C, Li S, Ding Y, Zhu D, Ying S, et al. Genetic and pharmacological targeting of GSDMD ameliorates systemic inflammation in macrophage activation syndrome. J Autoimmun. 2022;133:102929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.