

Summary
Tumor suppressor p53 plays a central role in tumor prevention. p53 protein levels and activity are under a tight and complex regulation in cells to maintain the proper function of p53. microRNAs play a key role in the regulation of gene expression. Here we report the regulation of p53 through microRNA miR-504. miR-504 acts as a negative regulator of human p53 through its direct binding to two sites in p53 3′-UTR. Overexpression of miR-504 decreases p53 protein levels and functions in cells, including p53 transcriptional activity, p53-mediated apoptosis and cell cycle arrest in response to stress, and furthermore, promotes tumorigenecity of cells in vivo. These results demonstrate the direct negative regulation of p53 by miR-504 as a mechanism for p53 regulation in cells, which highlights the importance of microRNAs in tumorigenesis.
Introduction
The tumor suppressor p53 plays a crucial role in maintaining genomic stability and preventing tumor formation (Levine et al., 2006; Vogelstein et al., 2000; Vousden and Prives, 2009). The p53 gene is the most frequently mutated gene in human tumors; over 50% of tumors harbor mutations in the p53 gene (Bennett et al., 1999). Disruption of normal p53 function is in many circumstances a prerequisite for the initiation and/or progression of tumors. p53 responds to various stress signals, which are detected and communicated to the p53 protein via a wide variety of enzymes that mediate the p53 protein modifications such as phosphorylation, acetylation, methylation, ubiquitination, summolation, and neddylation (Levine et al., 2006; Vousden and Prives, 2009). As a transcription factor, activated p53 selectively transcribes a set of its target genes to start several cellular responses. Depending upon the cell type, the transformed state of a cell and the type or degree of stress placed upon a cell, the p53 protein induces either cell cycle arrest, apoptosis or senescence to prevent the propagation of these damaged or mutant cells that could potentially become cancerous.
Recent studies show that the regulation of the expression of specific microRNAs (miRNAs) could be an additional mechanism for p53 in tumor suppression (He et al., 2007; Raver-Shapira et al., 2007). miRNAs are a class of endogenously expressed, small noncoding regulatory RNA molecules. Emerging evidence has demonstrated a key role of miRNAs in the posttranscriptional regulation of gene products (Bartel, 2004; Pillai et al., 2007; Plasterk, 2006). miRNAs pair with partially complementary sites in the 3′ untranslated regions (UTRs) of target mRNAs, leading to translational repression and mRNA degradation. Computational and biological analyses estimate that around 30% of all genes and the majority of genetic pathways are subject to regulation by multiple miRNAs (Bentwich et al., 2005; Griffiths-Jones et al., 2006; Lim et al., 2005). By targeting multiple transcripts, miRNAs play important roles in a wide array of biological processes including development and differentiation, cell proliferation, apoptosis and metabolism. Aberrant miRNA expression has been frequently observed in various human tumors, suggesting an important role of miRNAs in tumorigenesis (Caldas and Brenton, 2005; Calin and Croce, 2006; Kent and Mendell, 2006; Lu et al., 2005). Recent studies demonstrate that some miRNAs are direct targets of p53. For example, members of the miR-34 family were identified as direct p53 targets, which contain p53 DNA consensus binding elements. p53 induces the expression of miR-34 family, which in turn regulates apoptosis, cell cycle arrest, or senescence and contributes to tumor suppression (Bommer et al., 2007; He et al., 2007; Raver-Shapira et al., 2007). These results provide direct evidence that regulation of specific miRNAs is a mechanism for p53 in tumor suppression.
miRNAs are estimated to regulate the expression of over 30% of genes and their functions (Lewis et al., 2005; Lim et al., 2005). To investigate the possibility that some specific miRNAs might directly regulate p53 protein levels and function, which would be a mechanism for the regulation of p53, we screened for miRNAs which could potentially regulate p53 expression by performing an in silico search for putative binding sites of miRNAs in the 3′-UTR of p53. Through this approach, we identified miR-504 as a miRNA which could regulate p53 expression through its binding to two binding sites in human p53 3′-UTR. Experiments presented here demonstrate that overexpression of miR-504 reduces p53 protein levels, and impairs p53 functions, including p53-mediated transcriptional activation of its target genes, apoptosis and cell cycle arrest. Furthermore, miR-504 promotes tumorigenicity of cells in vivo. These results suggest the regulation of p53 by miR-504 as a mechanism for cells to regulate p53 activities and functions.
Results
miR-504 Negatively Regulates p53 Protein Levels
To search for potential miRNAs which directly target human p53, we performed an in silico search for putative binding sites of miRNAs in the 3′-UTR of the human p53 gene. TargetScan, an online computational algorithm (http://www.targetscan.org/), was employed for this purpose. We ranked the potential miRNAs produced by TargetScan according to the number of potential binding sites found in p53 3′-UTR. Multiple binding sites increase the efficiency of miRNA binding to its target gene as well as its inhibition of protein translation. For each seed a context score is obtained which is based on the types of seed, 3′ pairing contribution, position of the seed along the 3′-UTR, and the local AU content (Grimson et al., 2007). Three types of seed were selected, they are 1) 8mer seed, an A pairing with position 1 of miRNA followed by Watson-Crick (WC) base pairing from positions 2 to 8; 2) 7mer-m8 seed, WC base pairing from positions 2 to 8; and 3) 7mer-1A seed, an A pairing with position 1 of miRNA followed by WC base pairing from positions 2 to 7. Within the groups of miRNAs that have the same number of binding sites, we further ranked them according to the highest context score of the individual sites. The prediction for top five potential miRNAs targeting human p53 is shown in Table 1.
Table 1. The top five putative miRNAs targeting human p53.
| miRNA | Total putative binding sites |
8mer binding site |
7mer-m8 binding site |
7mer-1A binding site |
Context Score |
|---|---|---|---|---|---|
| miR-485-5p | 3 | 0 | 3 | 0 | -0.15 |
| miR-608 | 3 | 0 | 1 | 2 | -0.12 |
| miR-504 | 2 | 1 | 0 | 1 | -0.43 |
| miR-27α | 2 | 0 | 1 | 1 | -0.22 |
| miR-508 | 1 | 1 | 0 | 0 | -0.32 |
Data were downloaded from TargetScan (http://www.targetscan.org/), and the ranked list of potential miRNAs targeting for p53 were generated according to the number of potential binding sites found in the 3′UTR of human p53. 8mer, an A pairing with position 1 of miRNA followed by Watson-Crick (WC) base pairing from positions 2 to 8; 7mer-m8, WC base pairing from positions 2 to 8; 7mer-1A, an A pairing with position 1 of miRNA followed by WC base pairing from positions 2 to 7.
To examine whether these five miRNAs can regulate p53 protein levels, miRNA oligonucleotides, chemically modified double-stranded RNA oligonucleotides with mature miRNA sequences, were transfected into cells expressing endogenous wild type p53. Scrambled miRNA oligonucleotides were employed as a negative control. Three human cell lines expressing wild type p53 protein, including human colon HCT116 p53+/+, lung H460 and breast MCF7 cells, were employed for this experiment to avoid the effects caused by cell and tissue type specificity. The p53 protein levels were examined by Western-blot assays after transfection. As shown in Figure 1A &B, at 24 h after transfection p53 protein levels were greatly decreased (by ∼3-5 fold) in all three cell lines transfected with miR-504 oligonucleotides while no clear changes of p53 protein levels were observed for other miRNA oligonucleotides. The high expression of these five mature miRNAs in cells resulting from the transfection of miRNA oligonucleotides were confirmed by real-time PCR; over 100-500-fold induction of all these five mature miRNAs were observed in all three cell lines at different time points (24-48 h) after transfection (data not shown). These results clearly demonstrated that miR-504 could negatively regulate p53 protein levels.
Figure 1. miR-504 negatively regulates p53 protein levels in cells.
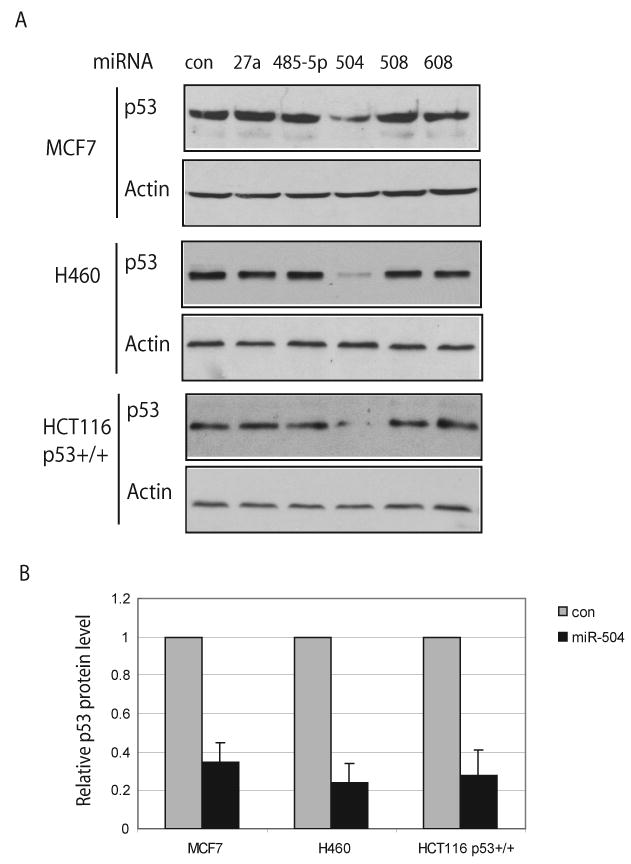
(A) p53 protein levels in cells transfected with different miRNA oligonucleotides. Human HCT116 p53+/+, H460 and MCF7 cells were transfected with various miRNA oligonucleotides (100 nM), and the p53 protein levels were measured at 24 h after transfection by Western-blot assays. Control (con) cells were transfected with scrambled miRNA oligonucleotides.
(B) Relative levels of the p53 protein in human cells transfected with miR-504 oligonucleotides. The p53 protein levels in (A) were normalized with actin. The expression levels of p53 in control cells transfected with scrambled miRNA oligonucleotides were designated as 1. Data are presented as mean±SD (n=3).
miR-504 Negatively Regulates p53 Through Binding to the 3′-UTR of Human p53
Computational analysis suggested that there are two putative binding sites for miR-504 in the 3′-UTR of human p53: an 8 mer seed CAGGGTCA (735-742 bp from the start of the 3′-UTR) and a 7 mer-1A seed AGGGTCA (1064-1070 bp from the start of the 3′-UTR) as shown in Figure 2A. To determine whether miR-504 binds to these two sites to repress p53 expression, a firefly luciferase reporter vector was constructed by inserting the 3′-UTR cDNA sequence of the human p53 gene containing these two putative binding sites into the 3′ end of the luciferase gene (Luc-WT in Figure 2B). The vector was transfected into HCT116 p53+/+ or H460 cells together with either miR-504 oligonucleotides or scrambled miRNA oligonucleotides. The pRL-SV40 vector expressing Renilla luciferase was co-transfected into cells to normalize the transfection efficiency. Results in Figure 2C showed that the expression of miR-504 greatly decreased (over 3-fold) the luciferase activities of the vectors containing the wild type p53 3′-UTR sequence with two putative miR-504 binding sites in HCT116 p53+/+ cells. Similar results were observed in H460 cells (Supplemental Fig 1).
Figure 2. miR-504 binding sites in human p53 3′-UTR mediates the down-regulation of p53 protein expression by miR-504.

(A) miR-504 sequences and its putative binding sites in the 3′-UTR of human p53. The drawing is not to scale.
(B) Luciferase reporter constructs containing wild type or mutant human p53 3′-UTR fused at the 3′ end of the firefly luciferase gene. Shown are luciferase reporter vectors with wild type human p53 3′-UTR (1), with mutant deletion of putative binding site 1 (2), site 2 (3), or both sites (4) for miR-504.
(C) The miR-504 binding sites in human p53 3′-UTR mediates the repression of luciferase activities in cells. HCT116 p53+/+ were transfected with 4 different luciferase constructs described in B together with miR-504 oligonucleotides (100 nM) or the scrambled miRNA oligonucleotides as a control (con). Luciferase activities were measured at 24 h after transfection. Data are presented as mean±SD (n=3).
To further investigate whether miR-504 represses human p53 expression through its binding to these two putative binding sites in the human p53 3′-UTR, we constructed three more luciferase vectors, which contain the mutant 3′-UTR sequence with deletion of either one of the two binding sites or deletion of both binding sites (Figure 2B). These vectors were transfected into HCT116 p53+/+ together with miR-504 oligonucleotides. As shown in Figure 2C, deletion of both binding sites abolished the activity of miR-504 in repressing luciferase activities in cells. Furthermore, the inhibitory effect on the luciferase activities was mainly contributed by the first putative binding site since its deletion resulted in the loss of over 70-80% of the inhibitory effect on luciferase activities in cells, whereas the deletion of the second putative binding site only resulted in around 20-30% loss of inhibitory effect. Similar results were also observed in H460 cells (Supplemental Fig 1). These results together demonstrate that miR-504 represses p53 protein expression through direct binding to these two binding sites in 3′-UTR of the human p53.
miR-504 Reduces p53 Protein Accumulation and Transcriptional Activity in Response to Stress
As a transcription factor, p53 mainly exerts its function through transcriptional regulation of its target genes (Levine et al., 2006; Vogelstein et al., 2000; Vousden and Prives, 2009). In response to various stress signals, including DNA damage, the half-life of p53 protein increases, leading to the accumulation of p53 protein in cells, which in turn leads to the transcriptional activation of selected p53 target genes and initiates various cellular responses.
To study the biological function of the regulation of p53 protein levels by miR-504, the protein levels and the transcriptional activity of the p53 protein in response to stress were examined in cells with miR-504 overexpression. HCT116 p53+/+ cells were transfected with miR-504 oligonucleotides and then treated with Etoposide, which acts as a stress signal to activate p53. As shown in Figure 3A, overexpression of miR-504 in HCT116 p53+/+ cells resulted in the clear inhibition of p53 protein accumulation after Etoposide treatment. Consistent with the decrease of p53 protein accumulation, the transcriptional activity of p53 also decreased; the expression of MDM2 and p21, two well-known p53 target genes, decreased under both non-stressed and stressed conditions. To further examine whether this down-regulation of MDM2 and p21 protein levels is p53 dependent, HCT116 p53-/- cells, isogenic cells for HCT116 p53+/+ with p53 deficiency (Bunz et al., 1998), were employed. As shown in Figure 3A, overexpression of miR-504 did not reduce the protein levels of MDM2 or p21 in HCT116 p53-/- cells, suggesting that the down-regulation of p21 and MDM2 by miR-504 was caused by the down-regulation of the p53 protein levels and function.
Figure 3. miR-504 decreases p53 protein accumulation and transcriptional activity in response to stress.
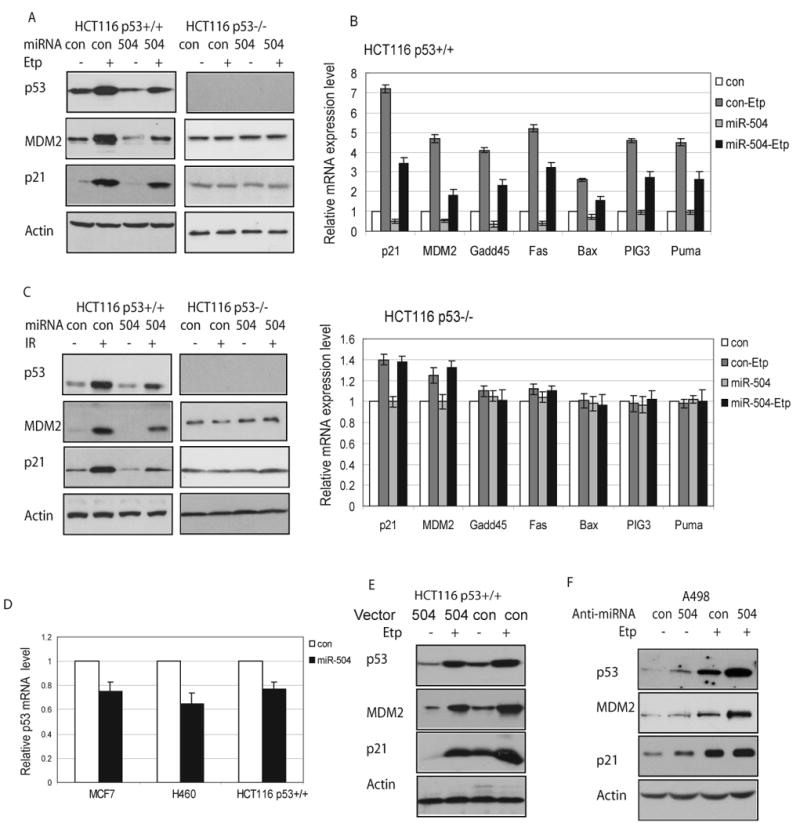
(A) Overexpression of miR-504 down-regulates protein levels of p53 and p53 target genes, MDM2 and p21, in HCT116 p53+/+ treated with Etoposide. HCT116 p53+/+ and p53-/- cells were transfected with 100 nM miR-504 oligonucleotides or scrambled miRNA oligonucleotides (con). At 24 h after transfection, cells were treated with Etoposide (Etp, 20 μM). Protein levels were analyzed at 8 h after Etoposide treatment.
(B) The mRNA expression patterns of p53 target genes in HCT116 cells with miR-504 overexpression. HCT116 p53+/+ and p53-/- cells were transfected with miR-504 oligonucleotides or control oligonucleotides (con). Cells were treated with Etoposide (Etp, 20 μM) at 24 h after transfection. The mRNA expression of p53 target genes was analyzed by real-time PCR at 8 h after Etoposide treatment. The expression of all genes was normalized to Actin. The gene expression levels in control cells (con) transfected with scrambled oligonucleotides were designated as 1. Data are presented as mean±SD (n=3).
(C) miR-504 expression down-regulates protein levels of p53, MDM2 and p21 in HCT116 p53+/+ treated with IR. Cells were transfected with miR-504 oligonucleotides, then treated with IR (10 Gy) at 24 h after transfection. Protein levels were analyzed at 8 h after IR.
(D) miR-504 down-regulates p53 mRNA levels in cells. MCF7, H460 and HCT116 p53+/+ cells were transfected with miR-504 oligonucleotides for 24 h before p53 mRNA levels were measured by real-time PCR. The levels of the p53 mRNA in control cells were designated as 1. Data are presented as mean±SD (n=3).
E) Stable ectopic miR-504 expression down-regulates protein levels of p53, MDM2 and p21 in cells. HCT116 p53+/+ cells with stable miR-504 overexpression by miR-504 expression vectors were treated with Etoposide (Etp, 20 μM), and protein levels were analyzed at 8 h after treatment. Control cells were stably transfected with control empty vectors.
F) Inhibition of endogenous miR-504 increases p53 protein levels and transcriptional activity. Human A498 cells were transfected with anti-miR-504 oligonucleotides (100 nM) 24 h before Etoposide treatment (Etp, 20 μM). Protein levels were measured at 8 h after Etoposide treatment.
The decreased p53 transcriptional activity by miR-504 was also demonstrated by the decreased expression of a group of p53 target genes at the mRNA level as detected by real-time PCR assays. The expression levels of a group of well-known p53 target genes were measured in HCT116 p53+/+ cells which were transfected with miR-504 oligonucleotides for 24 h before Etoposide treatment. These gene products are involved in the regulation of p53-mediated cell cycle arrest (p21 and GADD45), apoptosis (Fas, Bax, PIG3, Puma), and negative regulation of p53 (MDM2) (Levine et al., 2006). As shown in Figure 3B, miR-504 overexpression clearly reduced the transcriptional induction of p21, MDM2, GADD45, Fas, Bax, PIG, and Puma by ∼2-fold in HCT116 p53+/+ cells treated with Etoposide. Furthermore, miR-504 overexpression clearly decreased the basal expression levels of p21, MDM2, GADD45 and Fas by ∼2-fold in cells without Etoposide treatment. To confirm that the transcriptional activation of these genes in response to Etoposide is p53 dependent, HCT116 p53-/- cells were employed. The results in Figure 3B demonstrated that firstly the inductions of all these genes by Etoposide were p53-dependent, and secondly, miR-504 overexpression did not result in clear changes of expression levels of these genes in HCT116 p53-/- cells. These results demonstrated that the impact of miR-504 upon the expression of p53 downstream target genes was p53-dependent. Taken together, these results strongly suggest that miR-504 negatively regulates p53 protein expression and therefore the p53 transcriptional activity.
To study whether miR-504 can negatively regulate p53 protein levels and activity in response to other stress signals in addition to Etoposide, gamma-irradiation (IR) was employed. As shown in Figure 3C, miR-504 expression clearly inhibited the accumulation of p53 protein in HCT116 p53+/+ cells in response to IR, which led to the attenuated transcriptional activity of p53 towards both MDM2 and p21. No clear down-regulation of protein levels of MDM2 and p21 was observed in HCT116 p53-/- cells with miR-504 overexpression. Similar results were also obtained in MCF7, H460 and human osteosarcoma U2OS cells treated with Etoposide (Supplemental Fig 2). These results suggest that the negative regulation of p53 and its transcriptional activity by miR-504 is a common phenotype, which is observed in various cell types.
miRNAs negatively regulate target genes mainly through translational repression. For some target genes, miRNAs also function through degradation of their mRNA (Meister and Tuschl, 2004; Pillai et al., 2007). To investigate whether this is the case for miR-504, p53 mRNA levels were examined in HCT116 p53+/+, H460 and MCF7 cells with miR-504 overexpression. Results obtained from real-time PCR assays showed that in addition to the down-regulation of p53 at protein level, miR-504 also negatively regulated the mRNA levels of p53, but to a much lesser extent (Figure 3D), which suggest that miR-504 negatively regulated p53 expression at the level of both protein and mRNA, and translational repression was the main mechanism.
To exclude the possibility that synthetic mature miR-504 oligonucleotides might underscore the importance of primary miRNA in its native expressed form despite their optimized design criteria, we employed a miR-504 precursor vector and investigate if naturally expressed miR-504 has the same function in negative regulation of p53 in cells. This vector expresses the miR-504 precursor in its native context while preserving putative hairpin structures to ensure biologically relevant interactions with endogenous processing machinery and regulatory partners, which leads to properly cleaved miRNAs. HCT116 p53+/+ and p53-/- cells were stably transfected with miR-504 expression vectors and selected by puromycin. Ten individual cell clones with stable ectopic expression of mature miR-504 (over 50-fold higher miR-504 expression than control cells as measured by real-time PCR) were pooled together for further experiments. The negative regulation of p53 protein levels was also confirmed in HCT116 p53+/+ cells with stable miR-504 overexpression. As shown in Figure 3E, stable overexpression of miR-504 resulted in the clear decrease of protein levels of p53, MDM2 and p21 in HCT116 p53+/+ cells compared with control cells with stable transfection of empty vectors. The decreased p53 transcriptional activity towards a list of p53 target genes as listed in Fig 3B by miR-504 was demonstrated by real-time PCR assays in the HCT116 p53+/+ cells with stable miR-504 overexpression (Supplemental Fig 3). Similar as the observation made in HCT116 p53-/- cells, Etoposide did not result in the clear induction of MDM2 or p21 protein levels in HCT116 p53-/- cells stably transfected with miR-504 expression vectors (data not shown). These results clearly showed that stable expression of miR-504 in cells by expression vectors reduced p53 protein levels and activity.
To further test the role of miR-504 in negative regulation of p53, we inhibited endogenous miR-504 in cells and investigated the impact upon p53. Anti-miR-504 oligonucleotides, chemically synthesized single-stranded RNA oligonucleotides which completely match with mature miR-504 sequences, were employed to inhibit the function of endogenous miR-504 in cells. The endogenous miR-504 levels were measured in a panel of cultured human cell lines, including HCT116 p53+/+, MCF7, H460 and A498 cells, by real-time PCR assays. Among them, A498, human renal epithelial carcinoma cells expressing wild type p53 protein, exhibited 8-10-fold higher expression levels of miR-504 than H460, MCF7 cells and even higher than HCT116 p53+/+ cells (Supplemental Fig 4A). Therefore, A498 cells were transfected with anti-miR-504 oligonucleotides 24 h before Etoposide treatment. As shown in Figure 3F, anti-miR-504 treatment in A498 cells resulted in the clear induction (∼2-fold) of p53, p21 and MDM2 protein under both non-stressed and stressed conditions. In contrast, in HCT116 p53+/+, H460, or MCF7 cells which express low levels of endogenous miR-504, anti-miR-504 oligonucleotides had no clear effect on p53 protein levels (data not shown). To confirm that this effect of anti-miR-504 is resulted from the inhibition of endogenous miR-504 in cells, luciferase reporter assays were employed to investigate whether the anti-miR-504 oligonucleotides could specifically inhibit the function of miR-504. A498 cells were transfected with anti-miR-504 oligonucleotides 6 h before being transfected with miR-504 oligonucleotides and the luciferase reporter vectors containing the wild type human p53 3′-UTR (Luc-WT). Transfection of anti-miR-504 could relieve the inhibitory effect of miR-504 on the luciferase activities of the luciferase reporter vectors in cells, suggesting that anti-miR-504 oligonucleotides could specifically inhibit the function of miR-504 (Supplemental Fig 4B). We further co-transfected A498 cells with anti-miR-504 oligonucleotides and either wild type luciferase reporter vectors (Luc-WT) or mutant luciferase reporter vectors with deletion of both miR-504 binding sites (Luc-mut 1+2). Moderate but clear and consistent activation of luciferase activities were observed for wild type reporter vectors but not for mutant reporter vectors (Supplemental Fig 4C). These results together suggest that miR-504 can negatively regulate p53 protein levels and its transcriptional activity, and inhibition of endogenous miR-504 enhances p53 protein levels and its transcriptional activity.
miR-504 Reduces p53-mediated Apoptosis
The p53-mediated apoptosis is one of the main mechanisms for p53 in tumor suppression. To investigate the impact of miR-504 upon p53-mediated apoptosis, osteosarcoma U2OS cells and lung H460 cells were transfected with miR-504 oligonucleotides followed by Etoposide (20 μM) treatment. Apoptosis was measured at different time points after Etoposide treatment by Annexin V staining in a flow cytometry. Results in Figure 4A&B show that miR-504 overexpression significantly reduced Etoposide-induced apoptosis in both U2OS and H460 cell lines (p<0.05). The p53-dependent apoptosis induced by Etoposide in U2OS and H460 cells was confirmed by transfection of p53 siRNA oligonucleotides (Figure 4A&B), which clearly knocked down endogenous p53 (by over 80% as detected by Western-blot assays; data not shown) and resulted in a clear reduction of Etoposide-induced apoptosis in both cell lines. Co-transfection of miR-504 oligonucleotides together with p53 siRNA oligonucleotides did not further reduce apoptosis compared with transfection of p53 siRNA oligonucleotides only in cells. Taken together, these results clearly demonstrate that miR-504 reduces p53-mediated apoptosis in cells.
Figure 4. miR-504 reduces p53-mediated apoptosis.

(A) miR-504 reduces p53-mediated apoptosis in U2OS cells. U2OS cells were transfected with 100 nM miR-504 oligonucleotides (miR-504) or scrambled miRNA oligonucleotides (miR-con). Cells were treated with 20 μM Etoposide (Etp) at 24 h after transfection and apoptosis were measured at 24 and 36 h after Etoposide treatment. To show the p53-dependent apoptosis induced by Etoposide, cells were transfected with p53 siRNA oligonucleotides (si-p53; 100 nM) or co-transfected with miR-504 oligonucleotides and p53 siRNA (miR-504+si-p53) at 24 h before Etoposide treatment. For controls, cells were either transfected with control siRNA oligonucleotides (si-con) or control miRNA together with control siRNA (miR-con+si-con). Data are presented as mean±SD (n=3). *: p<0.05 (treated vs control).
(B) miR-504 reduces p53-mediated apoptosis in H460 cells. Cells were treated and apoptosis was measured as described in Fig 4A. Data are presented as mean±SD (n=3). *: P<0. 05 (treated vs control).
miR-504 Reduces p53-mediated Cell Cycle Arrest
Another main function of p53 is to induce cell cycle arrest in response to stress. It has been well-established that p53 induces G1 arrest in cells in response to stress (Deng et al., 1995; Kastan et al., 1992). To investigate whether down-regulation of p53 protein levels by miR-504 can result in the reduction of G1 arrest, HCT116 p53+/+ cells were transfected with miR-504 oligonucleotides and then treated with Etoposide at 24 h after transfection. Consistent with previous reports, results in Figure 5A showed that while a majority of the cells underwent a p53-independent G2 arrest, a fraction of the cells underwent p53-dependent G1 arrest in response to stress; Etoposide induced significantly higher G1 arrest in HCT116 p53+/+ cells than in HCT116 p53-/- cells (p<0.01) (Bunz et al., 1998; Moran et al., 2008). Furthermore, miR-504 overexpression in cells significantly reduced G1 arrest induced by Etoposide in HCT116 p53+/+ cells (p<0.01) but not in p53-/- cells (Figure 5A). Similar results were also observed in HCT116 cells with stable ectopic miR-504 expression: miR-504 overexpression by stable transfection of miR-504 expression vectors significantly reduced the G1 arrest induced by Etoposide in HCT116 p53+/+ cells compared with HCT116 p53+/+ cells stably transfected with control vectors (p<0.01; Figure 5B). Again, this reduction of G1 arrest was not observed in HCT116 p53-/- cells with stable miR-504 overexpression (data not shown). These results together clearly demonstrated that miR-504 reduces p53 function in regulating cell cycle arrest in response to stress.
Figure 5. miR-504 reduces p53-mediated cell cycle arrest.

(A) HCT116 p53+/+ (left panel) and p53-/- cells (right panel) were transfected with miR-504 oligonucleotides (miR-504; 100 nM) or scrambled miRNA oligonucleotides (con). At 24 h after transfection, cells were treated with Etoposide (Etp, 20 μM) and cell cycle arrest was analyzed in a flow cytometry at 36 h after Etoposide treatment. Data are presented as mean±SD (n=3). P<0.01 (G1 in #2 vs #4); p<0.01 (G1 in #2 vs #6).
(B) HCT116 p53+/+ cells with stable overexpression of miR-504 by miR-504 expression vectors (p53+/+-miR-504) and cells stably transfected with a control empty vector (p53+/+-Con) were treated with Etoposide (Etp, 20 μM) and cell cycle arrest was analyzed at 36 h after Etoposide treatment. Data are presented as mean±SD (n=3). P<0.01 (G1 in #2 vs #4).
miR-504 Promotes Tumor Growth in vivo
It has been well-established that loss of p53 function promotes tumor initiation and/or progression (Levine et al., 2006; Vogelstein et al., 2000). It has been reported that p53 loss promotes tumorigenicity of HCT116 cells in vivo (Buzzai et al., 2007; Yoon et al., 2009). To investigate whether the down-regulation of p53 protein levels and functions by miR-504 can promote tuorigenicity of HCT116 cells, HCT116 p53+/+ cells with stable ectopic miR-504 expression and control cells with stable transfection of control vectors were subcutaneously (s.c.) inoculated into nude mice and their tumorignicity was investigated. As shown in Figure 6, consistent with the effect of p53 loss on tumor growth as observed in HCT116 p53-/- tumors, which had a faster growth rate than HCT116 p53+/+ tumors in vivo, stable miR-504 overexpression in HCT116 p53+/+ cells promoted tumor growth in vivo compared with control cells stably transfected with control vectors. This promotion effect of miR-504 on tumorigenicity was not observed in HCT116 p53-/- cells with stable miR-504 overexpression, suggesting that miR-504 promotes tumorigenicity of cells through its negative regulation of p53 protein levels and functions.
Figure 6. miR-504 promotes tumorigenicity of cells in vivo.
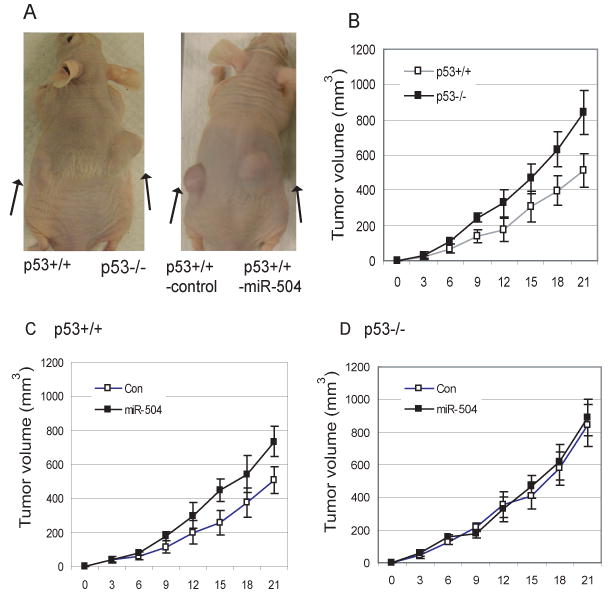
(A) Nude mice were inoculated (s.c.) with HCT116 p53+/+ or HCT116 p53+/+ cells stably transfected with control empty vectors (p53+/+-control) on the left flank, and were inoculated with HCT116 p53-/- cells or HCT116 p53+/+ cells with stable miR-504 overexpression by miR-504 expression vectors (p53+/+-miR-504) on the right flank, respectively (n=10 mice/group). Mice at day 21 after inoculation of tumor cells are presented.
(B) Nude mice were inoculated (s.c.) with HCT116 p53+/+ on the left flank and HCT116 p53-/-cells on the right flank, respectively (n=10 mice/group). Tumor volumes (mm3=1/2 (length × wildth2) were measured every 3 days after inoculation for 3 weeks. Data are presented as mean±SD (n=10).
(C) Nude mice were inoculated with HCT116 p53+/+ cells with stable miR-504 overexpression by miR-504 expression vectors on the right flank (miR-504) and control HCT116 p53+/+ cells stably transfected with empty vectors (Con) on the left flank. Data are presented as mean±SD (n=10).
(D) Nude mice were inoculated with HCT116 p53-/- cells with stable miR-504 overexpression on the right flank (miR-504) and control HCT116 p53-/- cells stably transfected with empty vectors (Con) on the left flank (n=10 mice/group). Data are presented as mean±SD (n=10).
Discussion
The miRNAs have been implicated in many critical biological processes. However, relatively few miRNA-target interactions have been experimentally validated, and the functions of most miRNAs remain to be discovered (Bartel, 2004; Kloosterman and Plasterk, 2006). Aberrant expression of miRNAs observed in various human cancers suggests that miRNAs may play an important role in tumorigenesis. For example, it has been reported that miR-15a and miR-16-1 were down-regulated in B-cell chronic lymphocytic leukemia (CLL) (Cimmino et al., 2005), and miR-143 and miR-145 were down-regulated in colorectal cancer (Cummins et al., 2006; Michael et al., 2003). Amplification or over-expression of the miR-17-92 cluster and miR-155 has been reported in lymphomas (He et al., 2005). In addition, more than 50% of human miRNA genes are located in cancer-associated genomic regions or at fragile sites, which suggests an important role of miRNAs for tumorigenesis (Calin et al., 2004).
The interactions between p53 and miRNAs have been recently demonstrated through the identification of several miRNAs regulated by p53 (He et al., 2007; Raver-Shapira et al., 2007). It has been shown that p53 regulates the expression of a set of specific miRNAs, including miR-34 family, which in turn regulates cellular apoptosis and senescence. For example, miR-34a has been shown to be a direct transcriptional target for p53. The p53 protein binds to the promoter of miR-34a, and increases the expression of miR-34a in response to stress. Inactivation of endogenous miR-34a strongly inhibits p53-dependent apoptosis. These findings strongly suggest that the regulation of miRNAs by p53 contributes to the function of p53 in tumor suppression. On the other hand, miRNAs are estimated to regulate the expression of over 30% of genes and their functions (Lewis et al., 2005; Lim et al., 2005), suggesting a possibility that some specific miRNAs might directly regulate p53 protein expression and function. In this study, we identified miR-504 as a direct negative regulator of p53. miR-504 can directly repress the p53 protein expression through its binding to the two binding sites in 3′-UTR of the human p53 gene, thereby negatively regulating p53 functions. We have demonstrated that overexpression of miR-504 inhibits the transcriptional activity of p53, and reduces the p53-mediated apoptosis and cell cycle arrest in response to stress. In addition to miR-504, a very recent study which was published during our submission of this study reported that miR-125b can directly target p53 and negatively regulate p53 function especially p53-mediated apoptosis (Le et al., 2009). Thus, in addition to many protein-encoding genes, miRNAs can either be regulated by p53 or regulate p53, and are involved in the composition of the complex p53 signaling pathway.
As a haplo-insufficient gene, a small change of p53 protein levels and activity could have a big impact upon tumorigenesis in both mice and humans. Loss of one allele of p53 gene is sufficient to promote tumorigenesis in mice. The Li-Fraumeni syndrome patients, who are heterozygous for the p53 gene, display a 50% cancer incidence by the age of 30 (Levine et al., 2006). The naturally existing single nucleotide polymorphisms in p53 (codon 72) and its key negative regulator, Mdm2 (SNP309), which can slightly alter p53 activity, have significant impacts upon tumorigenesis in humans (Bond et al., 2004; Hu et al., 2007a; Murphy, 2006). Therefore, p53 protein levels and activity are under a tight and complex regulation in cells in order to maintain its proper functions. Our findings that miR-504 can negatively regulate p53 functions in inducing apoptosis and cell cycle arrest especially in response to stress strongly suggest that miR-504 may contribute to tumorigenesis. Indeed, our results show that overexpression of miR-504 promotes tumorigenicity of cells in vivo. The regulation of miR-504 expression in cells and the biological significance of the regulation of p53 by miR-504 remain unclear. Interestingly, frequent DNA amplification of the locus where miR-504 is located (Xq27.1) has been reported in several different human tumors, including T cell non-Hodgkin's lymphomas and gastric carcinomas (Fukuda et al., 2000; Lilljebjorn et al., 2007; Nilsson et al., 2008; Renedo et al., 2001), suggesting that miR-504 might be overexpressed in some types of human tumors, which could contribute to tumorigenesis through its direct negative regulation of the p53. Considering that p53 function is frequently downregulated in a number of cancers without p53 mutations (Levine et al., 2006; Vogelstein et al., 2000), it would be interesting to investigate whether the miR-504 is overexpressed in these cancers, especially T cell non-Hodgkin's lymphomas and gastric carcinomas with DNA amplification in the region where miR-504 is located. Further studies are required to address the role of miR-504 in human cancers.
In summary, we have identified miR-504 as a direct negative regulator of p53. Our study extends the current understanding of miRNAs in the p53 pathway by revealing a role of miR-504 functioning upstream of p53, which is a mechanism for cells to monitor p53 protein levels and regulate p53 function. These findings that miRNAs not only mediate the downstream effect of p53 but also are involved in the upstream regulation of p53 further highlight an important role of miRNAs in human tumors.
Experimental Procedures
Cells and Cell Transfection
Human H460, MCF-7, U2OS, A498 cells were obtained from American Type Culture Collection (Manassas, VA). HCT116 p53+/+ and HCT116 p53−/− cells were generous gifts from Dr. B. Vogelstein at John Hopkins University. miRNA oligonucleotides, anti-miRNA oligonucleotides (Ambion, TX) and p53 siRNA oligonucleotides (Dharmacon, CO) were transfected into cells using Oligofectamine 2000 (Invitrogen). To establish stable cell lines with ectopic miR-504 expression, pEP-miR-504 expression vectors containing miR-504 precursor and its flanking 200 bp sequences (Cell Biolabs, CA) were transfected into HCT116 p53+/+ or p53-/- cells, and selected with puromycin (0.5 μg/ml) for 3-4 weeks. The expression of mature miR-504 was confirmed by real-time PCR assays. Over 10 individual clones of cells with confirmed ectopic mature miR-504 expression were pooled together for further experiments. Control cells were transfected with control empty vectors (pEP-mir-null vectors; Cell Biolabs) and selected by puromycin.
Construction of Luciferase Reporter Vectors
The human p53 3′-UTR sequences (565 bp, 557-1122 nt from the start of 3′-UTR) containing the two putative miR-504 binding sites were amplified by PCR using the following two primers: Forward primer 5′-ACTAGTGTTGGGCAGCTGGTTAGGTAGAGG-3′ and reverse primer 5′-AAGCTTAGGGGAAGGGTGGGGTG AAAAT-3′. These two primers contain HindIII and SpeI recognition sites at the 5′ end of the primers, respectively. Three additional mutant 3′-UTR sequence with deletion of either one of the two binding sites or deletion of both binding sites were synthesized by PCR according to the published method (Higuchi et al., 1988). For deletion of the first putative binding site (735-742 bp from the start of the 3′-UTR), the following pair of primers were employed in addition to the pair of primers used for amplification of wild type 3′-UTR: 1) 5′- GAAAAAAGAAATAGCATAAAACAAGTCT TGGTGG-3′; 2) 5′-GTTTTATGCT ATTTCTTTTTTCTTTTTTTTTTTTTTTTTTC-3′. For deletion of the second putative binding site (1064-1070 bp from the start of the 3′-UTR), the following pair of primers were employed in addition to the pair of primers used for amplification of wild type 3′-UTR: 1) 5′-GTAAAAGATGT TCCAGCTGGACGTGGTGGCTC 3′; 2) 5′-GTCCAGCTGGAACATC TTTTACATT CTGCAAGCAC-3′. These four PCR products were digested with HindIII and SpeI, and cloned into the 3′ end of the pMIR-luciferase reporter vector (Ambion) at HindIII and SpeI sites. The sequences of inserted fragments were confirmed by sequencing.
Luciferase Reporter Assay
Assays were performed as previously described (Feng et al., 2007a; Hu et al., 2007b). In brief, firefly pMIR-luciferase reporter vectors (100 ng) were transfected into cells in 6-well plates together with miR-504 oligonucleotides (100 nM) or scrambled miRNA oligonucleotides as a negative control by using Lipofectamine 2000. pRL-SV40 vectors (5 ng) which express Renilla luciferase (Promega) were co-transfected to normalize the transfection efficiency. For anti-miR-504 oligonucleotides, cells were transfected with anti-miR-504 (100 nM) for 6 h by using Oligofectamine before being transfected with miR-504 oligonucleotides together with luciferase reporter vectors. Luciferase activities were measured at 24 h after transfection by using the Dual-luciferase assay kit (Promega). Firefly luciferase activities were normalized to Renilla luciferase activities.
Western-blot Assay
Standard Western-blot assays were used to analyze protein expression. Antibody against MDM2, 2A10, was synthesized as previously described (Chen et al., 1996). Antibodies against p53 (FL393) and p21 (Ab-6) were purchased from Santa Cruz Biotechonology (Santa Cruz, CA), and Oncogene Research Products (Cambridge, MA), respectively. Band intensity on western blots was quantified by digitalization of the X-ray film and analyzed with Scion Image software (Scion Corporation, Maryland) and normalized to Actin.
Real-time PCR Analysis
A mirVana miRNA Isolation Kit (Ambion, TX) was employed to purify total RNA from cells. For miRNA detection, 10 ng total RNA was reverse transcribed into cDNA using specific primers designed for miRNA analysis (Applied Biosystem, CA). The miRNA expression levels were determined by real-time PCR using Taqman PCR master mixture (no AmpErase UNG) and specific primers designed for detection of mature miRNAs (Applied Biosystems). The expression of miRNA was normalized with the expression of U6 snRNA. For mRNA expression of p53 and p53 target genes, 1 μg cDNA was prepared with random primers using TaqMan reverse transcription kit (Applied Biosystems). Gene expression levels were determined by real-time PCR using Taqman PCR master mixture and primers. The expression of genes was normalized to Actin gene.
Cellular Apoptosis Analysis
Assays were performed as previously described (Feng et al., 2007b). In brief, cells were treated with Etoposide (20 μM), and at different time points (24-36 h) after treatment, both floating and attached cells were collected for analysis. Cells were washed with PBS, and resuspended in PBS containing 25 μg/ml Annexin V and 50 μg/ml propidium iodide before being analyzed in a flow cytometry (Beckman coulter).
Cell Cycle Analysis
Cells were trypsinized and fixed in 70% ethanol at −20 °C. Fixed cells were resuspended in PBS containing 25 μg/mL propidium iodide, 0.1% Triton, and 10 μg/mL RNase, and incubated for 30 min in the dark before being analyzed in a flow cytometry (Beckman coulter).
In vivo Tumorigenicity Assay
The 6-week-old female BALB/c nu/nu athymic nude mice (Taconic, Albany, NY) were employed. HCT116 cells with stable miR-504 expression (3×106 in 0.2 ml PBS) were injected subcutaneously (s.c.) into nude mice on the right side and their control cells with empty vectors were s.c. injected on the left side (10 mice/each group). After injection, mice were examined and tumor volumes were measured three times per week for tumor formation for 3 weeks. Tumor volume=1/2 (length × width2). Tumor samples were processed for routine histology examination. The expression of mature miR-504 in tumor samples was confirmed by real-time PCR analysis.
Supplementary Material
Acknowledgments
The work was supported by grants from National Institutes of Health (1R01CA143204-01 to Z.F.) and New Jersey Commission on Cancer Research (Z.F.). C.Z. was supported by New Jersey Commission on Cancer Research postdoctoral fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bennett WP, Hussain SP, Vahakangas KH, Khan MA, Shields PG, Harris CC. Molecular epidemiology of human cancer risk: gene-environment interactions and p53 mutation spectrum in human lung cancer. J Pathol. 1999;187:8–18. doi: 10.1002/(SICI)1096-9896(199901)187:1<8::AID-PATH232>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005;37:766–770. doi: 10.1038/ng1590. [DOI] [PubMed] [Google Scholar]
- Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–1307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, Bargonetti J, Bartel F, Taubert H, Wuerl P, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- Caldas C, Brenton JD. Sizing up miRNAs as cancer genes. Nat Med. 2005;11:712–714. doi: 10.1038/nm0705-712. [DOI] [PubMed] [Google Scholar]
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wu X, Lin J, Levine AJ. mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol Cell Biol. 1996;16:2445–2452. doi: 10.1128/mcb.16.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA, Jr, Sjoblom T, Barad O, Bentwich Z, Szafranska AE, Labourier E, et al. The colorectal microRNAome. Proc Natl Acad Sci U S A. 2006;103:3687–3692. doi: 10.1073/pnas.0511155103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995;82:675–684. doi: 10.1016/0092-8674(95)90039-x. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007a;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A. 2007b;104:16633–16638. doi: 10.1073/pnas.0708043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Kurihara N, Imoto I, Yasui K, Yoshida M, Yanagihara K, Park JG, Nakamura Y, Inazawa J. CD44 is a potential target of amplification within the 11p13 amplicon detected in gastric cancer cell lines. Genes Chromosomes Cancer. 2000;29:315–324. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1047>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–144. doi: 10.1093/nar/gkj112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network--another piece in the tumour-suppression puzzle. Nat Rev Cancer. 2007;7:819–822. doi: 10.1038/nrc2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ, Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005;435:828–833. doi: 10.1038/nature03552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R, Krummel B, Saiki RK. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Feng Z, Ma L, Wagner J, Rice JJ, Stolovitzky G, Levine AJ. A single nucleotide polymorphism in the MDM2 gene disrupts the oscillation of p53 and MDM2 levels in cells. Cancer Res. 2007a;67:2757–2765. doi: 10.1158/0008-5472.CAN-06-2656. [DOI] [PubMed] [Google Scholar]
- Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007b;450:721–724. doi: 10.1038/nature05993. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene. 2006;25:6188–6196. doi: 10.1038/sj.onc.1209913. [DOI] [PubMed] [Google Scholar]
- Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Le MT, Teh C, Shyh-Chang N, Xie H, Zhou B, Korzh V, Lodish HF, Lim B. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Lilljebjorn H, Heidenblad M, Nilsson B, Lassen C, Horvat A, Heldrup J, Behrendtz M, Johansson B, Andersson A, Fioretos T. Combined high-resolution array-based comparative genomic hybridization and expression profiling of ETV6/RUNX1-positive acute lymphoblastic leukemias reveal a high incidence of cryptic Xq duplications and identify several putative target genes within the commonly gained region. Leukemia. 2007;21:2137–2144. doi: 10.1038/sj.leu.2404879. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- Michael MZ, SM OC, van Holst Pellekaan NG, Young GP, James RJ. Reduced accumulation of specific microRNAs in colorectal neoplasia. Mol Cancer Res. 2003;1:882–891. [PubMed] [Google Scholar]
- Moran DM, Gawlak G, Jayaprakash MS, Mayar S, Maki CG. Geldanamycin promotes premature mitotic entry and micronucleation in irradiated p53/p21 deficient colon carcinoma cells. Oncogene. 2008 doi: 10.1038/onc.2008.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy ME. Polymorphic variants in the p53 pathway. Cell Death Differ. 2006;13:916–920. doi: 10.1038/sj.cdd.4401907. [DOI] [PubMed] [Google Scholar]
- Nilsson B, Johansson M, Heyden A, Nelander S, Fioretos T. An improved method for detecting and delineating genomic regions with altered gene expression in cancer. Genome Biol. 2008;9:R13. doi: 10.1186/gb-2008-9-1-r13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–126. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Plasterk RH. Micro RNAs in animal development. Cell. 2006;124:877–881. doi: 10.1016/j.cell.2006.02.030. [DOI] [PubMed] [Google Scholar]
- Raver-Shapira N, Marciano E, Meiri E, Spector Y, Rosenfeld N, Moskovits N, Bentwich Z, Oren M. Transcriptional activation of miR-34a contributes to p53-mediated apoptosis. Mol Cell. 2007;26:731–743. doi: 10.1016/j.molcel.2007.05.017. [DOI] [PubMed] [Google Scholar]
- Renedo M, Martinez-Delgado B, Arranz E, Garcia M, Urioste M, Martinez-Ramirez A, Rivas C, Cigudosa JC, Benitez I. Chromosomal changes pattern and gene amplification in T cell non-Hodgkin's lymphomas. Leukemia. 2001;15:1627–1632. doi: 10.1038/sj.leu.2402248. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Yoon CH, Lee ES, Lim DS, Bae YS. PKR, a p53 target gene, plays a crucial role in the tumor-suppressor function of p53. Proc Natl Acad Sci U S A. 2009;106:7852–7857. doi: 10.1073/pnas.0812148106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.