

Abstract
In the nervous system, neurons form in different regions, then they migrate and occupy specific positions. We have previously shown that RP2/sib, a well-studied neuronal pair in the Drosophila ventral nerve cord (VNC), has a complex migration route. Here, we show that the Hem protein, via the WAVE complex, regulates migration of GMC-1 and its progeny RP2 neuron. In Hem or WAVE mutants, RP2 neuron either abnormally migrates, crossing the midline from one hemisegment to the contralateral hemisegment, or does not migrate at al and fail to send out its axon projection. We report that Hem regulates neuronal migration through stabilizing WAVE. Since Hem and WAVE normally form a complex, our data argues that in the absence of Hem, WAVE, which is presumably no longer in a complex, becomes susceptible to degradation. We also find that Abelson Tyrosine kinase affects RP2 migration in a similar manner as Hem and WAVE, and appears to operate via WAVE. However, while Abl negatively regulates the levels of WAVE, it regulates migration via regulating the activity of WAVE. Our results also show that during the degradation of WAVE, Hem function is opposite to that of and downstream of Abl.
Keywords: Drosophila, Hem, Abl, WAVE, Neuron, Migration
Introduction
Cell migration is one of the most fundamental phenomena in biology displayed in simple organisms like yeast, to complex organisms like humans. It is involved in many biological processes including chemotaxis, tissue repair, immune responses and development (Keller, 2005; Luster et al, 2005; Vicente-Manzanares et al, 2005). Improper cell migration has been observed in many pathological situations (Ridley et al, 2003). In the nervous system, neurons and their precursor cells are formed in different regions, but they migrate and occupy specific positions in the mature nervous system. Their position is thought to be crucial in establishing proper synaptic connectivity. Neurons can migrate a few cell-lengths to several thousand cell-lengths - often taking very complex routes - by responding the internal of external signals (Wong et al, 2002). Perturbations in neuronal migration are known to cause neurodevelopmental defects such as smooth brain disease and mental retardation (Ghashghaei et al, 2007).
Though widely used as a model system to study development of the CNS, very little is known about neuronal migration in Drosophila. In Drosophila, the ventral nerve cord consists of segmentally repeated units, divided by the midline. Thus, each hemisegment contains ~320 neurons and ~ 30 glia. Neurons are generated from neuroblasts (NBs). A NB repeatedly divides to self-renew and produces a series of ganglion-mother-cells (GMCs). Each GMC divides asymmetrically to produce two neurons (or glia or a neuron and a glia) of different identities. The only description of an active neuronal migration in the Drosophila VNC comes from the NB4-2®GMC-1®RP2/sib lineage (Bhat, 2007). This is a typical and well-studied lineage formed from NB4-2, one of ~30 NBs in a hemisegment. It generates its first GMC, GMC-1, which divides asymmetrically into an RP2 motor neuron and its sibling cell known as sib (reviewed in Gaziova and Bhat, 2007). Our recent work on the migration has shown that GMC-1àRP2/sib undergo a complex 3-step migration and Wingless (Wg) signaling is necessary for the step 2 and step 3 migrations (Bhat, 2007). Recent work has shown that there is an active migration process going on in the optic lobe (Morante et al., 2011; Hasegawa et al., 2011). For example, medulla cortex cells in the optic lobe oerform two patterns of cell migrations to acquire their final position. First, neurons move to become arranged in columns below each neuroblast. Then, These neurons migrate laterally, intermingling with each other to reach their retinotopic position in the adult optic lobe. That these migratiosn are an active process is indicated by the involvement of eyeless, a Pax6 transcription factor (Morante et al., 2011). However, much remains to be elucidated to understand the underlying mechanisms responsible for complex migration patterns that occur during neurogenesis.
Polymerization of actin filaments (F-actin) to form filopodia and/or lamellipodia and depolymerization in the leading edge plays key roles in the regulation of cell migration (Raftopoulou and Hall, 2004). The Arp2/3 complex induces polymerization by promoting nucleation and by binding to the side of F-actin to elongate branched filaments. The Arp2/3 complex is activated by WASp family members, WASp and WAVE (Machesky and Insall, 1998; Symons et al, 1996; Suetsugu et al, 1999). However, WASp and WAVE are activated independently (Takenawa and Miki, 2001). WASp is autoinhibited, and could be directly activated by Cdc42. WAVE is not autoinhibited but forms a WAVE-complex with four other molecules: Abelson interactor (Abi), Hem, Sra-1 and HSPC300 (Higgs and Pollard, 2000; Ismail et al, 2009; Kim et al, 2000; Martinez-Quiles et al, 2001; Padrick et al, 2008; Rohatgi et al, 2001). Once activated by another GTPase, Rac, WAVE promotes actin polymerization through Arp2/3 (Miki et al, 2000; Eden et al, 2002). The exact mechanism by which WAVE is activated remains controversial. There is evidence suggesting that Rac activity causes dissociation of the WAVE complex and release of the WAVE-HSPC300 and the activation of WAVE (Eden et al, 2002). Other studies, however suggests that after Rac activation, WAVE complex is translocated to the membrane, where Abi recruits Ableson-tyrosine- kinase (Abl) into this complex, which then activates the WAVE complex (Leng et al, 2005).
The Hem protein (or Kette/dhem2/Hem-2/Nap1/Nap125) belongs to a highly conserved family, from invertebrates to mammals (Baumgartner et al, 1995). In humans, there two genes of the Hem family, Hem1 (NCKAP1L) and Hem2 (NCKAP1). Both Hem1 and Hem2 in humans generate two isoforms each. In Drosophila, however, there is only one Hem gene, which is homologous to the human Hem2. All the members of Hem family are predominantly expressed in the nervous system, where as the Hem-1 gene in humans is predominantly expressed in the hematopoietic cells (Weiner et al, 2006). On the other hand, in C. elegans, the Hem-2 protein, known as GEX-3 (and Hem in Drosophila) is expressed everywhere, and has an essential function in the migration of epithelial cells in embryos (Soto et al. 2002). In Drosophila, Hem is maternally expressed during early embryogenesis but then becomes specifically expressed in the brain and the VNC. Hem has six transmembrane domains, however, in Drosophila S2 cells, most of Hem is in the cytosol and only very little is present in the membrane (Bogdan and Klambt, 2003).
Hem is involved in processes such as apoptosis, formation and maturation of neuromuscular junction, axon-pathfinding, neuronal-differentiation, myoblast fusion etc (Hummel et al, 2000; Nakao et al, 2008; Schenck et al, 2004; Schroter et al, 2004; Suzuki et al, 2000; Yokota et al, 2007). These studies suggest that Hem dynamically regulates polymerization of F-actin. Hem binds to the first Src homology 3 (SH3) domain of Nck/Dock, an adaptor molecule containing one SH2 domain and three SH3 domains and links several receptor tyrosine kinases to the cytoskeleton (Li et al, 2001). Thus, Hem may link extracellular signals to the cytoskeleton. However, Hem is also part of the WAVE-complex and it may regulate the activity of this complex to promote polymerization of F- actin. It remains controversial how Hem regulates WAVE. Eden et al (2002) argued that WAVE is inhibited in the WAVE complex by Hem and PIP121. Upon activation by Rac1 or Nck, the WAVE complex is dissociated, releasing an active WAVE-HSPC300 to mediate actin nucleation (Eden et al, 2002). This conclusion is supported by the findings by Bogdan et al (2002) that loss of Hem function leads to an excess of F-actin in the cytosol; by reducing the WAVE gene dose on the other hand, the Hem mutant phenotype can be suppressed (Bogdan and Klambt, 2003). However, Kunda et al (2003) showed that Hem protects WAVE from proteasome-mediated degradation in cultured Drosophila cells. Furthermore, Schenck et al (2004) showed that the axon guidance defects in WAVE, Sra-1 and Hem mutants are similar to each other and loss of Hem leads to a reduction in WAVE.
Here, we show that Hem regulates neuronal migration during development of the VNC in Drosophila embryos. The loss of Hem activity results in migration defects of RP2/sib cell pairs. Moreover, Hem regulates neuronal migration through WAVE; not by inhibiting the WAVE activity, but by preventing WAVE degradation. Interestingly, we also find that Abl affects RP2 migration in a similar way as Hem and WAVE, and operates via WAVE. In this case, however, while Abl negatively regulates the level of WAVE protein, the regulation of migration by Abl appears to be via regulating the activity of WAVE. Our results also show that the Hem function in regulating the levels of WAVE is downstream of Abl.
Materials and Methods
Fly strains, genetics
All flies and crosses are kept/done at 22 °C unless otherwise indicated. The following strains were used: HemJ4-48, HemC3-20, Df(3L)ED230, insc22, nb796, Abl2, Abl1 FRT(w[hs])2A, UAS-Abl, Df(3L)st-j7, SCARΔ37, Df(2L)BSC32, UAS-WAVE, w;p{SCARK13811,W+} FRT40A/CyO, sca-Gal4, P{GAL4∷VP16-nos.UTR}MVD2, Sra-1EY06562, Df(3R)Exel6174, AbiEy20423, Df(3R)Exel7359, HSPC300EP506, HSPC300G19021, Df(2R)Exel6080, Wsp1 and Wsp3. Various mutant combinations are generated by standard genetics. To exclude possible maternal modifier effects of balancers (Bhat et al, 2007; Gaziova and Bhat, 2009), homozygous mutant embryos were also tested by out-crossing the balancer-bearing mutants to wild type and back-crossing the non-balancer bearing mutant adults. Staging of embryos was as described by Wieschaus and Nusslein-Volhard (1986).
Generating mosaic animals
Germline clones for WAVE were generated as described by Zallen et al (2002), by heat shocking hsFLP; SCAR13811FRT40A/ovoD FRT40A early stage larvae at 37°C for 1-hour and another heat shock 24-hr later. Adult germline clones carrying females were crossed to SCARΔ37 males and embryos (WAVEmat, zyg) were collected for analysis. Germline clones for Abl were generate by heat shocking hsFLP; Abl1 FRT(w[hs])2A/ovoD FRT(w[hs])2A early stage larvae at 37°C for 1-hour and another heat shock 24-hr later. Adult germline clone females were crossed to Abl2 males and embryos (Ablmat,zyg) were collected for analysis.
Generating Transgenic animals
To determine the antimorphic properties of the truncated Hem protein in HemJ4-48allele (ΔHemJ4-48), we synthesized the Hem truncated gene corresponding to the coding fragment of the first 489 amino acids of Hem by PCR. We used the following primers that carried specific restriction sites and a stop codon: 5’- ATAAGAATGCGGCCGCTAAACTATTGCACGCCTCCCAATACG-3’ and 5’- GCTCTAGATTAGTCCAGGCGGAATGGTC-3’. The PCR product was digested with NotI and XbaI and subcloned into NotI/XbaI digested pUAST and the cloned truncated Hem gene was sequenced. Several independent transgenic lines were generated and used for analysis.
Immunohistochemistry
Standard immunostaining procedures were used. The following antibodies were used: Eve (rabbit, 1:2000 dilution), Eve (mouse, 1:5), Zfh1 (mouse, 1:400), 22C10 (mouse, 1:4), LacZ (rabbit, 1:3000 or mouse, 1:400), BP102 (mouse 1:10) Fas II (mouse; 1:5), Sim (rat, 1:200), Wave (guinea pig 1:100). For confocal microscopy, cy5 and FITC-conjugated secondary antibodies were used. For light microscopy, alkaline phosphatase or DAB-conjugated secondary antibodies were used.
Sequencing of the mutant alleles and confirmation of the mutant homozygous identity by single embryo PCR
Mutant embryos were individually selected using the GFP-balancer as well as by using the visible mutant phenotype (that the Hem mutant embryos exhibit) under microscope. DNA from these single embryos was isolated and subjected to PCR using primers for the Hem gene. The PCR products were sequenced in both strands.
Western-blotting experiments
For each genotype examined, twenty embryos were picked under GFP microscope and homogenized in 40μL of Lysis buffer (0.15 M NaCl, 0.02 M Tris pH 7.5, 0.001M EDTA, 0.001 M MgCl2, 1% Triton-X-100, PIC). After centrifugation, the supernatant (37.5μL) was collected and 12.5μL of 4X Laemmli sample buffer was added. Out of this 50μL, 10μL was subjected for SDS-PAGE and Western analysis. Primary antibodies used were against: WAVE (guinea pig 1:1500), Hem (rabbit 1:1000), and Tubulin (Abcam, rabbit, 1:2000). X-ray films from various Western blotting analyses were scanned and the densities of the signals were determined by using AlphaEaseFC (AlphaInnotech, V6.0). Anti-Tubulin was used as loading control and intensity of the band were normalized against Tubulin signal. Several independent experiments were done and the intensity values followed the same trends with narrow variations in all the experiments.
Phosphatase treatment of embryo extract
Twenty embryos were collected, dechorionated and homogenized in 37.5μL of the Lysis buffer (see above). After centrifugation, supernatant (~37.5μL) was collected and incubated with Lambda protein phosphatase (Lambda PP, 100U) in NEB phosphatase buffer (5μL of 10X) and MnCl2 (5μL of 10 mM solution) in a total final volume of 50μL for 30 min at 30°C. For the control, lambda PP was omitted. Following the incubation, 16.6μL of 4X Laemmli buffer) was added, boiled for 10 min and the samples were run on an 8% separating SDS gel for Western analysis.
Real-time Polymerase chain reaction (PCR)
Embryos were collected, dechorionated and ~100 embryos were selected under microscope and RNA was isolated using RNAqueous Kit (Ambion). Synthesis of cDNA was performed with 1 μg of total RNA in a 20μL reaction using the reagents in the Taqman Reverse Transcription Reagents Kit from ABI (#N8080234). Reaction conditions were as follows: 25°C, 10 minutes, 48°C, 30 minutes and 95 °C, 5 minutes. Primers for real-time PCR were designed and were done by the Molecular Genomic Core facility at UTMB. Real-time PCR (in triplicates) were done using 2μL of cDNA in a total volume of 25μL with SYBR green using the SYBR Green PCR Master Mix (ABI #4364344). RpL32 was used as endogenous control. All PCR assays were performed in the ABI Prism 7500 Sequence Detection System and the conditions are as follows: 50 °C, 2 minutes, 95 °C, 10 minutes, 40 cycles of 95 °C, 15 seconds and 60 °C, 1 minute. Primers used: WAVE (Forward: 5’ ACGAAGAAGCCGGATACGG 3’, Reverse: 5’ GAAGCTGCTCGTAGGTGCTACC 3’), Abl (Forward: GCAATTTCATCGACGACCTCA, Reverse: GACTCTGCTCCAGACTATCGCC). Various p-values were calculated using the student t-test.
Rescue HemJ4-48 phenotype with WAVE
To rescue the migration defects in HemJ4-48 mutants with UAS-WAVE, UAS-WAVE (on the third chromosome) was introduced to the HemJ4-48 background by recombination. This UAS-WAVE was induced using sca-Gal4 and the embryos were examined using Eve and WAVE antibodies for the rescue of the migration defect.
Results
Hem mutants show a duplication-and-loss of RP2 phenotype
In a previous deficiency screen, we discovered that while in wild type a single RP2 motoneuron is present in each hemisegment (Fig. 1A, arrow), a deficiency that uncovers the Hem gene had a unique phenotype: duplication of the RP2 neuron in one hemisegment and missing in the contralateral hemisegment (Fig. 1B, arrows). We mapped this phenotype to mutations in the Hem gene. We examined two of the embryonic lethal alleles, HemJ4-48 and HemC3-20 (Hummel et al., 2000) and both showed this RP2 defect (Fig. 1C and D). This phenotype was also observed in embryos transheterozygous for the two alleles or transheterozygous for the mutant alleles and the Hem-deficiency [(3L)ED230; Fig. 1E]. The penetrance of the phenotype varied in the two alleles and in the deficiency (Fig. 1G). In HemJ4-48 we found a maximum of 45% of the hemisegment per embryo showing the defect, the average was 13% (total number of hemisegments examined, n=648). In HemC3-20 the maximum was 20% (4/20 hemisegments) and the average was 8% (n=96). In the Hem-deficiency the maximum number of hemisegments affected was 20% (4/20) and the average was 9% (n=114). Based on these results, HemJ4-48 allele is the strongest and HemC3-20 and the Hem-deficiency are of the same strength. This suggests that HemC3-20 behaves as a null allele, whereas HemJ4-48 behaves as an antimorphic allele. Consistent with this possibility, the penetrance of the defect was lower in embryos that are transheterozygous for HemJ4-48/Hem deficiency (Fig.1G).
Figure 1. RP2 defect in Hem mutants.
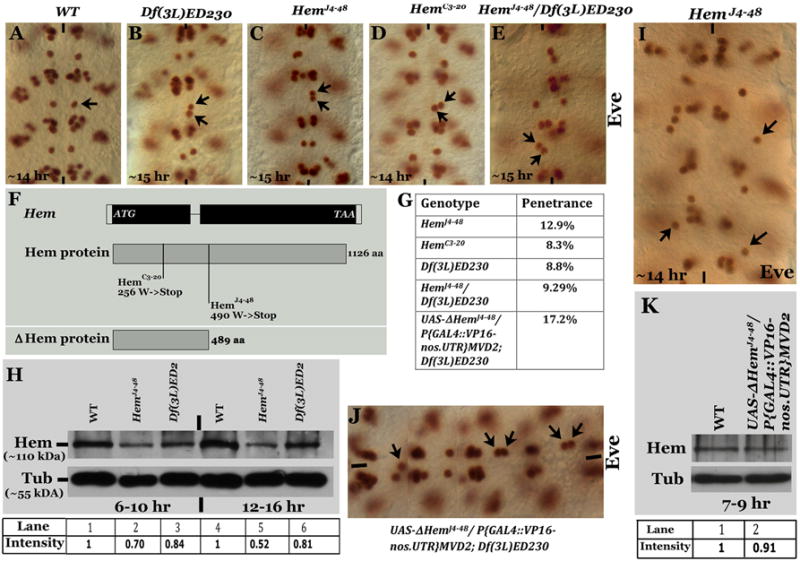
Embryos in panels A-E and I-J are stained with anti- Eve, anterior end is up in all Eve-stained panels except in panel J, where the anterior end is to the left. Midline is marked by vertical lines, RP2 is marked by arrow. Panel A: Wild type with one RP2 per hemisegment. Panels B-E: Different Hem mutant alleles. Panel F: Truncated Hem protein in the two Hem alleles and the truncated Hem produced in the transgenic UAS-DHem line. Panel G: Table with penetrance of the RP2 defect in different mutants. The number of embryos examined is shown within the bracket. Panel H: Western analysis for Hem in wild type (WT), HemJ4-48 and Hem-deficiency [Df(3L)ED2] embryos from 6-10 and 12-16-hrs of development. The intensity of the signals for Hem, normalized against the loading control Tubulin, is given below the Western. Panel I: HemJ4-48 embryo with RP2s located in the position of its parent GMC-1. Panel J: Hem-deficiency embryo with the truncated Hem expressed from a UAS-DHem transgene. This is a composite figure from three different photographs of the same embryo taken in different focal planes. Panel K: Western analysis of wild type embryos for wild type Hem expressing the truncated Hem from a UAS-DHem transgene. The intensity of the signals for Hem, normalized against the loading control Tubulin, is given below the Western.
We sequenced the Hem gene from these two alleles. In HemC3-20 there was a stop codon at amino-acid 256 in the Hem gene, while in HemJ4-48 the stop codon was at 490 (Fig. 1F). These molecular lesions are consistent with what has been previously reported (Hummel et al, 2000). Thus, the molecular lesion in the HemC3-20 allele is consistent with the possibility that this is a null allele, whereas the lesion in the HemJ4-48 allele is consistent with the possibility that it can produce a truncated protein that behaves as a dominant negative protein. The partial penetrance of the phenotype in the Hem-deficiency or Hem alleles is likely due to the maternal perdurance of the Hem protein in the embryo. Indeed, as shown in Fig. 1H, the wild type maternally deposited protein (~110 kDa) is still present in embryos that are 12-16 hrs old (the truncated Hem in the mutant is not recongnized by the antibody since it is raised against the portion of the protein beyond the truncation)., While the level of Hem protein in Hem deficiency is less than in wild type (Fig. 1H, l, compare lanes 1, 3, 4 and 6), in HemJ4-48, the level is much lower than in the deficiency or the wild type (Fig. 1H, compare lanes 1, 2, 4 and 5). This result suggests that the HemJ4-48 mutant (truncated) protein down-regulates the maternal wild type Hem protein. Perhaps the truncated Hem makes the maternal wild type Hem more degradable. Alternatively, this may be a maternal effect where the truncated Hem in the mutant suppresses maternal expression of the wild type protein. This is consistent with our previous findings that balancer chromosomes can have a maternal effect that influences the expressivity of a given mutant phenotype (Bhat et al, 2007; Gaziova and Bhat, 2009). In any case, the above result that the level of Hem in HemJ4-48 embryos is less than what it is in Hem deficiency embryos is consistent with the higher penetrance of the defect in HemJ4-48 embryos compared to Hem deficiency embryos.
We rarely observed HemJ4-48 embryos (~4% of HemJ4-48 mutant embryos, n=500) where the RP2 neurons are located in the position of a newly formed GMC-1 (Fig. 1I, see also Fig. 2M, N, P). This suggests that if the Hem function is eliminated at the GMC-stage, the GMC-1 and its progeny may not migrate at all. However, we could not generate germline mosaics for Hem because of its location on the chromosome, which is very close to the centromere. But, there is additional evidence in the proceeding sections that suggest that maternal and zygotic null for Hem will prevent the GMC-1 and its progeny from undertaking any migration.
Figure 2. The RP2 defect in Hem mutants is due to aberrant migration.
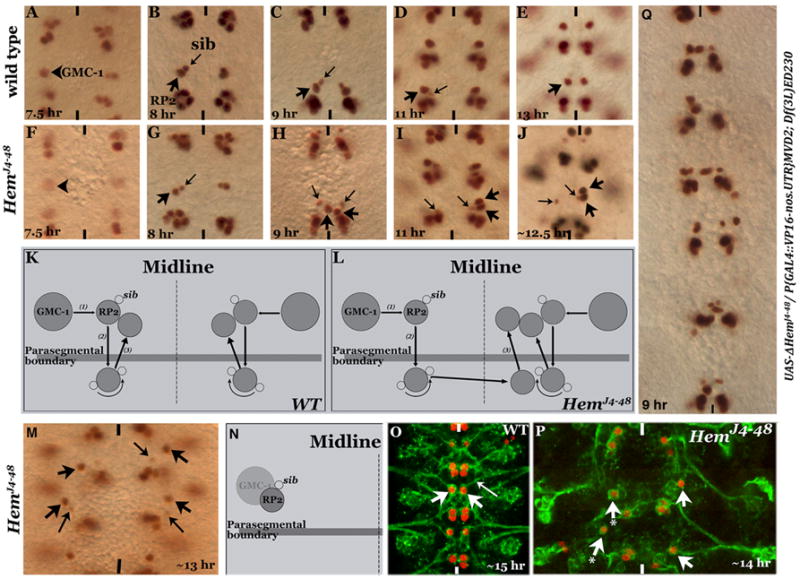
Embryos in panels A-J, M and Q are stained with anti-Eve, and in panels O and P are stained with anti-Eve and 22C10 antibodies. Anterior end is up, midline is marked by vertical lines. Panels A-E: Wild type embryos showing the 3-step RP2/sib migration. Panels F-J: Hem mutant embryos at the corresponding developmental stages. Panels K and L: Line drawings depicting RP2/sib migration in wild type and Hem mutants. Panel M: RP2 fails to migrate and is located at the position of its parent GMC-1. Panel N: Line drawing illustrating the migration defect. Panel O: Wild type embryo showing axon projection (22C10 positive) from an RP2, fasciculating with ISN. Panel P: Hem mutant embryo, RP2 fails to migrate from its location of formation; this RP2 has no discernible axonal projection (arrow-with-star). Panel Q: Hem-deficiency embryo with truncated Hem expressed from a UAS-DHem transgene.
The truncated Hem4-48 protein indeed behaves as an antimorphic protein
To determine if the truncated Hem in HemJ4-48 does behave as an antimorphic protein and down-regulates maternally deposited wild type Hem (see Fig. 1H), we generated transgenic lines carrying the HemJ4-48 truncated Hem (ΔHemJ4-48) under the UAS promoter. We expressed this transgene using a GAL4 driver (VP16-nos.UTR). The GAL4 driver was brought from the mother since there is maternal deposition of GAL4 with this driver. In these embryos, we rarely (<1%) found the same migration defect as HemJ4-48 embryos, the weak effect is likely due to the high maternal deposition of wild type Hem and the reduction in the levels of Hem by the transgene product was not sufficiently high to have an effect. Therefore, we induced the ΔHemJ4-48 transgene in the Hem-deficiency background and the penetrance of the defect was determined. The penetrance was now increased to ~17% (Fig. 1G and J, see also Fig. 2Q; deficiency alone had ~9% penetrance). Given the above results, we determined if the expression of the truncated protein down-regulates the levels of wild type Hem. Since we could not test this in Hem-deficiency background, we examined it in wild type embryos expressing the ΔHemJ4-48 transgene. While the effect was weak, we consistently observed a slight reduction in the levels of Hem in these embryos (Fig. 1K).
RP2 but not sib aberrantly migrates to the contralateral hemisegment in Hem mutants
We followed the development of the GMC-1->RP2/sib lineage in the mutant embryo. One possibility for the duplication-and missing-phenotype is that an RP2 aberrantly crosses the midline and resides next to the RP2 on the contralateral hemisegment. A detailed description of the migration pattern for the GMC-1, RP2 and sib cells has been previously published (Bhat, 2007) and summarized in Fig. 2K. Briefly, the GMC-1, once formed begins its migration perpendicular to and toward the midline, the Step 1 process of the migration. During Step 1, the GMC-1 divides to generate an RP2 and a sib, and both these cells continue their migration toward the midline. RP2 and sib cells stop Step 1 migration approximately two to three neuroectodermal cells away from the midline (Fig. 2B) and begin their Step 2 migration. In this step, RP2 and sib move in the posterior direction, parallel to the midline. By ~9.5 hours of development, both RP2 and sib cells have crossed the Wg expressing row 5 cells; these cells then stop their posterior migration and reside right posterior to the Wg stripe (Fig. 2C). By 11 hours of development, the sib rotates around the RP2 to reside closer to the midline (Fig. 2D). Soon after, the RP2 (but not the sib) migrates in the anterior direction (Step 3), parallel to the midline, crosses back the Wg row of cells and resides in the location where the RP2 and sib had initially started their posterior migration (Fig. 2E). The Step 3 migration of sib involves the rotational move around an RP2 but then it also follows the RP2, crosses the Wg-positive row of cells and resides slightly posterior and ventral to RP2 (see Bhat, 2007).
In the mutant, GMC-1 is formed in both hemisegments (Fig. 2F) and the GMC-1 normally divides into an RP2 and a sib; these cells also complete their step 1 and step 2 migrations (Fig. 2G and H). However, the RP2 from one of the hemisegments continues to migrate but towards and across the midline (Fig. 2H). By 11 hours of development, the aberrant migration across the midline to the contralateral hemisegment is complete (Fig. 2I). By 13 hours, such RP2 neurons complete step 3 migration together with the resident RP2 neuron and occupy the final position (Fig. 2J). The sib, however, stays in its own hemisegment (Fig. 2I and J) indicating that the aberrant migration is specific to the RP2 neuron and not to its sibling cell. This aberrant migration of RP2 in Hem mutants is summarized in Fig. 2L. Furthermore, when we induced the ΔHemJ4-48 transgene in Hem deficiency background using the VP16-GAL4 driver and examined younger stage embryos with Eve staining, we observed the strong migration defect (Fig. 2Q), also seen among HemJ4-48 mutant embryos (Fig. 1J).
As pointed out earlier, among the HemJ4-48 mutant embryos, in a small number of mutant embryos (4% of HemJ4-48 mutant embryos, N=500) RP2 neurons in about 50% of the hemisegments (n= 356 hemisegments) and sib cells in about 20% of the hemisegments fail to migrate. Instead, they stay in the same location as the parent GMC-1 (Fig. 2M and P). We examined such RP2s for their axon projections by staining with Mab22C10/MAPIB antibody. This antibody stains axon projection from neurons such as RP2. As shown in Figure 2O, in wild type an RP2 (arrow) sends its ipsilateral axon projection (long arrow). However, in HemJ4-48 mutants, the non-migrating RP2s had no axon projections although they had strong 22C10 staining in the cell body (Fig. 2P, arrow-with-a-star). These results suggest that proper migration is essential for sending out an axon projection. This result, however, does not exclude the possibility that the Hem-mediated pathway affects the ability of this neuron to generate a growth cone (axonogenesis) independent of the migration.
To obtain additional evidence for the mis-migration of RP2s, we examined embryos that are double mutant for Hem and inscuteable (insc) and Hem and numb. In insc mutants, the GMC-1 symmetrically divides into two RP2s (Fig. 3A; see also Buescher et al, 1998) where as in numb, it divides into two sibs (Fig. 3C; Buescher et al, 1998; Wai et al, 1999). Because we have never seen a neurogenic phenotype in Hem mutants and also that we have not seen two GMC-1s in Hem mutants, we reasoned that if the duplication in Hem mutants is due to migration defect, we should see hemisegments with four RP2 neurons in one hemisegment and missing in the contralateral hemisegment in Hem; insc mutants. Indeed, hemisegments with four RP2 neurons in one hemisegment and missing in the contralateral hemisegment were observed in HemJ4-48; insc double mutants (Fig. 3). But in HemJ4-48; numb double mutants, the numb-phenotype should be epistatic to Hem, with only two sibs per hemisegment, and it was (Fig. 3D). These results show that the abnormal migration in Hem mutants is indeed RP2-specific but not sib-specific.
Figure 3. The migration defect is specific to RP2 but not its sib.
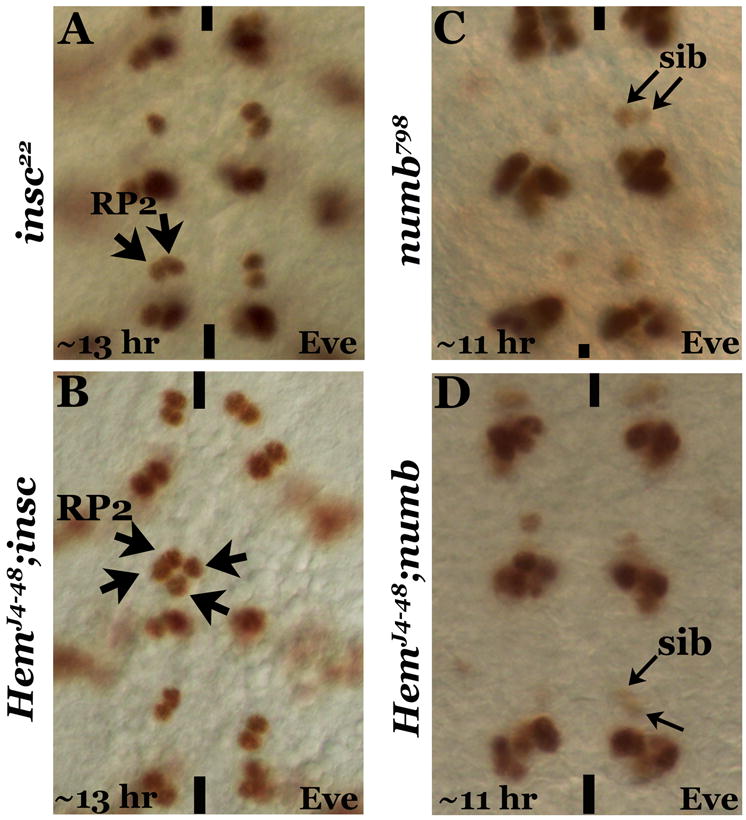
Embryos are stained with anti-Eve, anterior end is up, midline is marked by vertical lines. Panel A: insc mutant with the duplication of RP2. Panel B: HemJ4-48; insc double mutant showing four RP2s on one hemisegment and none on the contralateral hemisegment. Panel C: numb mutant showing the duplication of sib. Panel D: HemJ4-48; numb double mutant showing the numb-phenotype without any sib migration across the midline.
The aberrant RP2 migration in Hem mutants is not due to a midline defects, identity changes or commissural defects
We next examined if the abnormal migration of RP2 in Hem mutants is due to a midline defect by looking at the expression of Single-minded (Sim). Sim is expressed in all midline cells (Fig. 4A) and is involved in the specification of midline cells. When Hem embryos are double-stained with Sim and Eve antibodies, we observed normal expression of Sim in midline cells (Fig. 4B). We could even observe an RP2 right on the midline in the process of crossing (Fig. 4B), but not its sib.
Figure 4. The migration defect in Hem mutants is not related to midline defects, identity changes or commissural defects.
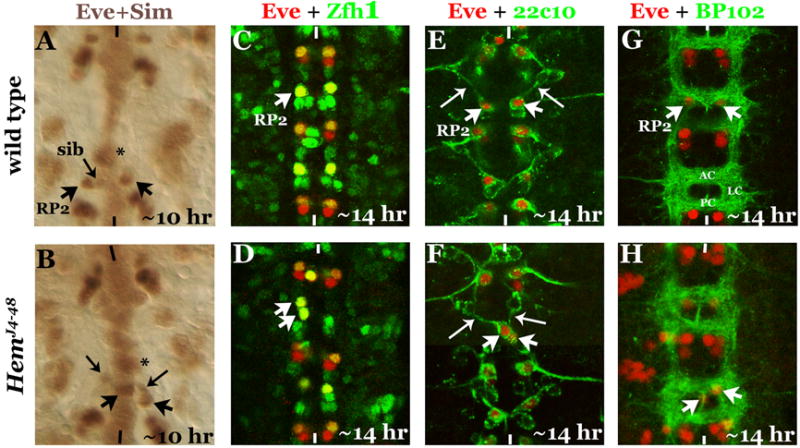
Anterior end is up, midline is marked by vertical lines. Panels A, B: Eve and Sim double-stained embryos. Star indicates a Sim-positive cell at the midline in the same focal plane as an RP2. The RP2 in the mutant is on the midline, its sib is in its normal location. Panels C, D: Embryos are double-stained with Eve and Zfh1. The mis-migrating RP2 expresses both Eve and Zfh1. Panels E, F: Embryos are double-stained with Eve and 22C10. Panel G, H: Embryos are double-stained with Eve and BP102. Although the commissural tracts in the mutant appear normal, the RP2 has crossed the midline in the mutant.
It was possible that the identity of the RP2 that crosses the midline has changed, which in turn induces it to migrate to the contralateral hemisegment. Therefore, we examined the mutant embryos with Zfh-1, an RP2-specific marker (Fig. 4C; a dividing GMC-1 and an occasional newly formed sib transiently expresses Zfh-1, Gaziova and Bhat, 2009). The abnormally migrated RP2 in Hem is still positive for Zfh-1 (Fig. 4D).
We next examined embryos double-stained with Eve and 22C10/MABIB. 22C10 stains the RP2 axonal projection that fasciculates with the Intersegmental Nerve bundle (ISN; Fig. 4E, long-arrow). In Hem mutants, the mis-migrated RP2 had a contra-ipsilateral axon projection, fasciculating with the ISN. This type of axon projection by an RP2 has never been documented before, in all those cases where the RP2 neurons are duplicated, both the neurons of the pair projected their axons ipsilaterally on its own hemisegment and never contralaterally. It is unlikely that the mis-migrated RP2 has changed its identity. An RP2 begins to project its axon 9-9.5 hours of age (data not shown). An RP2 in Hem mutants begins its abnormal migration ~9 hours of age. It appears likely that once a neuron begins its axonal projection, its growth cone is subjected to pathfinding cues and irrespective of the position of the cell body, the axon, as long as the cues are not affected, will project towards its usual target.
We further tested if the mis-migration of RP2 is tied to an abnormal commissural tract by visualizing the tracts with BP102. BP102 stains the anterior commissure (AC), the posterior commissure (PC) and the longitudinal connectives (LC)(Fig. 4G). An RP2 neuron normally resides in the armpit of AC (Fig. 4G). In Hem mutants, we observed RP2 mis- localized to the contralateral hemisegment even when the commissural tracts were more or less normal (Fig. 4H). These results are consistent with the conclusion that the mis-migration of RP2 is an active process.
WAVE/SCAR mediates the Hem-specific migration of RP2 neurons
The WAVE-complex, which activates Actin polymerization, has the following proteins: WAVE, Hem, Abi, Hspc300, Sra-1. In addition to the WAVE-complex, WASp is also part of the Hem-pathway. Therefore, we examined embryos mutant for some of these genes. WASp is located both in the membrane and in the cytoplasm and Hem is thought to activate WASp in the membrane (Bogdan and Klambt, 2003). However, WASp mutant embryos had no RP2 migration defects. While WASp is maternally deposited, migration defects were not observed in embryos that are mosaic for WASp (Ben-Yaacov et al, 2001). Thus, either its function is redundant or it is not required for migration. We also examined embryos mutant for Abi, Hspc300, Sra-1 using zygotic loss of function mutants. However, we did not find any migration defects in these mutants, which is likely due to maternal deposition of these gene products.
Abi is part of the WAVE-complex (Eden et al, 2002) and in vitro cell culture studies suggests Abi recruits Abl to the WAVE-complex, where Abl mediates WAVE-activation (Leng et al, 2005). When we examined Abi mutant embryos, no RP2 migration defect was observed. The absence of migration defect in Abi could be due to its maternal deposition. On the other hand, we found that Abl mutants have the same RP2 migration defect as Hem (Fig. 5B) with ~9% of the hemisegments showing the defect (n=600). This partial penetrance is likely due to the perdurance of the maternal Abl gene products. We generated Abl mosaic individuals lacking both the maternal and zygotic gene products. As shown in Figure 5C, in these embryos the RP2 neurons remained in the location where its parent GMC-1 forms, indicating that these neurons fail to migrate from the site of formation. This phenotype was nearly fully penetrant. As noted above, the same phenotype is also observed in HemJ4-48 embryos at a low frequency (see Fig. 1I and 2M, N, P).
Figure 5. Hem-like RP2 migration defects in embryos mutant for Abl and WAVE.
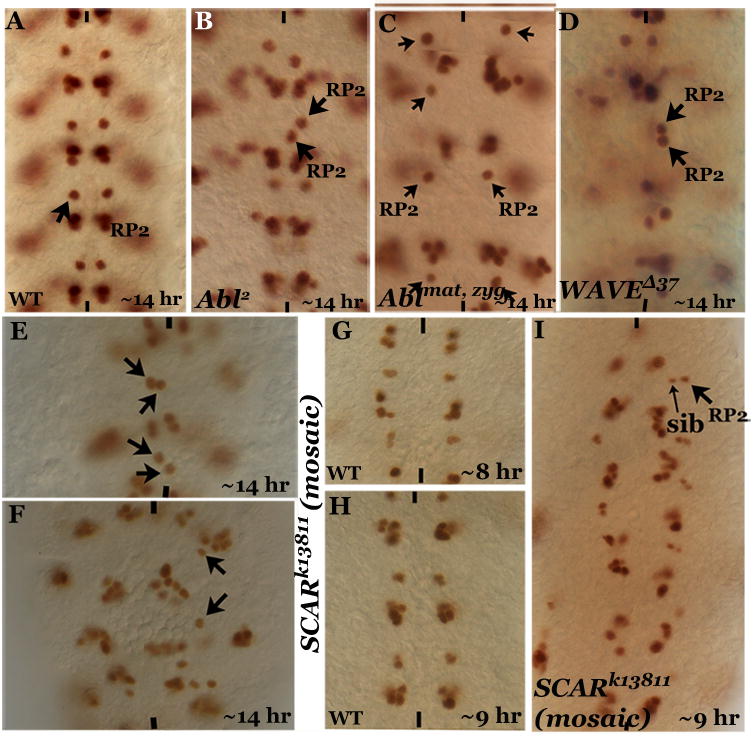
Embryos are stained with anti-Eve, anterior end is up, midline is marked by vertical lines. Panel A: Wild type with one RP2 per hemisegment. Panel B: Abl zygotic mutant showing the RP2 migration defect. Panel C: Abl maternal and zygotic mutant. Note that the RP2s fail to migrate from their position of formation and are placed farther apart. Panel D: WAVE zygotic mutant. Panels E, F: WAVE (also known as SCAR) zygotic and maternal mutants showing the abnormal midline crossing defect and failure of migration, respectively. Panels G, H: Wild type younger stage embryos. Panel I: WAVE zygotic and maternal mutant embryo, younger stage, showing the failure of migration of RP2 in an earlier stage.
We next examined if embryos mutant for WAVE had any migration defects. The RP2 midline-crossing defect was also observed in embryos mutant for WAVED37, which is an excision allele with the entire WAVE sequence excised downstream of the 5’ UTR of WAVE (Fig. 5D). A maximum of 15% of the hemisegments (n=600) were affected in WAVE mutant embryos. A similar penetrance was also observed in embryos that are homozygous for a deficiency that eliminates WAVE. WAVE is also maternally deposited (Zallen et al, 2002). Therefore, mosaic embryos for WAVE were generated and examined for the RP2 defects. A weaker WAVE allele, SCARk13811, was used since stronger alleles fail to yield mosaic embryos. In these mosaic embryos we found the same RP2 migration defect as in Hem mutants (Fig. 5E). We also found mosaic embryos where RP2s failed to migrate from their location of formation (Fig. 5F). This phenotype was similar to Abl maternal and zygotic mutant embryos (Fig. 5C) as well as a subset of HemJ4-48 embryos (Fig. 1I and 2N, Q). Consistent with this, we found younger stage mosaic embryos where the GMC-1 failed to perform stage 1 migration. Instead, it remained in its original NB4-2 location and divided into an RP2 and a sib (Fig. 5I). These results indicate that WAVE is necessary for the migration from the GMC-1-stage itself and in a WAVE null embryo there will be not be any migration.
WAVE protein level is down regulated in Hem mutant embryos
Since loss-of-function for WAVE had the same defect as Hem mutants, we determined if the level of WAVE is affected in Hem embryos by Western analysis. As shown in Figure 6A, in wild type a band of ~80-kDa was observed, which is higher than the previously reported molecular weight of ~68-kDa (Schenck et al, 2004) for WAVE. Therefore, we over- expressed WAVE from a UAS-WAVE transgene using the scabrous (sca)-GAL4 driver. As shown in Figure 6A (lane 2), the level of the 80-kDa band was increased by ~1.5 times the wild type in these embryos.; the level of WAVE was significantly reduced in WAVE-deficiency embryos compared to wild type (Fig. 6B, lanes 3, 7). Interestingly, with the reduced level, we could resolve the major band into 2 major ones and one minor band (Fig. 6B, lanes 3, 7; also 6C, lanes 1, 2) of the sizes ranging from 73-80 kDa range.
Figure 6. Regulation of WAVE by Hem.
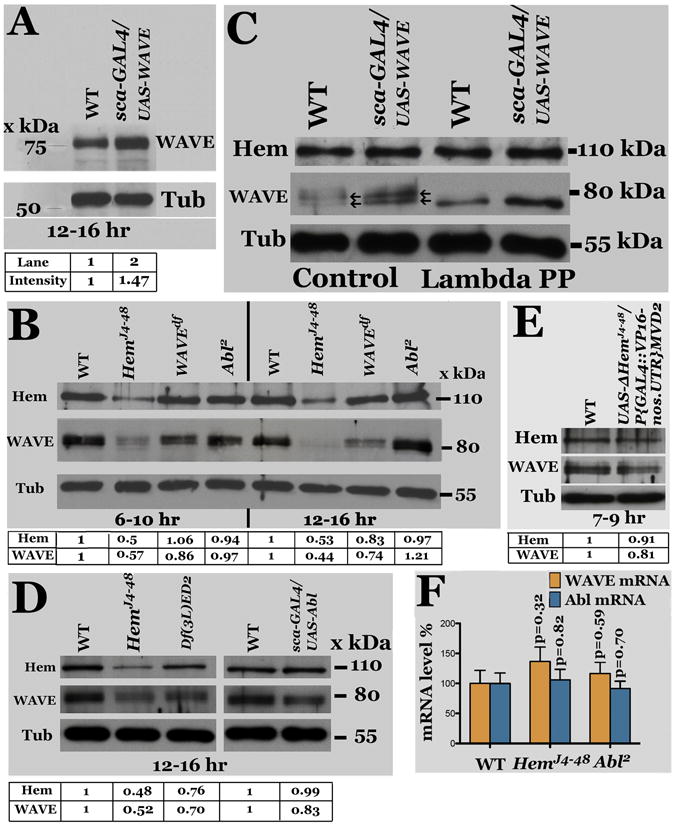
Panel A: Western analysis for WAVE using anti- WAVE antibody on extracts from 12-16 hr old wild type and sca-GAL4/UAS-WAVE embryos. Panel B: Western analysis for WAVE, Hem and Tubulin (Tub, as loading control) from 6-10 and 12-16-hr old wild type (WT), HemJ4-48, WAVE-deficiency, and Abl2 embryos. Panel C: The WAVE protein is phosphorylated. Western analysis of untreated and Lambda phosphatase-treated embryo extracts for Hem and WAVE. Panel D: Western analysis of extracts from Hem and Hem-deficiency embryos for Hem and WAVE. Panel E: Western analysis for WAVE and Hem in wild type embryos expressing the truncated UAS-DHem transgene. Panel F: qPCR for WAVE and Abl mRNA from 12-16-hr old Hem and Abl mutant embryos. The error bars indicate standard error, with corresponding p values (student t-test). The intensity of the signals for WAVE and Hem, normalized against the loading control Tubulin, is given below each Western.
It was possible that WAVE is phosphorylated and these three bands represent different levels of WAVE-phosphorylation. A previous paper mentions that WAVE2 is phosphorylated, moreover, this work was done using tissue culture cells (Lebensohn and Kirschner, 2009) and thus there is no in vivo evidence to show that WAVE is indeed phosphorylated in any organism. Therefore, we treated protein extracts from wild type as well as from embryos over-expressing WAVE with Lambda-phage phosphatase. As shown in Figure 6C, all the three bands in control-treated samples (lanes 1, 2) collapsed to one band of ~73 kDa size with the treatment (lanes 3, 4). This is closer to the predicted size of WAVE from the primary sequence.
We next examined the levels of WAVE in Hem mutant embryos. While Hem was unaffected with the over-expression of WAVE (Fig. 6C, lanes 2, 4) and unaffected or marginally affected in loss of function for WAVE (Fig. 6B, lanes 3, 7), as shown in Figure 6B (lanes 2, 6) and Figure 6D (lane 2), the level of WAVE was greatly reduced in HemJ4-48 embryos. The level was also reduced in Hem-deficiency embryos (Fig. 6D, lane 3), although the reduction was slightly lower compared to HemJ4-48. Furthermore, expression of the ΔHemJ4-48 truncated transgene in wild type background down-regulated WAVE (Fig. 6E, lane 2) compared to wild type (Fig. 6E, lane 1). These results indicate that Hem is necessary for maintaining a high level of WAVE, thus, the migration defect in Hem mutants could be due to the reduction in the levels of the WAVE protein.
We further examined if the reduction in the levels of WAVE protein in Hem mutant embryos is at the protein level or at the mRNA level by determining the mRNA level of WAVE in Hem embryos by quantitative PCR (qPCR). As shown in Figure 6F, a marginal increase in the levels of WAVE-mRNA was observed in Hem embryos (which is unlikely to be significant based on student t-test analysis). This result suggests that the reduction in the levels of WAVE in Hem is likely due to its enhanced degradation. This decrease seems unlikely due to a reduced translational rate given that the increase in mRNA is marginal, although we cannot entirely rule out this possibility Thus, it appears that Hem protects WAVE from degradation, and in the absence/reduction of Hem, more of WAVE is degraded.
In tissue culture experiments, Abl appears to mediate WAVE-activation in WAVE- complex (Leng et al, 2005). Our phenotypic analysis of Abl reveals that Abl have the same migration defects as WAVE and Hem mutants. We determined if the levels of WAVE and Hem are altered in Abl embryos. As shown in Figure 6B (lanes 4, 8), the levels of Hem was about the same as in wild type, however the level of WAVE was higher in 12-16 hr old Abl embryos (Fig. 6B, lane 8). By qPCR, there was no significant increase in the WAVE mRNA level in Abl mutant embryos (Fig. 6F). These results suggest that in Abl mutants, either that the WAVE is inactive or that Abl regulates RP2 migration independent of WAVE (see below).
Since Abl is maternally deposited to developing embryos, it was possible that the effect of zygotic loss-of-function for Abl on the levels of WAVE is not pronounced at the time of RP2 migration. Therefore, we generated Abl maternal and zygotic loss-of-function mutant embryos. We examined the level of WAVE in 6-10 hr old embryos (i.e., during the active migration of RP2 neurons)(Fig. 7A and B). Since these mutant embryos were smaller and become somewhat deformed by 12-16 hours of development (Fig. 7C; this difference in size is not apparent at 6-10-hr age), we also doubled the number for examining the level of WAVE in Western analysis (this small size embryo phenotype indicates that the Abl maternal and zygotic loss is significant). As shown in Figure 7A (lanes 3, 4; 6-10-hr old embryos) and Figure 7B (lanes 2, 3; 12-16-hr old embryos) we found that loss of both maternal and zygotic Abl function causes an increase in the levels of WAVE protein compared to wild type (lane 1) or Abl zygotic mutant embryos (lane 2).
Figure 7. Hem and Abl independently regulate WAVE and the migration of RP2.
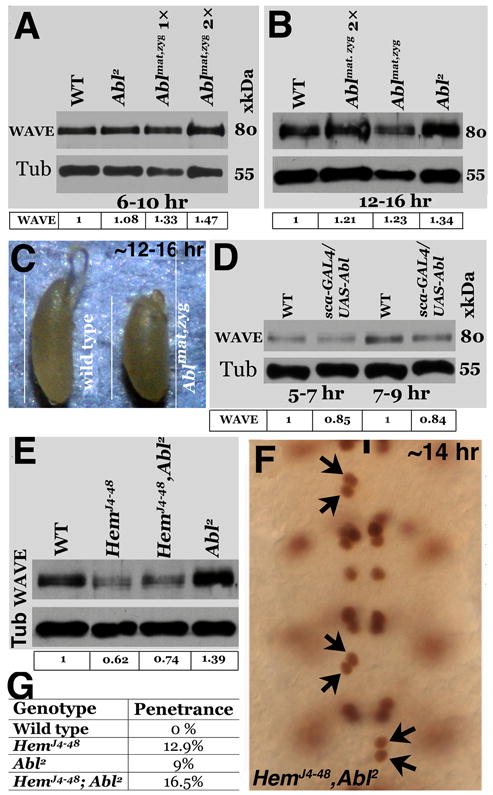
Panel A: Western analysis of embryo extracts for WAVE. Panels A and B: Western analysis of extracts for WAVE from Abl zygotic and maternal and zygotic mutant embryos from 6-10- hrs and 12-16-hrs of age. Panel C: Abl maternal and zygotic mutant embryos are of small size compared to wild type. Panel D: Gain-of-function for Abl (UAS-Abl X sca-GAL4) only marginally reduces the levels of WAVE between 5-9-hrs of age (during the time of RP2 migration). Panel E: The levels of WAVE in wild type (lane 1), Hem single mutant (lane 2), Hem, Abl (zygotic) double mutant (lane 3) and Abl zygotic mutant (lane 4). Panel F: Hem, Abl zygotic double mutant embryo stained with Eve. Panel G: Table showing the penetrance of the RP2 mis-migration defect. The intensity of the signals for WAVE, normalized against the loading control Tubulin, is given below each Western.
We examined this further by determining if over-expression of Abl from a UAS-Abl transgene using the sca-GAL4 driver down regulates WAVE. As pointed out above, in 12- 16-hr old embryos, gain-of-function for Abl down-regulates WAVE almost by half (Fig. 6D, lane 5), indicating Abl involvement in the regulation of WAVE protein levels. We also determined if the levels are down-regulated at earlier developmental time points, during the migration of RP2. As shown in Figure 7D (lanes 2, 4), the levels of WAVE are reduced in 5-9-hr old embryos compared to wild type (lanes 1, 3). Although Abl appears to down regulate WAVE, the effect on WAVE during the time of RP2 migration may not be significant enough to have an impact. This is also consistent with our finding that over-expression of Abl during the migration of RP2 has no effect on its migration (data not shown). However, these data show that Abl does mediate, directly or indirectly, the down regulation of the WAVE protein during development.
Given the opposing effects of Hem and Abl on WAVE, we determined the epistatic relationship between Hem and Abl with respect to WAVE protein levels and the migration of RP2. We recombined HemJ4-48 mutation with Abl2 and examined both the protein levels and migration defect in HemJ4-48, Abl2 double-mutant embryos. As shown in Figure 7E, Hem was epistatic to Abl in terms of WAVE levels as the WAVE level in the double mutant was similar to Hem single mutant (compare lane 3 with 2). Since our results suggest that WAVE may not be fully active in Abl mutants, and that the level of WAVE is down-regulated in Hem mutant embryos, the penetrance of the RP2 migration defect is expected to be enhanced in the double-mutant. Indeed, the frequency of the migration defect was enhanced in these double mutants (Fig. 7F and G). Given that there is maternal wild type Hem and Abl in these embryos, the enhancement was modest.
Expression of WAVE in Hem mutant embryos rescues the RP2 migration defect
Since WAVE is down-regulated in Hem embryos, we determined if the migration defects are due to this reduced levels of WAVE. We recombined a UAS-WAVE transgene into HemJ4-48 chromosome and induced this transgene with the sca-GAL4 driver. While the level of WAVE is barely detectable in HemJ4-48 embryos (Fig. 8B) compared to wild type (Fig. 8A), as shown in Figure 8C, the expression of UAS-WAVE in Hem embryos induced a high level of WAVE and completely rescued the RP2 migration defect (N, the number of mutant embryos examined, 30). This result shows that the migration defect in Hem mutant embryos is caused by the down-regulation of WAVE. Since Hem and WAVE normally form a complex, in the absence of Hem, WAVE, which is perhaps no longer in a complex, appears to be degraded.
Figure 8. Hem mediates RP2 migration via WAVE.
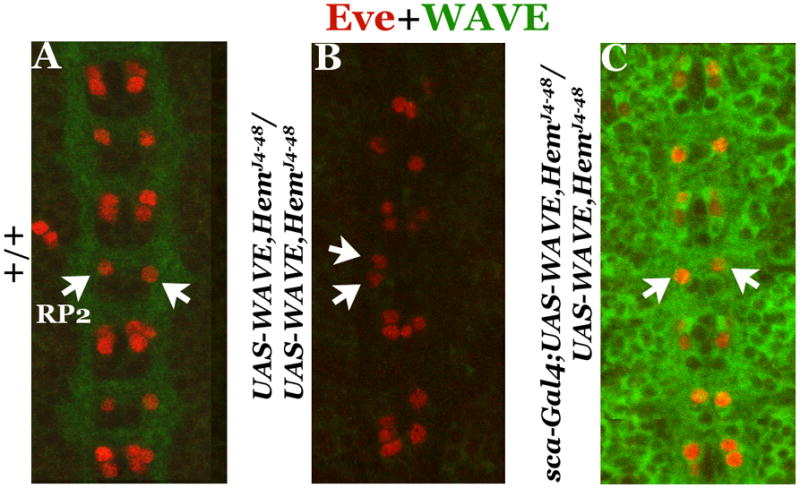
Embryos are double-stained with anti- Eve and anti-WAVE, anterior end is up, midline is indicated by vertical lines. Panel A: wild type. Panel B: Hem mutant that also carries the UAS-WAVE transgene (but not induced) on the same chromosome. Panel C: Hem mutant where the UAS-WAVE transgene is induced from the sca-GAL4 driver. Note that the level of WAVE is high and that the RP2 midline crossing migration defect is fully rescued.
Discussion
In this paper, we have shown that mutations in Hem, WAVE and Abl affect the migration of RP2 neurons in the developing embryonic VNC. Depending upon the strengths of the mutations, the cell either fails to migrate from its location of formation or mis-migrates across the midline into contralateral hemisegment. This defect is specific to RP2 but not its sibling cell. Indicating that proper migration is necessary for sending out its axonal projection, no axonal projection can be seen in those hemisegments where the RP2 had failed to migrate at all. Moreover, loss of migration in Hem mutants is due to a reduction in the levels of WAVE protein, and that Hem protects WAVE protein from degradation. While the loss-of-function for Abl gives the same migration defect as Hem or WAVE, and Abl mediates RP2 migration via WAVE, the effect of Abl on WAVE appears to be at regulating WAVE activity independent of Hem rather than the levels of WAVE. Our results also show that during the degradation of WAVE, Hem function is opposite to that of Abl and is downstream of Abl. These results provide novel insight into not only the problem of neuronal migration but also the functions of Hem and Abl in regulating WAVE during development.
Hem mediated regulation of neuronal migration
Several studies have suggested that Hem dynamically regulates polymerization of F- actin. Hem can play a crucial role in linking extracellular signals to the cytoskeleton. On the other hand, Hem is also part of the WAVE complex and it may regulate the activity of the WAVE complex to promote polymerization of F-actin. Our result that the migration defect in Hem mutants can be completely rescued by expression of WAVE from a transgene (Fig. 8) indicates that Hem regulates neuronal migration via WAVE.
How Hem regulates WAVE is controversial. Eden et al (2002) argued that Hem (together with PIP212) inhibits WAVE in the WAVE complex. Upon activation by Rac1 or Nck, the WAVE complex dissociates, releasing an active WAVE-HSPC300 to mediate actin nucleation (Eden et al, 2002). This conclusion was also supported by the findings by Bogdan et al (2002) that loss-of-function for Hem leads to an excess of F-actin in the cytosol. Moreover, a reduction in the WAVE gene dosage suppressed axon guidance defects in Hem mutant embryos (Bogdan and Klambt, 2003). But, in vitro studies using Drosophila tissue culture cells argue that Hem protects WAVE from proteasome-mediated degradation (Kunda et al, 2003). Our in vivo results are consistent with these studies and show that WAVE is protected by Hem and the above alternate model may be incorrect.
Hem regulates RP2 migration via regulating the levels of WAVE
The WAVE protein was first identified in Dictyostelium discoideum as a suppressor of mutations in the cAMP receptor (SCAR) (Bear et al, 1998) but it is present in flies to humans. All WAVEs contain a N-terminal WHD/SHD (WAVE/SCAR homologue domain), a central proline-rich region and a C-terminal VCA domain. WAVE protein regulates actin polymerization by mediating the signal of Rac to Arp2/3 in lamellipodia. It is involved in forming branched and cross-linked actin networks. Unlike WASp proteins, which are intrinsically inactive by autoinhibition and activated by directly binding to Cdc42, PIP2 etc (Kim et al, 2000), WAVE appears to be intrinsically active, at least in vitro (Eden et al, 2002; Machesky et al, 1999; Miki et al, 2000). However, the majority of WAVE is in the “WAVE complex” with four other proteins: Hem, Sra-1/PIR121/CYFIP, Abi and HSPC300/Brk1.
In the WAVE-complex, direct association between WAVE, Abi and HSPC300 represents the backbone of the complex. Hem binds to Sra-1 forming a sub-complex, which is able to bind to Rac through Sra-1 (Bogdan et al, 2004; Kitamura et al, 1997). The interaction between Abi and Hem is what binds Hem and Sra-1 into the complex (Derivery et al, 2009; Ismail et al, 2009). Hem and Sra-1 are sequentially recruited to the WAVE complex (Derivery et al, 2008). Free subunits and assembly intermediates of the WAVE-complex are usually not detected but supposedly degraded (Derivery and Gautreau, 2010). Also, previous studies suggest that depletion of one component leads to degradation of others (Kunda et al, 2003; Rogers et al, 2003; Schenck et al, 2004). Indeed, our result that in Hem mutants, the level of WAVE protein, but not the WAVE gene transcription, is drastically reduced supports this contention. Perhaps in the absence of Hem, WAVE complex is either not formed or partially formed, resulting in the degradation of WAVE and phenotypes such as mis- migration of neurons. When the levels of WAVE are supplemented using a WAVE transgene (UAS-WAVE), the migration defect in Hem mutants is promptly rescued.
While a complete lack of WAVE (or Hem) function causes an arrest in the migration of RP2, a reduction in the levels of WAVE due to a reduction in the levels of Hem causes abnormal migration. For example, the lowest level of WAVE is seen in the Hem allele that has the strongest penetrance. Moreover, since this mis-migration defect is rescued by expressing WAVE from a transgene, we can conclude that this mis-migration is also due to an effect on WAVE. It has been suggested that the WAVE-complex exists cytoplasmically and in membrane-bound forms (Suetsugu et al, 2006; Steffen et al, 2004). Through an interaction with Rac, WAVE gets recruited to the lamellipodia where actin polymerization required for membrane protrusion is initiated and regulated. The integrity of the complex is critical for it’s proper localization since removal of either WAVE or Abi prevents its translocation to the leading edge of the lamellipodia (Innocenti et al, 2004). It is possible that a reduction in the levels of WAVE in Hem mutant embryos causes non-translocation of the WAVE complex to the membrane, causing a non/mis-migration of RP2.
Ableson Tyrosine Kinase and WAVE function
Our results show that WAVE protein exists as three different molecular weight forms (Fig. 6). Treatment of the extract with phosphatase collapses these three forms into a single band (Fig. 6C), indicating that WAVE protein is phosphorylated, with varying degrees of phosphorylation to yield different molecular weight species. We carefully examined if there are any changes in the three different forms/their relative contributions in Hem and Abl mutants. However, we did not find any changes in their relative contributions and the levels of all the forms are reduced in Hem mutants. Therefore, it may be that the reduction in all the forms, or that the reduction in one or two of the forms is responsible for the migration defect.
In Abl mutants, the level of WAVE is modestly increased, which is the opposite to that of the effect of Hem on WAVE. Thus, it seems more likely that the activity of WAVE is affected in Abl mutants. Being a protein kinase, it was possible that Abl phosphorylates WAVE, thus affecting either its activity or level. However, no significant changes in the relative levels of the different phosphorylated forms of WAVE were observed in Abl mutants. It has been shown in vitro that Abl is recruited to WAVE2 by Abi following cell stimulation, triggering the translocation of Abl together with the WAVE complex to the leading edge of the membrane (Leng et al, 2005; Stuart et al, 2006). Thus, Abl might affect WAVE activity, either directly or indirectly, via the translocation of the WAVE complex to the membrane of an actively migrating RP2. It is also possible that Abl affects migration in a pathway that does not involve WAVE.
On the other hand, the effect of loss-of-function for Abl on WAVE levels is more pronounced in older embryos. These results indicate that Abl directly or indirectly regulates the levels of WAVE. Furthermore, though modest, ectopic expression of Abl does down- regulate WAVE (Fig. 6D and 7D). Interestingly, our results also show that Hem regulation of WAVE levels is downstream of the Abl regulation of WAVE since the Hem; Abl double mutants had the same levels of WAVE as Hem single mutants. It seems likely that in the absence of Hem, WAVE protein gets degraded, resulting in the loss of migration/abnormal migration. Whereas in Abl mutants, the most likely scenario is that the activity of WAVE is affected, resulting in the same migration defect.
Research highlights.
Hem, via WAVE, mediates migration of neuronal cells in the ventral nerve cord.
In Hem, WAVE or Abl, RP2 neurons abnormally migrate from one hemisegment to the contralateral, or do not migrate at all.
Such neurons fail to send out axon projection signifying the importance of migration for connectivity.
Hem regulates neuronal migration through stabilizing WAVE.
While Abl negatively regulates the levels of WAVE, it regulates migration via regulating the activity of WAVE.
Acknowledgments
We thank Drs. Manfred Frasch (Eve antibody), Eyal Schejter (WASp and WAVE mutant stocks), Jennifer Zallen (WAVE antibody, UAS-WAVE line), Christian Klämbt (Hem antibody), Zun Lai (Zfh1 antibody), Iowa Hybridoma Bank (antibodies against Eve, 22C10, BP102 and FasII), and the Bloomington stock center for various mutant stocks. We also thank the members of the Bhat lab for their help and comments on the work. This work is supported by an R01 grant from NIGMS-NIH to KB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baumgartner S, Martin D, Chiquet-Ehrismann R, Sutton J, Desai A, Huang I, Kato K, Hromas R. The HEM proteins: a novel family of tissue-specific transmembrane proteins expressed from invertebrates through mammals with an essential function in oogenesis. J Mol Biol. 1995;251:41–9. doi: 10.1006/jmbi.1995.0414. [DOI] [PubMed] [Google Scholar]
- Bear JE, Rawls JF, Saxe CL., 3rd SCAR, a WASP-related protein, isolated as a suppressor of receptor defects in late Dictyostelium development. J Cell Biol. 1998;142:1325–35. doi: 10.1083/jcb.142.5.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yaacov S, Le Borgne R, Abramson I, Schweisguth F, Schejter ED. Wasp, the Drosophila Wiskott-Aldrich syndrome gene homologue, is required for cell fate decisions mediated by Notch signaling. J Cell Biol. 2001;152:1–13. doi: 10.1083/jcb.152.1.1-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KM. Wingless activity in the precursor cells specifies neuronal migratory behavior in the Drosophila nerve cord. Dev Biol. 2007;311:613–22. doi: 10.1016/j.ydbio.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat KM, Gaziova I, Krishnan S. Regulation of axon guidance by slit and netrin signaling in the Drosophila ventral nerve cord. Genetics. 2007;176:2235–46. doi: 10.1534/genetics.107.075085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan S, Grewe O, Strunk M, Mertens A, Klambt C. Sra-1 interacts with Kette and Wasp and is required for neuronal and bristle development in Drosophila. Development. 2004;131:3981–3989. doi: 10.1242/dev.01274. [DOI] [PubMed] [Google Scholar]
- Bogdan S, Klambt C. Kette regulates actin dynamics and genetically interacts with Wave and Wasp. Development. 2003;130:4427–37. doi: 10.1242/dev.00663. [DOI] [PubMed] [Google Scholar]
- Buescher M, Yeo SL, Udolph G, Zavortink M, Yang X, Tear G, Chia W. Binary sibling neuronal cell fate decisions in the Drosophila embryonic central nervous system are nonstochastic and require inscuteable-mediated asymmetry of ganglion mother cells. Genes Dev. 1998;12:1858–70. doi: 10.1101/gad.12.12.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E, Fink J, Martin D, Houdusse A, Piel M, Stradal TE, Louvard D, Gautreau A. Free Brick1 is a trimeric precursor in the assembly of a functional wave complex. PLoS ONE. 2008;3:e2462. doi: 10.1371/journal.pone.0002462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E, Gautreau A. Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays. 2010;32:119–31. doi: 10.1002/bies.200900123. [DOI] [PubMed] [Google Scholar]
- Derivery E, Lombard B, Loew D, Gautreau A. The Wave complex is intrinsically inactive. Cell Motil Cytoskeleton. 2009;66:777–90. doi: 10.1002/cm.20342. [DOI] [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–3. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- Gaziova I, Bhat KM. Generating asymmetry: with and without self-renewal. Prog Mol Subcell Biol. 2007;45:143–78. doi: 10.1007/978-3-540-69161-7_7. [DOI] [PubMed] [Google Scholar]
- Gaziova I, Bhat KM. Ancestry-independent fate specification and plasticity in the developmental timing of a typical Drosophila neuronal lineage. Development. 2009;136:263–74. doi: 10.1242/dev.027854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghashghaei HT, Lai C, Anton ES. Neuronal migration in the adult brain: are we there yet? Nat Rev Neurosci. 2007;8:141–51. doi: 10.1038/nrn2074. [DOI] [PubMed] [Google Scholar]
- Hasegawa E, Kitada Y, Kaido M, Takayama R, Awasaki T, Tabata T, Sato M. Concentric zones, cell migration and neuronal circuits in the Drosophila visual center. Development. 2011;138:983–993. doi: 10.1242/dev.058370. [DOI] [PubMed] [Google Scholar]
- Higgs HN, Pollard TD. Activation by Cdc42 and PIP(2) of Wiskott-Aldrich syndrome protein (WASp) stimulates actin nucleation by Arp2/3 complex. J Cell Biol. 2000;150:1311–20. doi: 10.1083/jcb.150.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel T, Leifker K, Klambt C. The Drosophila HEM-2/NAP1 homolog KETTE controls axonal pathfinding and cytoskeletal organization. Genes Dev. 2000;14:863–73. [PMC free article] [PubMed] [Google Scholar]
- Innocenti M, Zucconi A, Disanza A, Frittoli E, Areces LB, Steffen A, Stradal TE, Di Fiore PP, Carlier MF, Scita G. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–27. doi: 10.1038/ncb1105. [DOI] [PubMed] [Google Scholar]
- Ismail AM, Padrick SB, Chen B, Umetani J, Rosen MK. The WAVE regulatory complex is inhibited. Nat Struct Mol Biol. 2009;16:561–563. doi: 10.1038/nsmb.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R. Cell migration during gastrulation. Curr Opin Cell Biol. 2005;17:533–41. doi: 10.1016/j.ceb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Kim AS, Kakalis LT, Abdul-Manan N, Liu GA, Rosen MK. Autoinhibition and activation mechanisms of the Wiskott-Aldrich syndrome protein. Nature. 2000;404:151–8. doi: 10.1038/35004513. [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Kitamura T, Sakaue H, Maeda T, Ueno H, Nishio S, Ohno S, Osada S, Sakaue M, Ogawa W, Kasuga M. Interaction of Nck-associated protein 1 with activated GTP-binding protein Rac. Biochem J. 1997;322:873–8. doi: 10.1042/bj3220873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13:1867–75. doi: 10.1016/j.cub.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. doi: 10.1016/j.molcel.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y, Zhang J, Badour K, Arpaia E, Freeman S, Cheung P, Siu M, Siminovitch K. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:1098–1103. doi: 10.1073/pnas.0409120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Fan J, Woodley DT. Nck/Dock: an adapter between cell surface receptors and the actin cytoskeleton. Oncogene. 2001;20:6403–17. doi: 10.1038/sj.onc.1204782. [DOI] [PubMed] [Google Scholar]
- Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Insall RH. Scar1 and the related Wiskott-Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr Biol. 1998;8:1347–56. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci U S A. 1999;96:3739–44. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Quiles N, Rohatgi R, Anton IM, Medina M, Saville SP, Miki H, Yamaguchi H, Takenawa T, Hartwig JH, Geha RS, Ramesh N. WIP regulates N- WASP-mediated actin polymerization and filopodium formation. Nat Cell Biol. 2001;3:484–91. doi: 10.1038/35074551. [DOI] [PubMed] [Google Scholar]
- Miki H, Yamaguchi H, Suetsugu S, Takenawa T. IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature. 2000;408:732–5. doi: 10.1038/35047107. [DOI] [PubMed] [Google Scholar]
- Morante J, Erclik T, Desplan C. Cell migration in Drosophila optic lobe neurons is controlled by eyeless/Pax6. Development. 2011;138:687–693. doi: 10.1242/dev.056069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao S, Platek A, Hirano S, Takeichi M. Contact-dependent promotion of cell migration by the OL-protocadherin-Nap1 interaction. J Cell Biol. 2008;182:395–410. doi: 10.1083/jcb.200802069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrick SB, Cheng H-C, Ismail AM, Panchal SC, Doolittle LK, Kim S, Skehan BM, Umetani J, Brautigam CA, Leong JM, Rosen MK. Hierarchical Regulation of WASP/WAVE Proteins. Molecular Cell. 2008;32:426–438. doi: 10.1016/j.molcel.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Developmental Biology. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Wiedemann U, Stuurman N, Vale RD. Molecular requirements for actin-based lamella formation in Drosophila S2 cells. J Cell Biol. 2003;162:1079–1088. doi: 10.1083/jcb.200303023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Nollau P, Ho HY, Kirschner MW, Mayer BJ. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway. J Biol Chem. 2001;276:26448–52. doi: 10.1074/jbc.M103856200. [DOI] [PubMed] [Google Scholar]
- Schenck A, Qurashi A, Carrera P, Bardoni B, Diebold C, Schejter E, Mandel JL, Giangrande A. WAVE/SCAR, a multifunctional complex coordinating different aspects of neuronal connectivity. 2004;274:260–270. doi: 10.1016/j.ydbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Schroter RH, Lier S, Holz A, Bogdan S, Klambt C, Beck L, Renkawitz-Pohl R. kette and blown fuse interact genetically during the second fusion step of myogenesis in Drosophila. Development. 2004;131:4501–4509. doi: 10.1242/dev.01309. [DOI] [PubMed] [Google Scholar]
- Soto MC, Qadota H, Kasuya K, Inoue M, Tsuboi D, Mello CC, Kaibuchi K. The GEX-2 and GEX-3 proteins are required for tissue morphogenesis and cell migrations in C. elegans. Genes Dev. 2002;16:620–632. doi: 10.1101/gad.955702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–59. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JR, Gonzalez FH, Kawai H, Yuan ZM. c-Abl interacts with the WAVE2 signaling complex to induce membrane ruffling and cell spreading. J Biol Chem. 2006;281:31290–7. doi: 10.1074/jbc.M602389200. [DOI] [PubMed] [Google Scholar]
- Suetsugu S, Kurisu S, Oikawa T, Yamazaki D, Oda A, Takenawa T. Optimization of WAVE2 complex-induced actin polymerization by membrane-bound IRSp53, PIP(3), and Rac. J Cell Biol. 2006;173:571–85. doi: 10.1083/jcb.200509067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suetsugu S, Miki H, Takenawa T. Identification of two human WAVE/SCAR homologues as general actin regulatory molecules which associate with the Arp2/3 complex. Biochem Biophys Res Commun. 1999;260:296–302. doi: 10.1006/bbrc.1999.0894. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Nishiyama K, Yamamoto A, Inazawa J, Iwaki T, Yamada T, Kanazawa I, Sakaki Y. Molecular Cloning of a Novel Apoptosis-Related Gene, Human Nap1 (NCKAP1), and Its Possible Relation to Alzheimer Disease. Genomics. 2000;63:246–254. doi: 10.1006/geno.1999.6053. [DOI] [PubMed] [Google Scholar]
- Symons M, Derry JMJ, Karlak B, Jiang S, Lemahieu V, McCormick F, Francke U, Abo A. Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell. 1996;84:723–734. doi: 10.1016/s0092-8674(00)81050-8. [DOI] [PubMed] [Google Scholar]
- Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–1809. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Webb DJ, Horwitz AR. Cell migration at a glance. J Cell Sci. 2005;118:4917–4919. doi: 10.1242/jcs.02662. [DOI] [PubMed] [Google Scholar]
- Wai P, Truong B, Bhat KM. Cell division genes promote asymmetric interaction between Numb and Notch in the Drosophila CNS. Development. 1999;126:2759–70. doi: 10.1242/dev.126.12.2759. [DOI] [PubMed] [Google Scholar]
- Weiner OD, Rentel MC, Ott A, Brown GE, Jedrychowski M, Yaffe MB, Gygi SP, Cantley LC, Bourne HR, Kirschner MW. Hem-1 complexes are essential for Rac activation, actin polymerization, and myosin regulation during neutrophil chemotaxis. PLoS Biol. 2006;4:e38. doi: 10.1371/journal.pbio.0040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieschaus E, Nusslein-Volhard C. Looking at embryos. In: Roberts DB, editor. Drosophila a Practical Approach. IRL Press; Oxford: 1986. pp. 199–207. [Google Scholar]
- Wong AH, Waugh JM, Amabile PG, Yuksel E, Dake MD. In vivo vascular engineering: directed migration of smooth muscle cells to limit neointima. Tissue Eng. 2002;8:189–99. doi: 10.1089/107632702753724969. [DOI] [PubMed] [Google Scholar]
- Yokota Y, Ring C, Cheung R, Pevny L, Anton ES. Nap1-regulated neuronal cytoskeletal dynamics is essential for the final differentiation of neurons in cerebral cortex. Neuron. 2007;54:429–45. doi: 10.1016/j.neuron.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallen JA, Cohen Y, Hudson AM, Cooley L, Wieschaus E, Schejter ED. SCAR is a primary regulator of Arp2/3-dependent morphological events in Drosophila. J Cell Biol. 2002;156:689–701. doi: 10.1083/jcb.200109057. [DOI] [PMC free article] [PubMed] [Google Scholar]