

Abstract
Background
It has been well documented that human adenovirus type 36 (Ad-36) is associated with obesity. However, the underlying molecular mechanism of Ad-36 inducing obesity remains unknown. We sought to investigate the effect of Ad-36 infection on Cidec, AMPK pathway and lipid metabolism in primary cultured human skeletal muscle cells.
Methods
Cidec/fat-specific protein 27 (FSP27), fatty acid oxidation, AMPK signaling and the abundance of proteins involved in lipid synthesis were determined in muscle cells infected with various doses (1.9–7.6 MOI) of Ad-36 and non-lipogenic adenovirus type 2 (Ad-2) as a negative control as well as an uninfected control. Cidec/FSP27 siRNA transfection was performed in Ad-36-infected muscle cells.
Results
Our data show that Ad-36 significantly reduced fatty acid oxidation in a dose-dependent manner (all P values are < 0.01), but Ad-2 did not affect fatty acid oxidation. Ad-36 substantially increased Cidec/FSP27, ACC, sterol regulatory element-binding protein 1c (SREBP-1c), SREBP-2 and 3-hydroxy-3-methylglutaryl-CoA reductase protein abundance, but significantly reduced AMPK activity, mitochondrial mass and uncoupling protein 3 (UCP3) abundance in comparison with control cells (all P values are <0.01). Oil Red O staining revealed that there was substantial fat accumulation in the Ad-36-infected muscle cells. Furthermore, Cidec/FSP27 siRNA transfection significantly reduced FSP27 expression and partially restored AMPK signaling, increased UCP3 and decreased SERBP 1c and perilipin proteins in Ad-36-infected muscle cells. Interestingly, neither Ad-36 nor Ad-2 affected peroxisome proliferator-activated receptor γ protein expression in muscle cells.
Conclusion
This study suggests that Ad-36 induced lipid droplets in the cultured skeletal muscle cells and this process may be mediated by promoting Cidec/FSP27 expression.
Keywords: Cidec/FSP27, fatty acid oxidation, siRNA, SREBP, UCP3
Introduction
Obesity has become the most prevalent chronic disorder that affects large populations throughout the world. Viral infections have been recognized as a possible cause of obesity, alongside the traditionally recognized causes that include genetic inheritance, and behavior/environmental causes such as diet, exercise, cultural practices and stress.1 Human adenovirus type 36 (Ad-36) is one of the 50 types of adenoviruses known to infect humans. It has recently been the focus of intense research due to a significant association with obesity.2 Ad-36 is capable of inducing adiposity in experimentally infected chickens, mice and non-human primates (marmosets).3 Ad-36 has also been reported to increase replication, differentiation, lipid accumulation and insulin sensitivity in fat cells and to reduce those cells’ leptin secretion and expression as well as enhance differentiation of preadipocytes.4 The presence of Ad-36 antibodies in humans is associated with increased body mass index.5,6 In contrast to the conventional association of obesity with insulin resistance, Ad-36-induced obesity appears to decrease plasma glucose, cholesterol, triglyceride and leptin as well as increase adiponectin concentrations.7 However, the precise mechanism of Ad-36 inducing obesity is largely unknown.
Obesity is largely due to the imbalance between energy intake and expenditure. It is believed to be a fat storage disease characterized by insulin resistance and a decreased capacity to oxidize lipids.8 Energy balance is regulated by interaction between various tissues such as adipose tissue, liver and skeletal muscle. Skeletal muscle is a major target of insulin action, and insulin-stimulated glucose uptake as well as fatty acid oxidation. Therefore, skeletal muscle plays a key role in determining nutrient oxidation rates and is the major site of glucose disposal and insulin action.9 The increased deposition of intramyocellular triacylglycerols (imTAGs) has received special interest, because several studies have shown a positive association between insulin resistance and imTAG storage.10 Accumulation of imTAG depends on the availability and uptake of fatty acids, the rate of fatty acid oxidation and the rate of synthesis and hydrolysis of TAG. Increased availability of plasma free fatty acid during lipid infusion or high-fat feeding is associated with development of insulin resistance and accumulation of imTAG in vivo.11
Recently identified as a set of novel proteins, cell death-inducing DNA fragmentation factor alpha-like effector A (Cidea) and fat-specific protein 27 (FSP27, also known as Cidec), which are associated with lipid droplets in adipocytes and their expression, can enhance the accumulation of triglycerides in adipocytes both in vivo and in vitro.12–14 Strikingly, Cidea and FSP27 expression was higher in insulin sensitive vs resistant obese humans. These studies indicate that Cidea and FSP27 define a novel, highly regulated pathway of triglyceride accumulation in mouse and human white adipose tissue and their expression is associated with insulin sensitivity in humans.15–17 Perilipin is a protein that coats lipid droplets in adipocytes, the fat-storing cells in adipose tissue. Perilipin behaves as a protective coating from the body’s natural lipases, such as hormone-sensitive lipase, which breaks down triglycerides into glycerol and free fatty acids for use in metabolism, a process called lipolysis.18 Knockdown of FSP27 in 3T3-L1 adipocytes was reported to result in the fragmentation of large lipid droplets to produce many small lipid droplets. Ectopic expression of FSP27 promoted lipid droplet formation when assessed in 3T3-L1 preadipocytes, COS cells and CHO cells.19 Similar to perilipin, Cidea and the related lipid droplet protein Cidec/FSP27, are controlled by peroxisome proliferator-activated receptor γ (PPARγ).20 However, regulation of endogenous Cidec/FSP27, perilipin and PPARγ expression in Ad-36-infected muscle cells is still unknown.
In a previous study, we reported that Ad-36 results in E4 orf1 virus gene expression, increases glucose uptake and GLUT4 protein abundance independent of insulin signaling in human skeletal muscle cells.21 Currently, no data are available about the effect of Ad-36 on lipid metabolism and its related cellular signaling such as AMPK pathway, perilipin and Cidec/FSP27 in primary skeletal muscle cell culture. The effect of Ad-36 on skeletal muscle cells is potentially of great significance. To extend our previous study, we evaluated the effects of Ad-36 on lipid metabolism and related cellular signaling in skeletal muscle. We hypothesize that Ad-36 could reduce fatty acid oxidation and increase de novo lipogenesis in muscle cells by altering cellular signaling pathways as the consequence of Ad-36-induced Cidec/FSP27 protein expression. To test this hypothesis, we measured fatty acid oxidation, mitochondrial mass, FSP27 expression and lipid metabolism-related cellular signaling in Ad-36 and Ad-2 infected primary cultured human skeletal muscle cells.
Methods
Bovine serum albumin (BSA) and the protease inhibitors, phenylmethylsulfonyl fluoride and all other reagent grade chemicals were purchased from Sigma (St Louis, MO, USA). Cell culture media was purchased from Cambrex (Walkersville, MD, USA). Fetal bovine serum was obtained from Hyclone (Logan, UT, USA). Anti-ACC, AMPK-p(Thr172) and uncoupling protein 3 (UCP3) antibodies were purchased from Upstate Biotechnology (Lake Placid, NY, USA). Anti-AMPK α antibody (Cell Signaling Biotechnology, Danvers, MA, USA). Cidec/FSP27 siRNA (sc-78016), negative control siRNA (sc-44230), anti-SREBP-1c and anti-SREBP-2 were ordered from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-FSP27 antibody was ordered from Thermo Inc. (Rockford, IL, USA) and perilipin antibody was ordered from ARP American Research (Belmont, MA, USA). Secondary horseradish peroxidase-conjugated antibody, protein A Sepharose and chemiluminescence reagents (ECL kit) were obtained from Amersham Life Science (Arlington, IL, USA). The nitrocellulose membrane, electrophoresis equipment, western blotting reagents and protein assay kits were from Bio-Rad (Hercules, CA, USA). [14C palmitate] and [r32P ATP] were obtained from NEN Life Science (Boston, MA, USA).
Human skeletal muscle cell culture
Muscle biopsy of the vastus lateralis, cell isolation and growth have been established in our laboratory as described previously.21
Infection of human skeletal muscle cells with Ad-36 virus
Cells were maintained in human skeletal growth media (SkGM Bullet Kit; Cambrex) containing 10 μg l−1 human epidermal growth factor, 500mgl−1 BSA, 500mg fetuin, 195 μg l−1 dexamethasone with 10% fetal bovine serum and no insulin until approximately 80% confluent. Antibiotics were removed before Ad-36 infection. Muscle cells were usually infected at a dose of 3.8 multiplicity of infection (MOI) Ad-36 and the same dose of Ad-2 as the negative control for 1 h; otherwise, the doses of viruses are indicated in the legends. After washing, we added fresh media (Ad-36 and Ad-2 were kindly provided by Dr Nikhil Dhurandhar, Pennington Biomedical Research Center). Virus gene expression had been confirmed in Ad-36-infected muscle cells.21
Western blotting analysis and Cidec/FSP27 siRNA transfection
At day 3 after Ad-36 infection, muscle cells were maintained in antibiotic-free medium with 1% fetal bovine serum and transfected with Cidec/FSP27 siRNA or a negative siRNA control. Cidec/FSP27 siRNA transfection was performed at dose 0, 0.2, 0.4 and 0.8 μM and 0.4 μM of negative siRNA control using Invitrogen Lipofectamine 2000 reagents (Carlsbad, CA, USA). Whole-cell lysates were prepared as previously described.22 Briefly, 50 μg of muscle cell lysates was subjected to SDS–polyacrylamide gel electrophoresis (PAGE), then transferred to nitrocellular membranes, and blocked with anti-pAMPK, AMPK α, ACC, Cidec/FSP27, perilipin, SERBP 1C, SERBP 2, 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMGR), UCP3 and β-actin antibodies. Signals were detected by enhanced chemiluminescence solution. The specific bands were quantized with scanning densitometry and the data were normalized by β-actin levels.
Reverse transcription and PCR (RT–PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen). Reverse transcription of RNA (1 μg) was performed using iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. PCR was performed using 1 μl of synthesized cDNA with Maxima SYBR Green qPCR Master Mix (Fermentas, Glen Burnie, MD, USA) and 100 nM (for Cidec/FSP27) or (for β-actin) of the respective primer pairs (IDT, San Diego, CA, USA) GCCTTCTCTACCCCAAGTCC and CAGGAAGAAGGGCTTGTCTG for Cidec/FSP27, GGACTTCGAGCAAGAGATGG and AGCACTGTGTTGGCGTACAG for β-actin). PCR products were visualized by 1.2% agarose gel electrophoresis and ethidium bromide staining.
Confocal microscopy
Immunofluorescence for Cidec/FSP27 and perilipin were performed on muscle cells at day 6 after infection (Ad-2 or Ad-36 at dose of 3.8 MOI) and uninfected control cells. Briefly, cells growing on glass cover slips were fixed in ice-cold 2% paraformaldehyde on ice, followed by 10 min of permeabilization in 0.2% Triton X-100 in phosphatebuffered saline (PBS). Reactions were blocked for 30 min with 0.2% BSA, followed by 60 min of incubation with a rabbit polyclonal anti-FSP27 (1:500 dilution) and perilipin antibody (1:500 dilution), FITC-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology) or Alexa Fluor 594-conjugated goat anti-mouse IgG (1:500 dilution; Invitrogen) were added for 30 min (1:500 dilution; Molecular Probes, Invitrogen (Eugene, OR, USA)). After washing, we obtained images on a Zeiss 510 Meta (Zeiss, Thornwood, NY, USA) Axiophot microscope equipped with a Nikon digital camera (Melville, NY, USA) and processed by using Metamorph imaging software, version 6.1 (Universal Imaging, Leica Microsystems Inc., Bannockburn, IL, USA).
Oil Red-O staining for intracellular lipid accumulation in the muscle cells
Oil Red-O (0.3%) in isopropyl alcohol solution was freshly made, mixed with one-half volume of H2O and filtered through 0.45 μm filter. To stain for triglycerides or neutral lipids, we first washed cells in the monolayer three times with PBS and then fixed them in 4% formaldehyde solution in PBS (30 min). After extensive washes, we stained fixed cells with the freshly prepared Oil Red O solution for 10 min at room temperature, followed by six washes with water. Imaging was performed using a Nikon Eclipse TS100 inverted microscope with Nikon digital camera (model DXM1200F) and ACT-1 version 2 software (Melville, NY, USA).
Mitochondrial mass assay
For quantification of mitochondrial content in the virus-infected cells, we used MitoTracker Green probe (Molecular Probes) described by Pendergrass et al.23 MitoTracker Green probe preferentially accumulates in mitochondria and provides an accurate assessment of mitochondrial mass. Cells were washed with PBS and incubated at 37 °C for 30 min with 40 nM MitoTracker Green FM (Molecular Probes). Cells were harvested using trypsin/EDTA and re-suspended in PBS. The relative fluorescence intensity was detected with excitation and emission wavelengths of 490 and 516nm using a fluorescence microplate spectrophotometer (Biotex, Winooski, VT, USA) and values corrected for total protein (mg ml−1).
Fatty acid oxidation assay
Fatty acid oxidation was measured as described by Hulver et al.24 In brief, at day 6 after infection, muscle cells were incubated in a bicarbonate-buffered Krebs-Ringer medium containing 5mM glucose, 0.5mM palmitate, 2% BSA and 0.5 μCi of [1-14C] palmitate. After 3 h, we added 0.5 ml of NaOH to the center of 0.5 tube in the incubation well (to trap CO2), and terminated cellular reactions by addition of 0.5 ml of 1 N H3SO4 to the media. CO2 was collected for 30 min. Acid-soluble products (ASM) were extracted from the media with chloroform/methanol/NaCl. Incorporation of radioactivity into CO2, ASM and total lipids was measured using a liquid-scintillation spectrometer. Portions of cell lysates were used for measuring protein concentration, and results were expressed relative to nmolmg−1 protein per h. All assays were performed in triplicate.
AMPK activity assay
AMPK activity assays were carried out according to Carling’s method.25 In brief, 200 μg of cell lysates were immunoprecipitated with anti-AMPK α antibody and protein A beads. Assays were performed at 30 °C for 10 min in 50 μl reaction mixtures containing a reaction buffer (40mmoll−1 HEPES (pH 7.0), 80mmoll−1 NaCl, 5mmol l−1 magnesium acetate, 1mmoll−1 dithiothreitol, 200 μmol l−1 each of AMP and ATP, and 2 μCi [γ-32P] ATP) with or without 200 μmol l−1 SAMS peptide (Upstate Biotechnology). The reaction mixtures were spotted onto P81 cation exchange papers, washed three times with 1% phosphoric acid and measured using a scintillation counter. AMPK activity was expressed as [32P] incorporated per microgram of protein.
Statistical analysis
The data are presented as mean ± s.e.m. Statistical differences were determined by analysis of variance or by paired or unpaired Student’s t-test, as appropriate. An interaction between the viral gene expression and fatty acid oxidation as well as cell signaling pathways was anticipated.
Results
Adenovirus infection resulted in virus gene expression in human skeletal muscle cells
Ad-36 and Ad-2 infection resulted in corresponding virus gene expression in human skeletal muscle cells. The E4 orf1 of Ad-36 gene expression was increased in a dose- and time-response manner (Figures 1a and b). Ad-2 gene expression also was increased in a dose-dependent manner (Figure 1c).
Figure 1.

Ad-36 E4 orf1 and Ad-2 E4 orf1 gene expression in virus-infected human skeletal muscle (HSKM) cells. Muscle cells were grown to 80% confluence and infected with various doses of virus or incubated at different times as indicated. (a) Ad-36 E4 orf1 gene expression of dose response and (b) the time course experiments in Ad-36-infected muscle cells. (c) Ad-2 E4 orf1 gene expression in Ad-2-infected muscle cells. (Modified from Wang et al.21 with permission.)
Ad-36 increased FSP27 and perilipin protein abundance in muscle cells
Ad-36 induced ectopic expression of FSP27 and promoted lipid droplet formation in Ad-36-infected muscle cells. Next, we determined if Cidec/FSP27 and perilipin protein abundance were altered in Ad-36-infected cells in comparison with control cells. The results showed that Ad-36 substantially increased Cidec/FSP27 content in a dose-dependent manner and the fold changes were from 7.3, 9.2 to 11.8 at doses of 1.9, 3.8 and 7.6 MOI (P<0.001). Ad-2 only slightly increased Cidec/FSP27 content (P=NS). A similar effect on perilipin protein abundance with Ad-36 infection was observed in muscle cells. However, a lower dose of Ad-36 infection did not significantly affect perilipin abundance (Figure 2).
Figure 2.
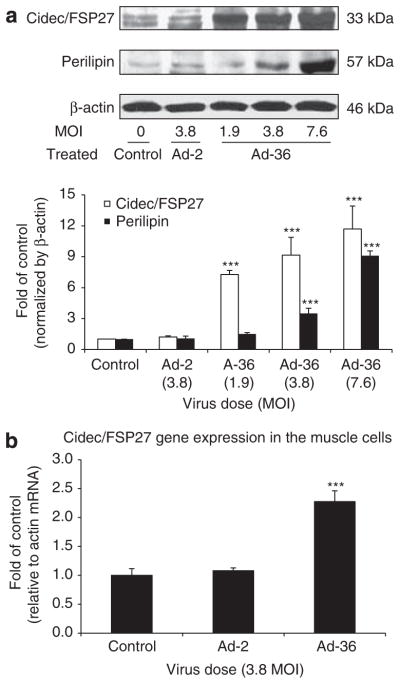
Effect of Ad-36 on Cidec/FSP27, perilipin protein abundance and Cidec/FSP27 gene expression in human skeletal muscle (HSKM) cells. Cidec/FSP27 and perilipin protein abundance were measured in the muscle cells at day 6 after infection with doses of Ad-36 from 1.9 to 7.6 MOIs as indicated. Lysates (50 μg) were subjected to SDS–PAGE, and transferred to nitrocellulose membranes. Cidec/FSP27 and perilipin were detected with specific antibodies against Cidec/FSP27 and perilipin. Panel a shows Cidec/FSP27 and perilipin protein content in control, Ad-2 and Ad-36 infected muscle cells. Panel b reveals gene expression results of Cidec/FSP27 in control, Ad-2, Ad-36 infected muscle cells. The results were normalized by β-actin and represented as mean ± s.e.m. of three separated studies. ***P<0.001, Ad-36 vs control.
Ad-36 increased de novo lipogenesis in skeletal muscle cells
Sterol regulatory element-binding proteins (SREBPs) have been established as physiological regulators of lipid synthesis. The data show that Ad-36 dramatically increased SREBP-1c, SERBP2, HMGR and ACC protein abundance in a dose-dependent manner (P<0.001), but Ad-2 did not significantly affect expression of these proteins (Figure 3).
Figure 3.

The effects of Ad-36 on SREBP-1c, SREBP-2, HMGR and ACC protein levels in the muscle cells. Muscle cells were infected with various doses of Ad-36 and 3.8 MOI of Ad-2 at day 6 after infection. The cell lysates were subjected to SDS–PAGE and the indicated proteins were detected with corresponding antibodies. Protein levels were normalized by β-actin and data are presented as mean±s.e.m. from three independent experiments. Mean±s.e.m. of three individual experiments. ***P<0.001, Ad-36 vs control.
Ad-36 reduced fatty acid oxidation in muscle cells by altering AMPK signaling, but not PPARγ signaling
Fatty acid oxidation data reveal that Ad-36 significantly decreased fatty acid oxidation from 100±7%, 62±9%, 56±11% and 43±5% when compared to control when evaluating, 1.9, 3.8, 7.6 doses of Ad-36, respectively (P<0.01 and P<0.001, Figure 4). However, Ad-2 did not significantly affect fatty acid oxidation when compared with control (P=NS). AMPK activity and AMPK protein abundances were measured in the muscle cells infected by Ad-2 or Ad-36 viruses. AMPK activities were significantly lower in Ad-36-infected cells than in uninfected control cells in both basal and AMP-stimulated conditions (P<0.05, P<0.01 and P<0.001, respectively in Figure 5a). Western blotting analysis revealed that phosphorylation of AMPK and AMPK α isoform protein abundance were reduced when compared with control cells. However, there was no significant difference of AMPK-p and AMPK α protein abundance between Ad-2 and control cells (Figure 5b). Interestingly, neither Ad-36 nor Ad-2 alters PPARγ protein expression (Figure 5c).
Figure 4.
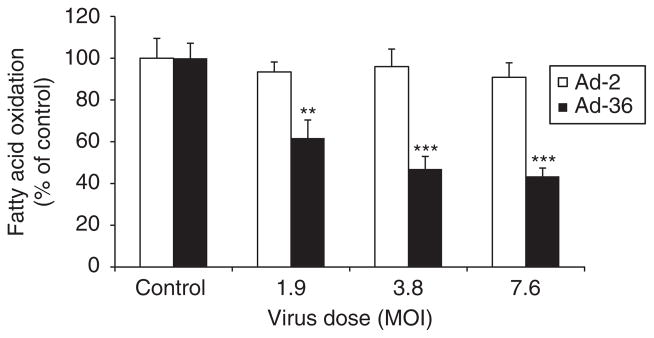
Ad-36 reduced fatty acid oxidation in the muscle cells. Muscle cells were infected with various doses of Ad-2 and Ad-36 as shown. At day 6 after infection, fatty acid oxidation of human skeletal muscle cells (myoblasts) was measured using [14C]-labeled palmitate in a 24-well plate for 3 h as described in the Methods section. Cells were incubated in oxidation media containing 14C labeled. The assay was carried out in triplicate and the data represent three independent experiments. **P<0.01 and ***P<0.001, virus infected vs control.
Figure 5.
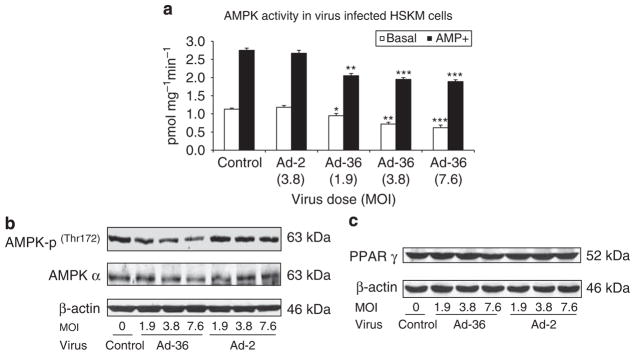
Ad-36 affects AMPK signaling in muscle cells. At day 6 after virus infection, cells were harvested and 200 μg cell lysates were immunoprecipitated with antibody against AMPK α and protein A. AMPK activity was carried out by adding 200 μM of [γ32P]ATP and SAMS peptide with or without 200 μg AMP and incubated at 32 °C for 10 min. Reaction was terminated by spotting a 20 μl aliquot onto a 1 cm2 piece of P-81 phosphocellulose paper and washing for 3×5min in 1% phosphoric acid. Samples were then air-dried, incorporation of γ-32P was quantified and AMPK activity is expressed as incorporated ATP (pmols) per mg protein per min. (a) AMPK activity in virus-infected and control muscle cells. Assay was conducted in triplicate. Mean±s.e.m., *P<0.05, **P<0.01 and ***P<0.001, Ad-36 vs control. (b) AMPK-p and AMPK α protein as well as PPAR γ (c) abundance was measured by western blot in virus-infected and non-infected control muscle cells.
Effect of Ad-36 on mitochondrial mass and UCP3 protein abundance in the muscle cells
We further measured mitochondrial mass and UCP3 protein abundance in the muscle cells and noted that Ad-36 substantially reduced mitochondrial mass in a dose-dependent manner, but Ad-2 slightly decreased mitochondrial mass in comparison with control cells (Figure 6a). In addition, Ad-36 significantly reduced UCP3 abundance as well (Figure 6b).
Figure 6.
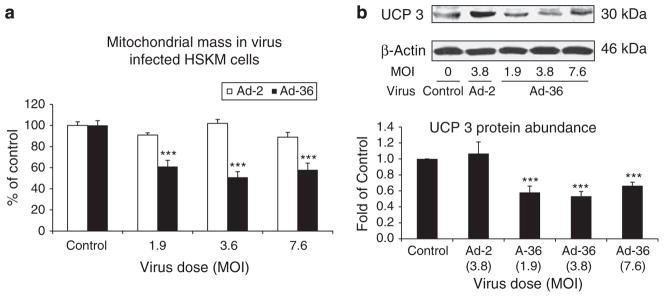
Effect of Ad-36 on mitochondrial mass and UCP3 protein abundance in the muscle cells. Mitochondrial mass was measured at day 6 after viral infections in the muscle cells using a MitoTracker Green FM kit. (a) The effect of the viruses on mitochondrial mass in the muscle cells. (b) UCP3 abundance measured by western blotting. Data represent mean±s.e.m. of three independent experiments. ***P<0.001, virus vs control.
Knockdown Cidec/FSP27 could partially attenuate the effect of Ad-36 on AMPK signaling, UCP3 and SREBPs as well as reduce lipid droplets accumulation in muscle cells
To test if knockdown of Cidec/FSP27 gene expression could attenuate the effect of Ad-36 on AMPK signaling and lipid metabolism, we performed of Cidec/FSP27 siRNA transfection in Ad-36-infected muscle cells. Ad-36 infection significantly increased Cidec/FSP27 gene expression, and Cidec/FSP27 siRNA significantly reduced Ad-36-induced Cidec/FSP27 gene expression at dose of 0.4 and 0.8 μM (P<0.001, Figure 7a). Indeed, we observed that knockdown of Cidec/FSP27 could partially restore AMPK phosphorylation, increase UCP3 protein abundance and reduce SREBP-1c and perilipin protein contents in the Ad-36-infected muscle cells with Cidec/FSP27 siRNA in a dose-dependent manner (Figure 7b). To further verify whether Cidec/FSP27 and perilipin were primarily localized around lipid droplets in muscle cells in Ad-36-infected muscle cells, and whether Cidec/FSP27 knockdown could reduce lipid droplets in Ad-36-infected muscle cells with Cidec/FSP27 siRNA in a transfection, we determined immunofluorescence localization of Cidec/FSP27 and perilipin proteins in virus-infected and control cells using confocal microscopy. We confirmed that Ad-36 significantly increased Cidec/FSP27 and perilipin protein expression around lipid droplets in the muscle cells, but Ad-2 did not alter these protein levels when compared with control cells (Figures 8a–c). Oil Red O staining showed Ad-36-induced lipid accumulation in the cells and there were no lipid droplets deposited in the control and Ad-2 infected muscle cells (Figure 8d). Moreover, Cidec/FSP27 siRNA transfection greatly reduced Cidec/FSp27 and perilipin contents as well as lipid droplets in Ad-36-infected muscle cells.
Figure 7.

Effect of Cidec/FSP27 siRNA transfection on Cidec/FSP27 gene expression, and AMPK-p, AMPK α, UCP3, SREBP-1c and perilipin protein expression in Ad-36-infected human skeletal muscle (HSKM) cells. At day 3 after Ad-36 infection, Cidec/FSP27 siRNA transfection was performed as described in Methods section. At 72 h after siRNA transfection, RNA was isolated and RT–PCR was performed described in Methods section. (a) Cidec/FSP27 mRNA levels. Data were presented from three separated experiments, mean±s.e.m.; ***P<0.001, Ad-36 vs control, ###P<0.001, FSP27 siRNA vs Ad-36, respectively. (b) The same treated muscle cells were homogenized and lysates were subjected to SDS–PAGE. Cidec/FSP27, AMPK-p, AMPK α, UCP3, SREBP-1c, perilipin and β-actin were measured by western blotting. Data were presented after normalized by β-actin.
Figure 8.

At day 3 after virus infection at a dose of 3.8 MOI, then transfected with or without 0.4 μM Cidec/FSP27 siRNA or negative siRNA for 72 h. Confocal immunofluorescence and Oil Red O staining were performed in muscle cells. Immunofluorescence shows localization of Cidec/FSP27 (a, green), perilipin (b, red), Cidec/FSP27 and perilipin colocalization (c, greenish yellow). Oil Red O staining (d) illustrates that Ad-36 infection results in substantial lipids accumulation in the muscle cells (pointed by arrows). However, Cidec/FSP27 siRNA transfection greatly reduces lipid droplets accumulated in the muscle cells.
Discussion
The data demonstrate that Ad-36 increased Cidec/FSP27, perilipin, SERBP 1c, SERBP2, HMGR and ACC protein abundance, and greatly reduced mitochondrial mass and UCP3 protein expression in primary skeletal muscle culture. Furthermore, Ad-36 substantially reduced fatty acid oxidation and decreased AMPK activity, but Ad-2 did not have these effects in the primary human skeletal muscle culture when compared with control culture. Thus, accumulation of fat in Ad-36-infected muscle cells may be associated with reprogramming cellular signaling and significantly elevating Cidec/FSP27 protein, which resulted in inhibition of fatty acid oxidation and increase of de novo lipogenesis.
AMPK is activated by external metabolic stresses and subsequently orchestrates a complex downstream signaling cascade that mobilizes the cell for efficient energy production. AMPK has emerged as a key kinase driving lipid oxidation in skeletal muscle, and this function has important implications for exercise adaptation as well as metabolic defects associated with obesity.26 Ad-36 significantly reduced fatty acid oxidation and decreased AMPK activity and phosphorylation of AMPK in the muscle cells. It has been reported that AMPK stability and enzymatic activity were increased in Cidea−/− adipocytes differentiated from mouse embryonic fibroblasts or preadipocytes. This indicates that AMPK may be regulated by Cidea-mediated ubiquitin-dependent proteosome degradation.17 Studies show that animals with deficiency in Cidea, Cideb and Cidec/FSP27 all display lean phenotypes with higher energy expenditure and are resistant to diet-induced obesity.18,19 Cide proteins, localized to lipid droplets and endoplasmic reticulum, may control lipid metabolism in adipocytes and hepatocytes through regulating AMPK stability and influencing lipogenesis or lipid droplet formation. Thus, the effect of Ad-36 on AMPK activity may be mediated by elevated Cidec/FSP 27 protein. The expression of Cide proteins is controlled at both transcriptional and post-translational levels and positively correlates with the development of obesity, liver steatosis and insulin resistance in both rodents and humans.27 Our observation revealed that Cidec/FSP27 negatively regulates AMPK signaling in Ad-36-infected muscle cells. Recently Keller et al.28 reported that FSP27 binds to lipid droplets and regulates their enlargement based on the findings that overexpression of FSP27 is sufficient to increase apoptosis of 293 T and 3T3-L1 cells. More physiological levels of expression stimulate spontaneous lipid accumulation in several cell types without induction of adipocyte genes. Increased triacylglycerol is likely due to decreased β-oxidation of nonesterified fatty acids. Altered flux of fatty acids into triacylglycerol may be a direct effect of FSP27 function. Conversely, stable knockdown of FSP27 during adipogenesis of 3T3-L1 cells substantially decreases lipid droplet size, increases mitochondrial and lipid droplet number and modestly increases glucose uptake and lipolysis.19,29 Similar to these findings, localization of Cidec/FSP27 and perilipin to lipid droplets in the Ad-36-infected muscle cells indicates Ad-36 has a key role in the pattern of lipid droplet accumulation in human muscle cells.
Ad-36 dramatically reduced muscle mitochondrial mass and UCP3 protein abundance. These effects could contribute to lowering of fatty acid oxidation in the muscle cells because fatty acid oxidation occurs in the mitochondria and UCP3 is a mitochondrial anion carrier protein with highly selective expression in skeletal muscle. Experimental evidence showed increases in muscle levels of UCP3 in a wide variety of in vitro and in vivo experimental systems are consistent with decreased reactive oxygen species production, increased fatty acid oxidation, reduction of intramuscular triglyceride and lipid derivatives. UCP3 action in skeletal muscle may enhance cellular health and longevity, metabolic flexibility and insulin sensitivity.29 We showed that knockdown of Cidec/FSP27 using Cidec/FSP27 siRNA could partially restore AMPK signaling and increase UCP3 protein abundance in the Ad-36-infected muscle cells. It is well documented that the PPARγ regulates expression of adipocyte-specific genes Cidec/FSP27 and perilipin, which results in the promotion of intracellular fat storage.20 However, we showed neither Ad-36 nor Ad-2 alters PPARγ protein abundance in muscle cells (Figure 5c). Thus, Ad-36 may directly promote Cidec/FSP27 expression without affecting PPARγ signaling pathway. Our findings indicated that Cidec/FSP27 negatively regulates mitochondrial mass, AMPK signaling and UCP3 in the Ad-36-infected muscle cells.
Ad-36 promoted de novo lipogenesis in the muscle cells by increasing SERBP 1c, SERBP 2, HMGR and ACC protein expression. SREBP-1c and SERBP 2 are the members of the family of transcription factors that stimulate sterol and fatty acid biosynthesis in animal cells.30 Human SREBP-1c, mapped to chromosome 17p11.2, is expressed in liver, intestine, skeletal muscle and adipocytes.31 SREBPs have been established as physiological regulators of lipid synthesis.32,33 SREBP-1 may be selectively involved in the activation of genes associated with fatty acid metabolism, whereas SREBP-2 is more specific in cholesterol homeostasis, primarily because of its selective activation of the squalene synthase gene.34 It was reported that glucose upregulated SREBP-1c precursor and nuclear proteins, and the knockdown of SREBP-1 mRNA using an RNA interference technique, totally abrogated the glucose-induced upregulation of lipogenic enzymes. This evidence indicates that SREBP-1c increased intramuscular lipid accumulation associated with muscle insulin resistance in obesity or type 2 diabetes could arise partly from de novo fatty acid synthesis in skeletal muscle.35 In the cholesterol biosynthetic pathway, the SREBPs directly activate transcription of the genes encoding 3-hydroxy-3-methylglutaryl (HMG) coenzyme A synthase, HMGR, farnesyl diphosphate synthase and squalene synthase.36,37 It was well established that activation of AMPK suppresses hepatic SREBP-1 mRNA and nuclear SREBP-1 protein.38 We observed that Ad-36 significantly increased HMGR abundance in the muscle. AMPK increases lipolysis and reduces lipid synthesis by decreasing SREBPs expression, inhibiting ACC and HMGR activities. Knockdown of FSP27 with siRNA technique increased AMPK phosphorylation and UCP3 content as well as decreased perilipin and SREBP-1c protein abundance in Ad-36-infected muscle cells (Figure 8). Therefore, increasing SREBPs, ACC and HMGR expression by AD-36 may result from decreased AMPK activity mediated by elevated Cidec/FSP27 expression. However, Ad-36 does not affect PPARγ signaling pathway in muscle cells.
Conclusion
Our study suggests that the dual effects of Ad-36 on lipid metabolism by reducing fatty acid oxidation and increasing de novo lipogenesis, result in fat accumulation in muscle cells that may be modulated by promoting Cidec/FSP27 expression.
Acknowledgments
We thank Dr Nikhil Dhurandhar for kindly providing adenovirus 2 and adenovirus 36. This project used facilities that are supported in part by COBRE (NIH P20-RR021945) and CNRU (NIH 1P30-DK072476) center grants from the National Institutes of Health.
Abbreviations
- SREBP-1c
sterol regulatory element-binding protein 1c
- HMGR
3-hydroxy-3-methylglutaryl-oA reductase
- UCP3
uncoupling protein 3
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Van Ginneken V, Sitnyakowsky L, Jeffery JE. Infectobesity: viral infections (especially with human adenovirus-36: Ad-36) may be a cause of obesity. Med Hypotheses. 2009;72:383–388. doi: 10.1016/j.mehy.2008.11.034. [DOI] [PubMed] [Google Scholar]
- 2.Dhurandhar N, Atkinson R, Ahmed A. Obesity of infectious origin–a review. Growth Genet Horm. 2004;20:33–39. [Google Scholar]
- 3.Whigham LD, Israel BA, Atkinson RL. Adipogenic potential of multiple human adenovirus in vivo and in vitro in animals. Am J Physiol Regul Inter Comp Physiol. 2006;290:R190–R194. doi: 10.1152/ajpregu.00479.2005. [DOI] [PubMed] [Google Scholar]
- 4.Rogers PM, Fusinski KA, Rathod MA, Loiler SA, Pasarica M, Shaw MK, et al. Human adenovirus Ad-36 induces adipogenesis via its E4 orf-1 gene. Int J Obes (Lond) 2008;32:397–406. doi: 10.1038/sj.ijo.0803748. [DOI] [PubMed] [Google Scholar]
- 5.Greenway F. Virus-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2006;290:R188–R189. doi: 10.1152/ajpregu.00607.2005. [DOI] [PubMed] [Google Scholar]
- 6.Vangipuram SD, Sheele J, Atkinson RL, Holland TC, Dhurandhar NV. A human adenovirus enhances preadipocyte differentiation. Obes Res. 2004;12:770–777. doi: 10.1038/oby.2004.93. [DOI] [PubMed] [Google Scholar]
- 7.Atkinson RL, Dhurandhar NV, Allison DB, Bowen RL, Israel BA, Albu JB, et al. Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids. Int J Obes (Lond) 2005;29:281–286. doi: 10.1038/sj.ijo.0802830. [DOI] [PubMed] [Google Scholar]
- 8.Schutz Y, Flatt JP, Jequier E. Failure of dietary intake to promote fat oxidation: a factor favoring the development of obesity. Am J Clin Nutr. 1989;50:307–314. doi: 10.1093/ajcn/50.2.307. [DOI] [PubMed] [Google Scholar]
- 9.Dauncey MJ, Gilmour RS. Regulatory factors in the control of muscle development. Proc Nutr Soc. 1996;55:543–559. doi: 10.1079/pns19960047. [DOI] [PubMed] [Google Scholar]
- 10.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, et al. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46:983–988. doi: 10.2337/diab.46.6.983. [DOI] [PubMed] [Google Scholar]
- 11.Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2002;5:545–549. doi: 10.1097/00075197-200209000-00014. [DOI] [PubMed] [Google Scholar]
- 12.Keller P, Petrie JT, Rose PD, Gerin I, Wright WS, Chiang S-H, et al. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem. 2008;283:14355–14365. doi: 10.1074/jbc.M708323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang L, Zhao M, Xu Z, Yokoyama KK, Li T. Molecular cloning and characterization of CIDE-3, a novel member of the cell-death-inducing DNA-fragmentation-factor (DFF45)-like effector family. Biochem J. 2003;370:195–203. doi: 10.1042/BJ20020656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nishino N, Tamori Y, Tateya S, Kawaguchi T, Shibakusa T, Mizunoya W, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest. 2008;118:2808–2821. doi: 10.1172/JCI34090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keller P, Petrie JT, De Rose P, Gerin I, Wright WS, Chiang SH, et al. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem. 2008;283:14355–14365. doi: 10.1074/jbc.M708323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puri V, Virbasius JV, Guilherme A, Czech MP. RNAi screens reveal novel metabolic regulatos: RIP140, MAP4k4 and the lipid droplet associated fat specific protein (FSP) 27. Acta Physiol (Oxf) 2008;192:103–115. doi: 10.1111/j.1748-1716.2007.01786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, et al. Downregulation of AMP-activated protein kinase by Cidea-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. June Perilipin, a major hormonally regulated adipocyte- specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266:11341–11346. [PubMed] [Google Scholar]
- 19.Puri V, Konda S, Ranjit S, Aouadi M, Chawla A, Chouinard M, et al. Fat specific protein 27: a novel lipid droplet protein that enhances triglyceride storage. J Biol Chem. 2007;282:34213–34218. doi: 10.1074/jbc.M707404200. [DOI] [PubMed] [Google Scholar]
- 20.Puri V, Ranjit S, Konda S, Nicoloro SMC, Straubhaar J, Chawla A, et al. Cidea is associated with lipid droplets and insulin sensitivity in humans. Proc Natl Acad Sci USA. 2008;105:7833–7838. doi: 10.1073/pnas.0802063105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang ZQ, Cefalu WT, Zhang XH, Yu Y, Qin J, Son L, et al. Human adenovirus type 36 enhances glucose uptake in diabetic and nondiabetic human skeletal muscle cells independent of insulin signaling. Diabetes. 2008;57:1805–1813. doi: 10.2337/db07-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang ZQ, Bell-Farrow AD, Sonntag WE, Cefalu WT. Effect of age and caloric restriction on insulin receptor binding and glucose transporter levels in aging rats. Exp Gerontology. 1997;32:671–684. doi: 10.1016/s0531-5565(97)00054-5. [DOI] [PubMed] [Google Scholar]
- 23.Pendergrass W, Wolf N, Poot M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry. 2004;A61:162–169. doi: 10.1002/cyto.a.20033. [DOI] [PubMed] [Google Scholar]
- 24.Hulver MW, Berggren JR, Cortright RN. Skeletal muscle lipid metabolism with obesity. Am J Physiol Endocrinol Metab. 2003;284:E741–E747. doi: 10.1152/ajpendo.00514.2002. [DOI] [PubMed] [Google Scholar]
- 25.Carling D. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 26.Osler ME, Zierath JR. Minireview: adenosine 5′-monophosphate-activated protein kinase regulation of fatty acid oxidation in skeletal muscle. Endocrinology. 2008;149:935. doi: 10.1210/en.2007-1441. [DOI] [PubMed] [Google Scholar]
- 27.Gong J, Sun Z, Li P. CIDE proteins and metabolic disorders. Curr Opin Lipidol. 2009;20:121–126. doi: 10.1097/MOL.0b013e328328d0bb. [DOI] [PubMed] [Google Scholar]
- 28.Keller P, Petrie JT, De Rose P, Gerin I, Wright WS, Chiang SH, et al. Fat-specific protein 27 regulates storage of triacylglycerol. J Biol Chem. 2008;283:14355–14365. doi: 10.1074/jbc.M708323200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Be’zaire V, Seifert EL, Harper M-E. Uncoupling protein-3: clues in an ongoing mitochondrial mystery. FASEB J. 2007;21:312–324. doi: 10.1096/fj.06-6966rev. [DOI] [PubMed] [Google Scholar]
- 30.Tarling E, Salter A, Bennett A. Transcriptional regulation of human SREBP-1c (sterol-regulatory-element-binding protein-1c): a key regulator of lipogenesis. Biochem Biochem Soc Trans. 2004;32 (Part 1):107–109. doi: 10.1042/bst0320107. [DOI] [PubMed] [Google Scholar]
- 31.Wong J, Quinn CM, Brown AJ. SREBP-2 positively regulates transcription of the cholesterol efflux gene, ABCA1, by generating oxysterol ligands for LXR. Biochem J. 2006;400:485–491. doi: 10.1042/BJ20060914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pai JT, Guryev O, Brown MS, Goldstein JL. Differential stimulation of cholesterol and unsaturated fatty acid biosynthesis in cells expressing individual nuclear sterol regulatory element-binding proteins. J Biol Chem. 1998;273:26138–26148. doi: 10.1074/jbc.273.40.26138. [DOI] [PubMed] [Google Scholar]
- 33.Shimano H. SREBPs: physiology and pathophysiology of the SREBP family. FEBS J. 2009;276:616–621. doi: 10.1111/j.1742-4658.2008.06806.x. [DOI] [PubMed] [Google Scholar]
- 34.Horton JD, Shimomura I, Brown MS, Hammer RE, Goldstein JL, Shimano H. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–2339. doi: 10.1172/JCI2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guillet-Deniau I, Pichard AL, Koné A, Esnous C, Nieruchalski M, Girard J, et al. Glucose induces de novo lipogenesis in rat muscle satellite cells through a sterol-regulatory-element-binding-protein- 1c-dependent pathway. J Cell Sci. 2004;117 (Part 10):1937–1944. doi: 10.1242/jcs.01069. [DOI] [PubMed] [Google Scholar]
- 36.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 37.Guan G, Dai P, Shechter I. Differential transcriptional regulation of the human squalene synthase gene by sterol regulatory element-binding proteins (SREBP) 1a and 2 and involvement of 5′ DNA sequence elements in the regulation. J Biol Chem. 1998;273:12526–12535. doi: 10.1074/jbc.273.20.12526. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Craddock L, Hong S, Liu ZM. AMP-activated protein kinase suppresses LXR-dependent sterol regulatory element-binding protein-1c transcription in rat hepatoma McA-RH7777 cells. J Cell Biochem. 2009;106:414–426. doi: 10.1002/jcb.22024. [DOI] [PubMed] [Google Scholar]