

Background: Collagen VI amino acid substitutions are common, and it is difficult to determine if they are pathogenic.
Results: COL6A2 p.D871N chains are abnormal and cannot assemble. Alternatively spliced chains lacking the mutation cannot functionally substitute.
Conclusion: COL6A2 p.D871N is a recessive mutation.
Significance: Protein studies reveal the consequences of amino acid substitutions in the globular domains of collagen VI.
Keywords: Collagen, Extracellular Matrix Protein, Genetic Disease, Mitochondria, Muscular Dystrophy, Bethlem Myopathy, Autosomal Recessive, Collagen VI, Mutation
Abstract
Bethlem myopathy and Ullrich congenital muscular dystrophy (UCMD) sit at opposite ends of a clinical spectrum caused by mutations in the extracellular matrix protein collagen VI. Bethlem myopathy is relatively mild, and patients remain ambulant in adulthood while many UCMD patients lose ambulation by their teenage years and require respiratory interventions. Dominant and recessive mutations are found across the entire clinical spectrum; however, recessive Bethlem myopathy is rare, and our understanding of the molecular pathology is limited. We studied a patient with Bethlem myopathy. Electron microscopy of his muscle biopsy revealed abnormal mitochondria. We identified a homozygous COL6A2 p.D871N amino acid substitution in the C-terminal C2 A-domain. Mutant α2(VI) chains are unable to associate with α1(VI) and α3(VI) and are degraded by the proteasomal pathway. Some collagen VI is assembled, albeit more slowly than normal, and is secreted. These molecules contain the minor α2(VI) C2a splice form that has an alternative C terminus that does include the mutation. Collagen VI tetramers containing the α2(VI) C2a chain do not assemble efficiently into microfibrils and there is a severe collagen VI deficiency in the extracellular matrix. We expressed wild-type and mutant α2(VI) C2 domains in mammalian cells and showed that while wild-type C2 domains are efficiently secreted, the mutant p.D871N domain is retained in the cell. These studies shed new light on the protein domains important for intracellular and extracellular collagen VI assembly and emphasize the importance of molecular investigations for families with collagen VI disorders to ensure accurate diagnosis and genetic counseling.
Introduction
Bethlem myopathy (MIM 158810) and Ullrich congenital muscular dystrophy (UCMD2; MIM 254090) sit at opposite ends of a clinical spectrum of muscular dystrophies caused by mutations in the extracellular matrix protein collagen VI (1, 2). Bethlem myopathy is a relatively mild disorder that usually becomes apparent in early childhood. Its characteristic features include proximal muscle weakness and wasting, and contractures commonly involving the finger, elbow, and ankle joints. Patients usually remain ambulant in adulthood and only rarely require mechanical ventilation at night. By contrast, UCMD is a serious congenital disorder. Severe muscle weakness, joint contractures and joint hypermobility mean that many patients lose independent ambulation by their teenage years and can succumb to early respiratory failure without effective interventions (2). A major challenge for diagnosing and counseling UCMD and Bethlem myopathy patients is that both recessive and dominant mutations are found in patients across the entire clinical spectrum (2–6) and so molecular diagnosis and an understanding of the biochemical consequences of mutations is crucial for each patient and their family.
Collagen VI is an abundant and widely expressed extracellular matrix protein that forms a microfibrillar network closely associated with basement membranes (7). The most common isoform is a heterotrimer containing one α1(VI), one α2(VI) and one α3(VI) subunit, encoded by the COL6A1, COL6A2, and COL6A3 genes, respectively. Humans have two recently identified additional chains α5(VI) and α6(VI) that are expressed at lower levels than the major isoform chains and show tissue specificity (8, 9). Mutations have not yet been identified in the α5(VI) and α6(VI) chains, and the function of these minor collagen VI chains is currently unknown. The collagen VI chains have a central triple helical region composed of Gly-X-Y amino acid repeats which are essential for the helical structure. This triple helix is flanked by globular N- and C-terminal regions; the predominant modules in these regions are ∼200 amino acid A-domains that show homology to the type A-domains of von Willebrand factor (10). The α1(VI), α2(VI), and α3(VI) chains each have two C-terminal A-domains (C1 and C2); α1(VI) and α2(VI) have 1 N-terminal A-domain (N1), while α3(VI) has up to 10 N-terminal A-domains depending on alternative splicing (11, 12). The correct structures of the triple helix and the globular A-domains are essential for collagen VI assembly which is a complex process beginning with the intracellular association of the three chains at the C-terminal end and folding of the triple helix to form the collagen VI monomer. Monomers come together to form antiparallel overlapping dimers which then align to form tetramers, the secreted form of collagen VI. In the final step secreted collagen VI tetramers assemble end-to-end into beaded extracellular matrix microfibrils (13).
Most structural collagen VI mutations are in the triple helical regions of the three chains and we have a good understanding of how the mutations affect collagen VI assembly and some insights into the genotype/phenotype relationships. Mutations toward the N terminus of the triple helix, including glycine substitutions that interrupt the Gly-X-Y repeat and in-frame deletions, are dominant, and the disease severity tends to correlate with the effect of the mutation on collagen VI assembly (14, 15). Mutations that disrupt tetramer and microfibril formation are likely to produce a more severe phenotype than those that prevent dimer formation or those that have little effect on microfibril formation (3, 15–17). Glycine substitutions toward the C-terminal end of the triple helix are recessively inherited (14, 18, 19). They prevent the chains assembling into triple helical monomers (20) leading to collagen VI haploinsufficiency in heterozygous carriers and a collagen VI muscular dystrophy in homozygous individuals. By contrast, much less is known about the consequences of amino acid substitutions in the N- and C-terminal globular A-domains. Some of these are recessive disease causing mutations (3), some are dominant mutations (17), and some are found in unaffected individuals and are unlikely to be pathogenic. However, in the absence of detailed biochemical studies it is often not possible to provide a molecular diagnosis for patients with amino acid substitutions in the A-domains or advise them about the expected course of their disorder.
A molecular diagnosis of recessive Bethlem myopathy has been reported in only three families (4, 6). A family with myosclerosis myopathy, a disorder with considerable clinical overlap with Bethlem myopathy, also has recessive collagen VI mutations (5). All these mutations are in COL6A2 and all involve changes in the C2 A-domain on at least one allele. Although some functional studies were undertaken in these families our understanding of the molecular pathology of recessive Bethlem myopathy mutations remains limited. We have identified a homozygous recessive COL6A2 C2 domain p.D871N mutation in a Bethlem myopathy patient and have done detailed studies in patient muscle biopsy and fibroblasts as well as transfected cells to understand the effect of the mutation on the mutant C2 domain and the mutant α2(VI) chain and the consequences for collagen VI intracellular and extracellular protein assembly.
EXPERIMENTAL PROCEDURES
Ethical Standards
This study was approved by the Royal Children's Hospital Human Research Ethics Committee in accordance with the National Health and Medical Research Council's National Statement on Ethical Conduct in Human Research (2007).
Muscle Biopsy and Staining
Frozen sections (7 μm) were cut from sartorius muscle biopsies and stained for cytochrome c oxidase (COX) or succinate dehydrogenase (SDH) according to standard protocols (21). Some sections were immunostained with a collagen VI antibody (clone VI-26, Millipore, 1:1000 dilution) following antigen retrieval at pH 9. Specific staining was visualized using an Envision FLEX detection and amplification system (Dako). For electron microscopy muscle samples were fixed in 2.5% cacodylate-buffered glutaraldehyde containing 1% osmium tetroxide, dehydrated in an alcohol series, and embedded in Epon 812. Ultrathin sections were observed in a Zeiss Libra 120 PLUS EF transmission electron microscope.
RNA and DNA Extraction, PCR, and Sequencing
Total RNA was extracted from control and patient fibroblasts using RNeasy (Qiagen). RNA was reverse transcribed using murine leukemia virus reverse transcriptase and an oligo(dT) primer according to the manufacturer's protocol (GeneAmp RNA PCR kit; Applied Biosystems). The resulting cDNA was used as a template for PCR amplification and sequencing of the entire coding regions of the α1(VI) and α2(VI) chains and the region coding for α3(VI) domains N2-C5, as described previously (17). Genomic DNA was extracted from patient fibroblasts and from blood samples obtained from both parents. COL6A2 exon 28 containing the mutation was PCR amplified and sequenced using the forward primer 5′-AATGGAAGGGCACAGGTGCG-3′ and the reverse primer 5′-CTTAGCACCATGGACGGGGT-3′.
Collagen VI Biosynthetic Labeling and Immunoprecipitation
Patient and control fibroblasts were grown to confluence in 10 cm2 dishes and incubated overnight in DMEM with 10% FBS containing 0.25 mm sodium ascorbate. Fibroblasts were radiolabeled for 18 h with 100 mCi/ml [35S]methionine (Tran35S-label 1032 Ci/mmol; ICN Pharmaceuticals, Inc.) in 750 μl of serum-free and methionine-free DMEM containing 0.25 mm sodium ascorbate. The cell and medium fractions were harvested, and collagen VI was immunoprecipitated with an α3(VI) N1 domain antibody and analyzed under reducing and nonreducing conditions as described previously (3). For pulse-chase analysis, the fibroblasts were radiolabeled, as above, for 30 min, and chased for 0, 0.5, 1, 2, 3, 4, 6, or 8 h in serum-free DMEM containing 0.25 mm sodium ascorbate and 10 mm l-methionine. Collagen VI was immunoprecipitated from cell and medium fractions with an α3(VI) N1 domain antibody as described above. In some experiments, fibroblasts were pre-incubated for 2 h with the protease inhibitors 5 μm Z-Leu-Leu-Leu-al (MG132, Sigma), 5 μm clasto-Lactacystin β-lactone (Sigma), 10 μm trans-epoxysuccinyl-l-leucylamido(4-guanidino)butane (E64, Sigma), or 1 μg/ml pepstatin A (Sigma), then labeled and chased in the presence of the same protease inhibitors. Cell layer samples were immunoprecipitated with an α2(VI) N1 domain antibody and analyzed as above.
Fibroblast Immunostaining
Fibroblasts were grown to confluence in four-well chamber slides and supplemented daily with 0.25 mm sodium ascorbate for 2 days. The extracellular matrix was stained for collagen VI, and the cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) using methods described previously (3).
Negative Staining Electron Microscopy
Confluent fibroblasts in 10-cm2 dishes were incubated for 18 h in serum-free DMEM containing 0.25 mm sodium ascorbate. The medium was collected and clarified by centrifugation. Microfibrils were deposited on a carbon-coated electron microscopy grid, stained for 30 s with 1% aqueous uranyl acetate, blotted, and quickly air dried. Grids were observed at 300kV using a Tecnai F30 (FEI, Eindhoven) and micrographs were acquired with an Utrascan 1000 (Gatan, Pleasanton, CA).
Constructing Expression Vectors Containing the α2(VI) Wild Type and p.D871N Mutant C2 Domains
Wild type and mutant α2(VI) C2 domains (nucleotides 2464–3057; amino acid residues 822–1019) were amplified from control and UCMD65 cDNA using a forward primer containing a 5′ PvuII restriction enzyme site followed by the StrepII tag sequence (22), and a reverse primer containing a 3′ HindIII restriction enzyme site. PCR products were purified and sequentially digested with PvuII and HindIII (New England Biolabs). Digested products were purified from a 1% (w/v) agarose gel and ligated into the episomal expression vector pCEP4-BM40-HisEK that had also been digested with PvuII and HindIII. This vector has previously been modified from the pCEP4 vector (Invitrogen) to contain a cleavable BM40 secretory signal peptide at the N terminus of the multiple cloning site (23). Selected clones were sequenced with a pCEP4 specific primer (Invitrogen) to ensure no errors had been introduced during PCR.
Expressing Recombinant α2(VI) C2 Domains
Human embryonic kidney 293-EBNA cells were grown in 10 cm2 wells in DMEM with 10% FBS until 90% confluent. Cells were transfected with wild type or mutant α2(VI) C2 domain expression constructs using FuGene HD (Roche Applied Sciences), then selected with 250 μg/ml Hygromycin B (Roche Applied Sciences). Cells were passaged twice to ensure complete selection, and then 2 × 106 cells were plated in 10 cm2 wells containing 2 ml DMEM with 10% FBS containing 250 μg/ml Hygromycin B. After 24 h, the medium was removed to 1.5-ml tubes, and protease inhibitors were added to the final concentrations: 20 mm NEM and 1 mm AEBSF. Medium was cleared of cellular debris by centrifugation at 13,000 × g for 30 min at 4 °C. The cell layer was rinsed twice in PBS, lysed in 500 μl of lysis buffer (150 mm NaCl; 50 mm Tris-HCl, pH 7.5; 5 mm EDTA; 1 mm AEBSF; 20 mm NEM; 1% Nonidet P-40), and fractionated by centrifugation at 13,000 × g for 30 min at 4 °C. The soluble cell lysate was retained, and the insoluble pellet was resuspended in 100 μl of urea/thiourea buffer (7 m urea, 2 m thiourea, 30 mm Tris pH 8.5, 4% CHAPS).
Western Blotting of Recombinant α2(VI) C2 Domains
Aliquots of the insoluble cell fraction, the soluble cell fraction, and the medium harvested from transfected 293-EBNA cells and an untransfected control were incubated at 65 °C for 10 min in 1× NuPage LDS sample buffer (Invitrogen) containing 2 m urea. Proteins were separated on NuPage Novex 4–12% Bis-Tris precast acrylamide gels (Invitrogen) alongside a Precision Plus Protein Standard (Bio-Rad) then transferred to nitrocellulose membranes (GE Healthcare). Membranes were blocked in 5% BSA (w/v) in PBS for 1 h at room temperature, probed with a 1:1000 dilution of Strep-tag antibody (Qiagen) for 1 h at room temperature, washed four times in PBS and then incubated at room temperature for 1 h in PBS containing a 1:10,000 dilution of horseradish peroxidase-conjugated anti-mouse antibody (Dako). Proteins were detected by incubating membranes with Amersham Biosciences Plus ECL Western blotting detection reagents (GE Healthcare Life Sciences) for 5 min and visualized by exposure to Amersham Biosciences high performance chemiluminescent film (GE Healthcare Life Sciences).
Protein Homology Modeling
A three-dimensional protein homology model of the human α2(VI) C2 domain (amino acids 831–1014) was generated using the EasyModeller 4.0 graphical user interface (24) to the MODELLER 9.12 program. The best template for the model, αXβ2 integrin (1N3Y.pdb), was identified by BLAST searching the PDB sequence database. The final model was visualized using Discovery Studio Visualizer (Accelrys.com).
RESULTS
Case Report
UCMD65 (Fig. 1), is a 61-year-old male with slowly progressive proximal limb muscle weakness. He has no family history of muscle disease but his parents are second-degree relatives. From the first to third decade our patient complained of clumsiness while playing sport or running but could walk and work normally. In his thirties, he developed gait disturbance with bilateral genu valgus deformities and underwent a tibial osteotomy. When he was 40 years old, he noticed decreased proximal muscle strength and muscle atrophy in his arms that made combing his hair difficult. At age 60, muscle weakness had progressed to his legs, he had difficulty walking, and had begun to use an orthopaedic walker. Simultaneously, he complained about joint contractures in his upper and lower limbs, which restricted his movements, and occasional difficulty swallowing liquids. A neurological examination at age 61 showed proximal weakness with symmetrical muscle atrophy involving all limbs, absent osteotendinous reflexes, and joint contractures in elbows, wrists, fingers, and knees. No skin alterations or sensory disturbances were noted. His gait was unsteady. Electromyography revealed reduced amplitude and frequency of the motor unit potentials in vastus medialis, iliopsoas, deltoids, and right tibialis anterior muscles. Blood tests were normal except for mildly elevated creatine phosphokinase (CK, 149–161 units/liter). Muscle magnetic resonance imaging (MRI) showed generalized muscle atrophy with diffuse muscular fatty infiltration, predominantly in proximal limb muscles. Our patient's clinical presentation thus suggested a mitochondrial myopathy, Emery-Dreifuss muscular dystrophy, or Bethlem myopathy.
FIGURE 1.
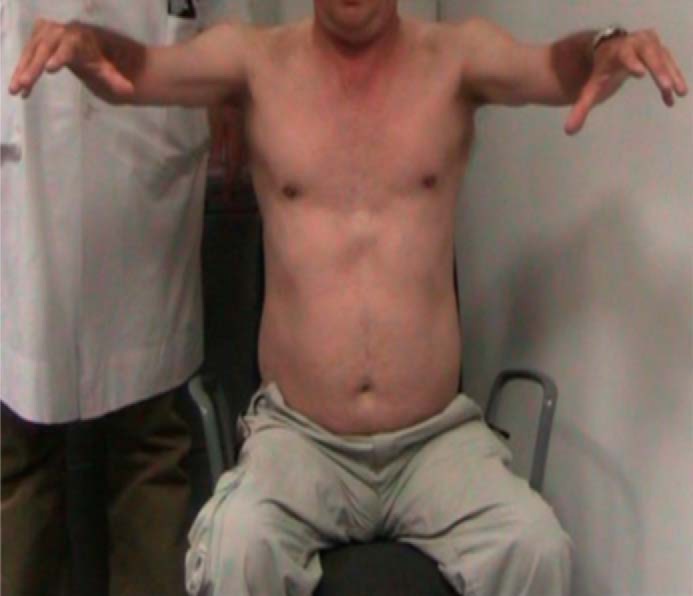
Clinical features. Patient UCMD65 at 61 years showing finger contractures and limb muscle atrophy.
Abnormal Mitochondria and Absent Collagen VI in UCMD65 Muscle Biopsy
To explore UCMD65 muscle pathology we stained frozen muscle sections for mitochondrial markers cytochrome c oxidase (COX) (Fig. 2a) and succinate dehydrogenase (SDH) (Fig. 2b). In normal muscle virtually all fibers have COX staining with type I fibers staining more intensely than type II reflecting their increased number of mitochondria. UCMD65 muscle had COX negative fibers indicating mitochondrial dysfunction; however, we did not see fibers with increased SDH staining suggesting the disorder was not a primary mitochondrial myopathy (Fig. 2b). Consistent with this, we screened for the common mitochondrial DNA mutations found in MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes), m.3243A>G, m.11084A>G, m.3271T>G, and m.3291T>G; MERRF syndrome (myoclonic epilepsy with ragged red fibers), m.8344A>G and m.8356T>C; and Leigh syndrome, m.8993T>C, and found no alterations (data not shown).
FIGURE 2.

UCMD65 muscle pathology. a, UCMD65 had COX negative fibers (*) indicative of a mitochondrial myopathy. b, SDH staining was normal suggesting there was no mitochondrial proliferation. c, electron microscopy revealed frequent mitochondrial paracrystalline inclusions (second panel from left). The panel on the right shows an enlargement of the boxed area to visualize the paracrystalline inclusions. Normal mitochondria in a control sample are shown in the panel on the left. d, immunostaining with a collagen VI specific antibody shows abundant collagen VI surrounding each muscle fiber in the control (nonspecific myopathy); however, collagen VI was not detected in UCMD65 muscle. UCMD65 also had many central nuclei (black arrows).
Transmission electron microscopy confirmed the mitochondrial abnormality detected by COX staining. We saw frequent mitochondria with paracristalline inclusions (Fig. 2c) which are typically found in muscle from patients with mitochondrial myopathies. Having ruled out the most common mitochondrial myopathies, we reasoned that since mitochondria with abnormal cristae are present in skeletal muscle from collagen VI knock-out mice (25), UCMD65 may have a collagen VI disorder, and so we stained skeletal muscle with a collagen VI antibody. There was abundant collagen VI staining around each muscle fiber in the control biopsy (nonspecific myopathy) but collagen VI was not detected in UCMD65 muscle (Fig. 2d), confirming that this patient has a collagen VI disorder. UCMD65's clinical presentation, at the mild end of the collagen VI disorder spectrum, indicates that he has Bethlem myopathy; however, absent collagen VI staining is usually associated with severe Ullrich congenital muscular dystrophy (26–28) and has not been reported in Bethlem myopathy.
UCMD65 Has a Homozygous COL6A2 Mutation
To characterize the mutation responsible for Bethlem myopathy in this patient we RT-PCR amplified and sequenced the entire coding regions of the α1(VI) and α2(VI) chains and the region coding for domains N3-C5 of the α3(VI) chain. UCMD65 had a homozygous c.2611G>A mutation in α2(VI) causing a p.D871N amino acid substitution. We confirmed the mutation by sequencing genomic DNA PCR products from UCMD65 and his unaffected mother and father who were both heterozygous carriers of the mutation (Fig. 3a). The mutation is in the α2(VI) C2 domain and will be present in the most common α2(VI) splice variant, but not the minor α2(VI) C2a form which has an alternative smaller C2 domain (Fig. 3b).
FIGURE 3.
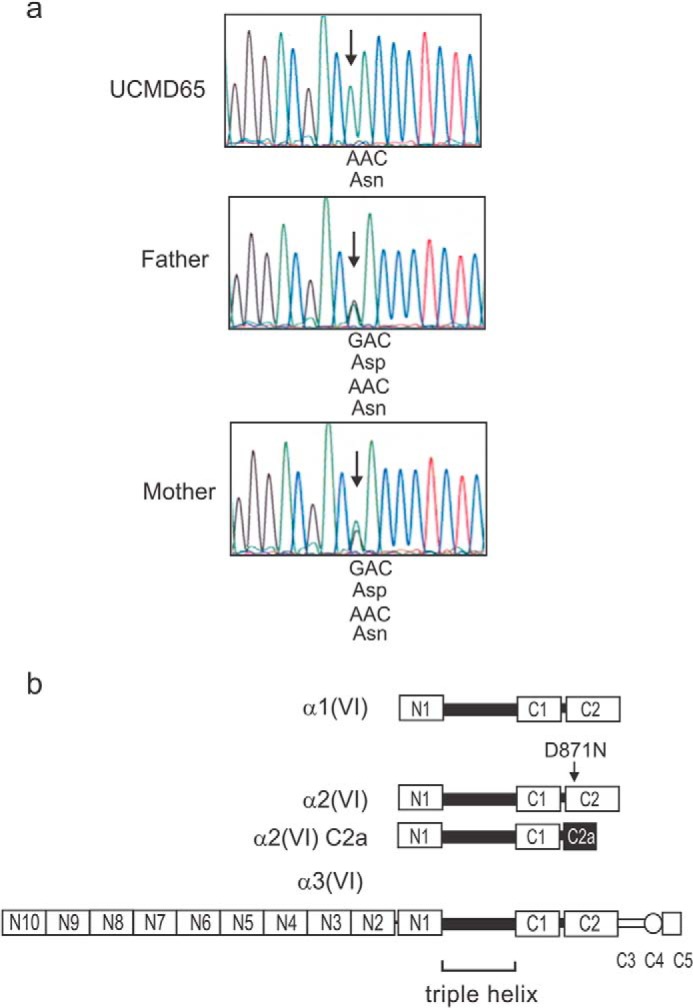
UCMD65 has a homozygous recessive α2(VI) mutation. a, genomic DNA PCR and sequencing. UCMD65 has a homozygous c.2611G>A change causing a D871N amino acid substitution. The unaffected parents, who are second degree relatives, are both heterozygous carriers of the mutation. b, schematic representation of the three collagen VI chains, α1(VI), α2(VI), and α3(VI) showing the central triple helix and the N- and C-terminal globular A-domains (N10-N1, C1, and C2). The α3(VI) chain has three additional C-terminal domains (C3-C5). A minor splice variant of the α2(VI) chain, α2(VI) C2a, which has a shorter C-terminal domain than the major α2(VI) chain, is also shown. One α1(VI), one α2(VI) and one α3(VI) chain associate together to form the collagen VI triple helical monomer, the building blocks of collagen VI dimers and tetramers.
The α2(VI) p.D871N Mutation Interferes with Collagen VI Assembly, Secretion, and Microfibril Formation
We then examined the effects of the mutation on collagen VI production by UCMD65 fibroblasts. In overnight biosynthetic labeling and immunoprecipitation experiments we consistently found that UCMD65 cells secreted less collagen VI than control fibroblasts (Fig. 4, a and b); however, we noted that under reducing conditions, which allow us to visualize the individual collagen VI chains, the α2(VI) C2a band was a similar intensity in control and UCMD65 medium (Fig. 4a). This suggested that the alternatively spliced α2(VI) C2a chains were able to assemble normally with α1(VI) and α3(VI) to form collagen VI monomers, but assembly and secretion of α2(VI) chains with the p.D871N mutation were impaired. Consistent with this, using quantitative RT-PCR we confirmed that the α1(VI), α2(VI) C2, and α2(VI) C2a mRNAs were all expressed at normal levels (data not shown). As in control cells, collagen VI tetramers were the major secreted form of collagen VI (Fig. 4b) and there was no intracellular accumulation of collagen VI triple helical monomers or dimers (Fig. 4b) indicating that the mutation most likely prevented incorporation of the chains into monomers. Outside the cell collagen VI tetramers connect end-to-end to form microfibrils which can be deposited into the extracellular matrix surrounding cells. Control fibroblasts accumulate an extensive collagen VI extracellular matrix but, by contrast, the UCMD65 extracellular matrix contained only a very small amount of collagen VI (Fig. 4c).
FIGURE 4.
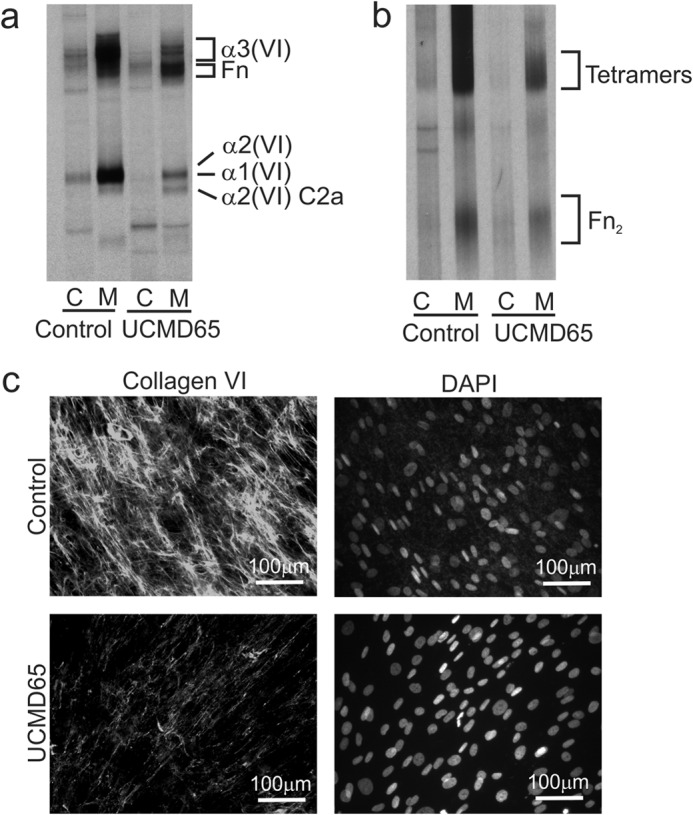
Collagen VI production in UCMD65 fibroblasts. Control and UCMD65 fibroblasts were labeled overnight with [35S]methionine and collagen VI was immunoprecipitated from the cell (C) and medium (M) with an α3(VI) N1 domain antibody. a, immunoprecipitated collagen VI was analyzed under reducing conditions on 3–8% Tris acetate gels to resolve the α1(VI), α2(VI), and α3(VI) chains. When compared with the control, UCMD65 fibroblasts secrete much less collagen VI, and the α2(VI) C2a chain is a major component of the secreted collagen VI. Contaminating fibronectin (Fn) is indicated. b, non-reduced immunoprecipitated collagen VI was analyzed on 2.4% acrylamide, 0.5% agarose gels to visualize collagen VI monomers, dimers, and tetramers. Collagen VI tetramers and fibronectin dimers (Fn2) are indicated. There was no accumulation of collagen VI monomers or dimers in UCMD65 cells indicating that dimer and tetramer assembly were not affected. c, immunostaining of the collagen VI matrix deposited by fibroblasts. Control and UCMD65 fibroblasts were grown for 2 days post-confluence in the presence of sodium ascorbate and stained with a collagen VI antibody. The cell nuclei were stained with DAPI. Control fibroblasts deposited an extensive collagen VI network; however, the collagen VI matrix was fine and sparse in UCMD65 cultures.
Having established that the homozygous α2(VI) p.D871N mutation ultimately leads to a severe collagen VI matrix deficiency, we then examined all the stages of collagen VI assembly in detail to establish the molecular mechanisms responsible for this deficiency. We first used pulse-chase experiments where cells were biosynthetically radiolabeled for 30 min, chased for up to 8 h and collagen VI immunoprecipitated with an α3(VI) chain antibody, to establish the time-course of collagen VI intracellular assembly and secretion. In control cells, α1(VI) and α2(VI) chains were co-immunoprecipitated with α3(VI) from 0.5 h (Fig. 5a). On non-reduced gels collagen VI triple helical monomers, dimers and tetramers were all present at 0.5 h of chase (Fig. 5b) indicating that once the three chains come together further assembly is rapid. Collagen VI was present in the medium at 1 h of chase and most of the collagen VI synthesized during the 30 min pulse was secreted by two 2 hours of chase (Fig. 5, a and b). By contrast, collagen VI assembly and secretion was reduced in UCMD65. Some α1(VI) and α2(VI) C2a chains co-immunoprecipitated with α3(VI) at 0.5 h of chase (Fig. 5c) but these bands were very feint when compared with the α3(VI) chains and the relative intensities of these bands in the control cells (compare intracellular α3(VI) with α1(VI) and α2(VI) in Fig. 5, a and c). These data confirm that assembly of the mutant α2(VI) chains into disulfide bonded monomers with α1(VI) and α2(VI) was indeed disrupted in UCMD65 and suggest that assembly of the minor α2(VI)-C2a chain without the mutation proceeded normally.
FIGURE 5.

Pulse-chase analysis of collagen VI assembly and secretion. Control (a and b) and UCMD65 (c and d) fibroblasts were labeled with [35S]methionine for 30 min and chased for up to 8 h. Collagen VI in the cell and medium was immunoprecipitated with an α3(VI) N1 domain antibody and analyzed under reducing conditions on 3–8% Tris acetate gels to resolve the α1(VI), α2(VI), and α3(VI) chains (a and c) and non-reducing conditions to compare chain assembly (b and d). In the control, α1(VI) and α2(VI) chains were co-precipitated with α3(VI) from 0.5 h and collagen VI appeared in the medium from 1 h of chase. By contrast, in UCMD 65 co-precipitating α1(VI) and α2(VI) chains were barely visible in the cell throughout the chase period, and small amounts of secreted collagen VI first appeared in the medium at 2 h of chase. To allow the collagen VI produced by UCMD65 to be clearly seen the gels shown in panels c and d were exposed for three times longer than the control gels in panels a and b. Collagen VI α1(VI), α2(VI), α3(VI), and α2(VI) C2a chains, and contaminating fibronectin (Fn) are indicated in panels a and c. Collagen VI monomers, dimers, tetramers, and contaminating fibronectin dimers (Fn2) are marked in panels b and d.
Following secretion, collagen VI tetramers associate end-to-end to form microfibrils. We visualized and quantitated microfibril assembly in the medium of control and UCMD65 fibroblasts using negative staining electron microscopy (Fig. 6, a–d). Around 60% of the collagen VI microfibrils in the medium of control cells contained more than 5 tetramers (Fig. 6e); however, there were very few of these large microfibrils in UCMD65 medium and around 40% of the microfibrils contained only a single tetramer (Fig. 6, b–e), indicating that end-to-end assembly of the tetramers secreted by UCMD65 fibroblasts was severely compromised.
FIGURE 6.
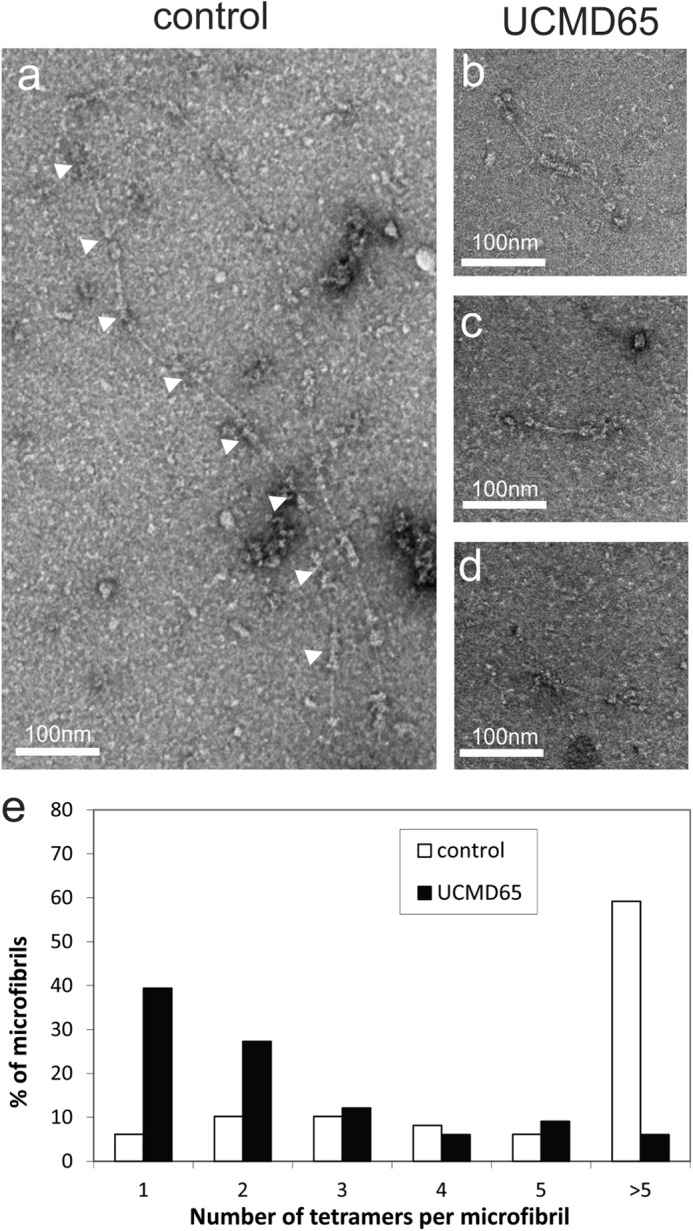
Collagen VI microfibril formation is compromised in UCMD65. Negative staining electron micrographs of collagen VI microfibrils in the medium from control (a) and UCMD65 fibroblasts (b–d). The white arrowheads in a indicate end-to-end association of the tetramers. e, collagen VI secreted into the medium of control (white bars) and UCMD65 (black bars) fibroblast was visualized by negative staining electron microscopy, and the ability of the tetramers to associate end-to-end was quantitated. The occurrence of microfibrils containing 1–5 or >5 tetramers is shown as a percentage of the total number of microfibrils. Tetramer-tetramer association was severely compromised in UCMD65 compared with the control.
α2(VI) p.D871N Chains Are Degraded by the Proteasome
We then looked at the fate of the mutant α2(VI) p.D871N chains in pulse-chase experiments. UCMD65 fibroblasts were preincubated for 2 h with and without inhibitors of lysosomal or proteasomal protein degradation, biosynthetically radiolabeled for 30 min, then chased for up to 6 h. The mutant α2(VI) p.D871N and alternatively spliced α2(VI)-C2a chains were precipitated with an antibody raised to the α2(VI) N1 domain and analyzed under reducing conditions. In the absence of protease inhibitors the mutant α2(VI) chains were almost completely eliminated by 6 h of chase (Fig. 7); however, mutant α2(VI) chains were protected by MG132 and clasto-Lactocystin β-lactone (Fig. 7) indicating that they are normally degraded by proteasomal proteases. The lysosomal protease inhibitors E64 and pepstatin A did not protect the mutant α2(VI) p.D871N chains from degradation. This is consistent with the conclusion that mutant α2(VI) p.D871N chains are selectively degraded by the proteasome.
FIGURE 7.
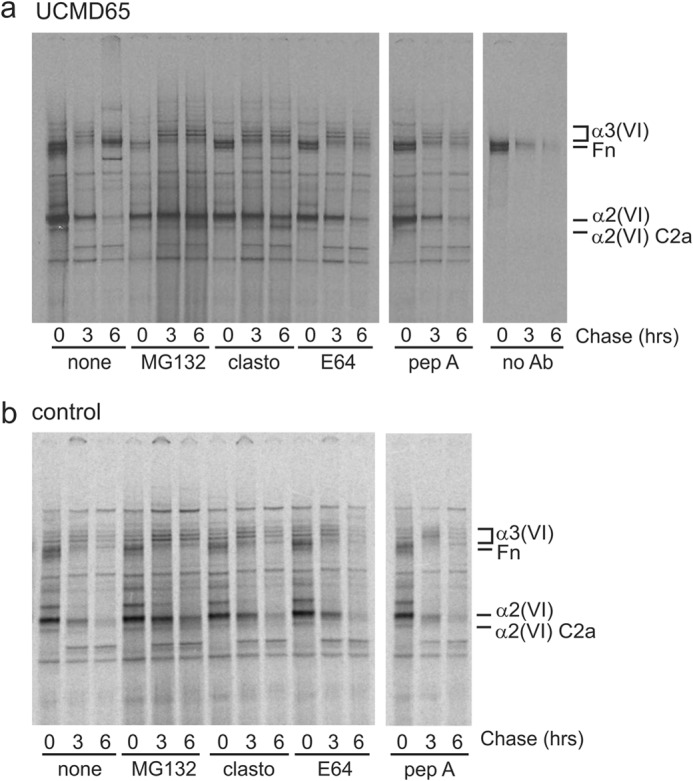
Pulse-chase analysis of α2(VI) chain degradation. UCMD65 (a) and control fibroblasts (b) were labeled with [35S]methionine for 30 min and chased for up to 6 h. Intracellular collagen VI was immunoprecipitated with an α2(VI) N1 domain antibody that recognizes the α2(VI) and the α2-C2a chains, and analyzed under reducing conditions on 3–8% Tris acetate gels. When used, proteasome inhibitors MG132 and clasto-Lactocystin β-lactone (clasto), and lysosomal enzyme inhibitors E64 or pepstatin A (pep A) were added 2 h before the experiment and included in the labeling and chase medium. In the absence of inhibitors (none) UCMD65 α2(VI) chains are almost completely eliminated by 6 h of chase; however, α2(VI) chain degradation is almost completely prevented by the proteasome inhibitors MG132 and clasto-Lactocystin β-lactone. The lysosomal protease inhibitors E64 and pepstatin A do not protect UCMD65 α2(VI) chains from degradation. The no primary antibody control (no Ab) lanes show the fibronectin that binds non-specifically to the protein A-Sepharose beads. Protease inhibitors do not affect clearance of α2(VI) chains from control fibroblasts.
Mutant α2(VI) C2 Domains Are Not Secreted
To explore the effect of the p.D871N mutation on protein folding we expressed recombinant Strep II tagged wild-type and mutant α2(VI) C2 domains in HEK293-EBNA cells. We separated secreted proteins, soluble intracellular proteins and intracellular proteins that had formed aggregates by SDS-PAGE and detected recombinant C2 domains by immunoblotting with a Strep II antibody. Recombinant wild-type domains were efficiently secreted as expected for a correctly folded protein (Fig. 8a). By contrast, C2 domains containing the p.D871N mutation were predominantly found in the soluble intracellular fraction and were not detected in the medium. This indicates that the mutant domains are recognized as abnormal by endoplasmic reticulum quality control mechanisms and are retained in the cell.
FIGURE 8.
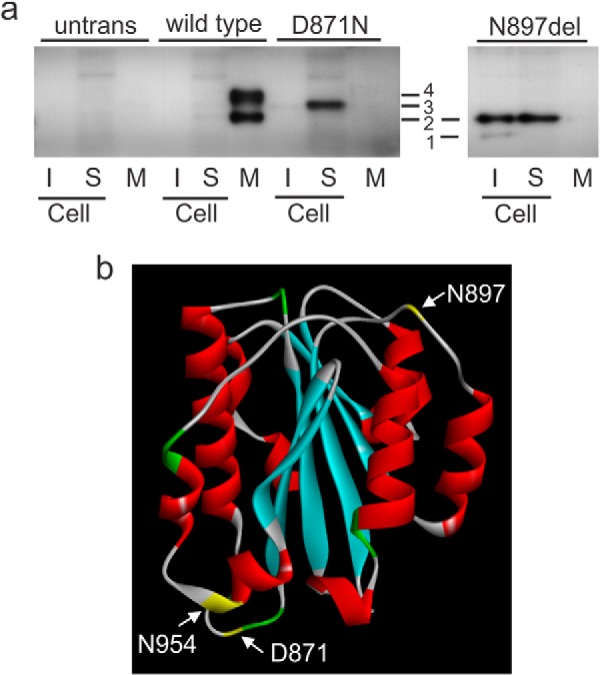
The mutant α2(VI) C2 domain is misfolded. a, StrepII-tagged wild-type C2 domains, mutant C2 D871N domains, and mutant C2 N879del domains were expressed in 293-EBNA cells. Proteins in the insoluble cell fraction (I), soluble cell fraction (S), and the medium (M) were separated on acrylamide gels, transferred to nitrocellulose, and the recombinant proteins detected using a Strep-tag antibody. Similar fractions from untransfected cells (untrans) were also analyzed. Wild-type C2 domains are predominantly in the medium indicating that they have folded correctly. By contrast, D871N C2 domains are not secreted and are mainly in the soluble intracellular fraction, indicating that the domains are misfolded and retained in the endoplasmic reticulum. The C2 domain contains two potential N-linked glycosylation sites, N897LT and N954DS. When the N897LT site is deleted (N879del) only one N-linked glycosylation is possible and cells expressing this C2 domain show two bands, an unglycosylated form (band 1) and a form substituted with one N-linked oligosaccharide (band 2). The N879del C2 domain is misfolded and retained intracellularly in both the soluble and insoluble fractions. The single D871N band (band 3) migrated more slowly than band two suggesting it has two N-linked oligosaccharides. This band can also be seen in the wild type soluble fraction on longer exposures. The larger wild-type C2 domain (band 4) migrated more slowly than band 3 with 2 N-linked oligosaccharides; as this protein was in the medium it is likely that one or both of the oligosaccharides has been modified by the addition of further sugars as the protein was transported through the Golgi. b, homology model of the wild-type α2(VI) C2 domain with the amino acids colored according to structure with β-sheets in blue and α-helices in red. Residues Asn-897 and Asn-954 (yellow) are located in loop regions on the outside of the folded protein structure consistent with our data showing that both can be substituted with N-linked oligosaccharides. The D871N mutation (yellow) is also in an external loop.
The C2 domain contains 2 potential N-linked glycosylation sites, N897 in the sequence NLT and N954 in the sequence NDS, both are located in loop regions on the outside of the folded protein structure (Fig. 8b) and our data indicate that both can be substituted with an N-linked carbohydrate. When we expressed C2 domains with a deletion of Asn-897 we saw an unglycosylated form and a larger form that is glycosylated at N954 (Fig. 8a). We also detected two forms of the wild-type C2 domain (Fig. 8a). The smaller faster migrating band co-migrated with the glycosylated Asn-897 band indicating that it is carrying one N-linked oligosaccharide. The single D871N band migrated more slowly than the smaller wild-type band suggesting it is glycosylated at both Asn-879 and Asn-954, and this band can also be seen in the wild-type soluble fraction on longer exposures (not shown). The larger wild-type C2 domain migrated more slowly than the form containing two N-linked oligosaccharides. As this protein is in the medium it is likely that one or both of the high mannose N-linked oligosaccharides that were added in the endoplasmic reticulum have been modified by the addition of further sugars as the protein was transported through the Golgi. This form was not seen in the D871N cells and this is further confirmation that the C2 D871N domain is recognized as abnormal and retained in the endoplasmic reticulum.
DISCUSSION
To our knowledge this is the first patient with a molecular diagnosis of Bethlem myopathy shown to have mitochondria with paracristalline inclusions by electron microscopy. Similar mitochondrial changes are seen in collagen VI knock-out mice (25), and have also been reported in a patient with an autosomal dominant benign limb-girdle myopathy with contractures who fitted the clinical criteria for Bethlem myopathy (29). This patient did not have a molecular diagnosis as the report was prior to the discovery that collagen VI mutations were the underlying cause of Bethlem myopathy (30). The patient also had COX negative fibers similar to our patient and had similar mild and very slowly progressing muscle wasting and weakness. Together these data suggest that COX negative fibers and structural mitochondrial abnormalities might be common in Bethlem myopathy muscle.
UCMD65, has a homozygous COL6A2 p.D871N mutation which is in the α2(VI) C2 A-domain. Our data show that the mutant α2(VI) C2 domain is misfolded and the mutant α2(VI) chains are not able to assemble normally with α1(VI) and α3(VI). When compared with control fibroblasts, UCMD65 fibroblasts secrete much less collagen VI and the minor alternatively spliced α2(VI) C2a product, which does not contain the mutation, is a major component of the UCMD65 secreted assemblies. While the α2(VI) C2a chain is clearly able to assemble with α1(VI) and α2(VI), the kinetics are slower than assembly of the major α2(VI) splice form and collagen VI secretion is delayed by approximately one hour. The α2(VI) p.D871N mutation was reported previously in a patient with recessive Bethlem myopathy who had a premature stop codon that resulted in nonsense-mediated mRNA decay on the other COL6A2 allele (4). In that case the authors showed collagen VI secretion was reduced but didn't detect the α2(VI) C2a chain when they immunoprecipitated collagen VI with an α1(VI) antibody and then immunoblotted with an antibody that recognizes all three collagen VI chains. Two factors could explain these seemingly contradictory findings. Firstly, the patient in the earlier report would have been making half the normal amount of the α2(VI) C2a chain because of mRNA decay of the products from one allele, and secondly, it is likely that radiolabeling is a more sensitive technique for detecting minor protein products than immunoblotting.
Our data fit closely with the demonstrated effects of other α2(VI) C2 amino acid substitutions on collagen VI biosynthesis and intracellular assembly. For example, a homozygous α2(VI) p.R876S mutation destabilizes the mutant chain and the small amount of secreted collagen VI contains the α2(VI) C2a chain (31). We reported a patient with recessive α2(VI) C2 domain mutations, p.L873P and p.N897del, and showed that the mutant chains were unstable and only small amounts of collagen VI were secreted (3). At the time we didn't recognize that the secreted collagen VI contained the α2(VI) C2a chain but more recent analyses clearly show that this is the case (data not shown). Our data showing that the p.N897del C2 domain is misfolded and retained in the cell suggests that the consequences of all these α2(VI) C2 domain amino acid substitutions, p.L873P, p.N897del and p.R876S are very similar to the p.D871N mutation reported here. It is surprising then, that the clinical presentation of these patients is quite different. UCMD65 reported here (p.D871N; p.D871N) and BM2 (p.Q819X; p.D871N; (4)) both have classical mild Bethlem myopathy and retained independent ambulation well into adulthood, while UCMD2 (p.L873P; p.N897del; (3)) and UC15 (p.R876S; p.R876S; (31)) had a more severe disorder consistent with UCMD and lost ambulation at 10 and 11 years of age, respectively. While we don't have a definitive explanation for why mutations that have a similar effect on collagen VI biosynthesis and assembly have different clinical outcomes it is possible that uncharacterized modifying genes are expressed differently in these patients or that subtle differences in the ability of the mutant chains to be incorporated into tetramers and microfibrils modulate the phenotype.
The role of the minor α2(VI) C2a splice variant in normal tissues has not yet been defined. It assembles with the other collagen VI chains into triple helical monomers more slowly than α2(VI) chains and outside the cell tetramers containing the C2a splice variant cannot assemble end-to-end into microfibrils efficiently and only small amounts of this collagen VI isoform are deposited into the extracellular matrix. These data emphasize the critical importance of the α2(VI) C2 domain in monomer and microfibril assembly. Further, electron microscopy of the collagen VI fibrils deposited by the patient with compound heterozygous mutations, α2(VI) p.Q819X and p.D871N, in which only α2(VI) C2a chains are likely to be incorporated into secreted collagen VI molecules, revealed that the microfibrils in the extracellular matrix did not develop the extensive interconnections seen control microfibrils (4). This finding suggests that the α2(VI) C2a domain is also important in promoting interactions between collagen VI microfibrils and raises the possibility that inclusion of this domain in collagen VI tetramers might modulate collagen VI microfibril formation and interactions with other extracellular matrix molecules.
We explored how the p.D871N mutation and N897del mutations affected the structure and trafficking of the α2(VI) C2 domain by expressing wild-type and mutant C2 domains in transfected mammalian cells. Our experiments are the first confirmation that the two consensus sites for N-linked glycosylation in the C2 domain are both used. While wild-type C2 domains were efficiently secreted, both mutant domains were retained in the cell suggesting that they are misfolded and held in the ER by quality control mechanisms. This probable misfolding likely explains why the mutant α2(VI) chains containing the abnormal domain are not able to assemble with α1(VI) and α3(VI) chains. Interestingly, collagen VI monomer assembly proceeds in a patient with an α2(VI) C1 domain mutation (p.V619_I620del2) which leads to misfolding of the C1 domain (32). The clear implication from these studies is that the α2(VI) C2 domain might be more important in the interactions leading to triple helix formation than the C1 domain which is immediately C-terminal to the triple helical domain.
Mutant misfolded proteins are removed from the ER by ER-associated degradation (ERAD) and/or autophagy (33–36). We investigated the fate of the mutant α2(VI) chains and found they were eliminated from patient fibroblasts by proteolysis that could be prevented by two drugs that inhibit proteasomal degradation. Lysosomal protease inhibitors did not slow degradation of the mutant α2(VI) chains. This indicates that the mutant α2(VI) chains likely remain soluble and are transported out of the ER for degradation by the proteasome (ERAD), and that the alternative autophagy/lysosomal pathway is not activated. Collagen type I proα1(I) chains that are unassembled because they have a mutation in the C-terminal propeptide, the domain critical for intracellular chain association, are also retro-translocated to the cytoplasm for degraded by the proteasome (37, 38). This may be a common pathway for degrading collagen subunits with mutations that prevent the initial step of collagen assembly, chain recognition and triple helix formation.
Our detailed study into the functional consequences of this α2(VI) C2 domain amino acid substitution p.D871N sheds new light on the requirements for intracellular and extracellular collagen VI assembly and the cellular degradation pathway responsible for eliminating the mutant chains that are unable to assemble. It also emphasizes the critical importance of molecular diagnosis for each family as patients with very different clinical presentations can have similar mutations that affect collagen VI protein in similar ways. Further detailed studies on molecular mechanisms of the collagen VI disorders are needed to understand the factors contributing to this clinical enigma.
Acknowledgment
We thank Anastasia Kurniawan for technical assistance.
This work was supported in part by Project Grant 491252 and Research Fellowship 436903 (to S. R. L.) from the National Health & Medical Research Council of Australia, the Murdoch Childrens Research Institute, and the Victorian Government's Operational Infrastructure Support Program.
- UCMD
- Ullrich congenital muscular dystrophy
- DAPI
- 4′,6-diamidino-2-phenylindole
- COX
- cytochrome c oxidase
- SDH
- succinate dehydrogenase.
REFERENCES
- 1. Allamand V., Briñas L., Richard P., Stojkovic T., Quijano-Roy S., Bonne G. (2011) ColVI myopathies: where do we stand, where do we go? Skelet Muscle 1, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bönnemann C. G. (2011) The collagen VI-related myopathies: muscle meets its matrix. Nat. Rev. Neurol. 7, 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baker N. L., Mörgelin M., Peat R., Goemans N., North K. N., Bateman J. F., Lamandé S. R. (2005) Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum. Mol. Genet. 14, 279–293 [DOI] [PubMed] [Google Scholar]
- 4. Gualandi F., Urciuolo A., Martoni E., Sabatelli P., Squarzoni S., Bovolenta M., Messina S., Mercuri E., Franchella A., Ferlini A., Bonaldo P., Merlini L. (2009) Autosomal recessive Bethlem myopathy. Neurology 73, 1883–1891 [DOI] [PubMed] [Google Scholar]
- 5. Merlini L., Martoni E., Grumati P., Sabatelli P., Squarzoni S., Urciuolo A., Ferlini A., Gualandi F., Bonaldo P. (2008) Autosomal recessive myosclerosis myopathy is a collagen VI disorder. Neurology 71, 1245–1253 [DOI] [PubMed] [Google Scholar]
- 6. Foley A. R., Hu Y., Zou Y., Columbus A., Shoffner J., Dunn D. M., Weiss R. B., Bönnemann C. G. (2009) Autosomal recessive inheritance of classic Bethlem myopathy. Neuromuscul. Disord. 19, 813–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Keene D. R., Engvall E., Glanville R. W. (1988) Ultrastructure of type VI collagen in human skin and cartilage suggests an anchoring function for this filamentous network. J. Cell Biol. 107, 1995–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fitzgerald J., Rich C., Zhou F. H., Hansen U. (2008) Three novel collagen VI chains, α4(VI), α5(VI), and α6(VI). J. Biol. Chem. 283, 20170–20180 [DOI] [PubMed] [Google Scholar]
- 9. Gara S. K., Grumati P., Urciuolo A., Bonaldo P., Kobbe B., Koch M., Paulsson M., Wagener R. (2008) Three novel collagen VI chains with high homology to the α3 chain. J. Biol. Chem. 283, 10658–10670 [DOI] [PubMed] [Google Scholar]
- 10. Whittaker C. A., Hynes R. O. (2002) Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol. Biol. Cell 13, 3369–3387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stokes D. G., Saitta B., Timpl R., Chu M. L. (1991) Human α3(VI) collagen gene. Characterization of exons coding for the amino-terminal globular domain and alternative splicing in normal and tumor cells. J. Biol. Chem. 266, 8626–8633 [PubMed] [Google Scholar]
- 12. Zanussi S., Doliana R., Segat D., Bonaldo P., Colombatti A. (1992) The human type VI collagen gene. mRNA and protein variants of the α3 chain generated by alternative splicing of an additional 5-end exon. J. Biol. Chem. 267, 24082–24089 [PubMed] [Google Scholar]
- 13. Timpl R., Chu M. L. (1994) Microfibrillar collagen type VI in Extracellular Matrix Assembly and Structure (Yurchenco P. D., Birk D., Mecham R. P., eds), pp. 207–242, Academic Press, Orlando [Google Scholar]
- 14. Butterfield R. J., Foley A. R., Dastgir J., Asman S., Dunn D. M., Zou Y., Hu Y., Flanigan K. M., Swoboda K. J., Winder T. L., Weiss R. B., Bonnemann C. G. (2013) Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum. Mutat. 34, 1558–1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pace R. A., Peat R. A., Baker N. L., Zamurs L., Mörgelin M., Irving M., Adams N. E., Bateman J. F., Mowat D., Smith N. J., Lamont P. J., Moore S. A., Mathews K. D., North K. N., Lamandé S. R. (2008) Collagen VI glycine mutations: perturbed assembly and a spectrum of clinical severity. Ann. Neurol. 64, 294–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamandé S. R., Shields K. A., Kornberg A. J., Shield L. K., Bateman J. F. (1999) Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J. Biol. Chem. 274, 21817–21822 [DOI] [PubMed] [Google Scholar]
- 17. Baker N. L., Mörgelin M., Pace R. A., Peat R. A., Adams N. E., Gardner R. J., Rowland L. P., Miller G., De Jonghe P., Ceulemans B., Hannibal M. C., Edwards M., Thompson E. M., Jacobson R., Quinlivan R. C., Aftimos S., Kornberg A. J., North K. N., Bateman J. F., Lamandé S. R. (2007) Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann. Neurol. 62, 390–405 [DOI] [PubMed] [Google Scholar]
- 18. Jimenez-Mallebrera C., Maioli M. A., Kim J., Brown S. C., Feng L., Lampe A. K., Bushby K., Hicks D., Flanigan K. M., Bonnemann C., Sewry C. A., Muntoni F. (2006) A comparative analysis of collagen VI production in muscle, skin and fibroblasts from 14 Ullrich congenital muscular dystrophy patients with dominant and recessive COL6A mutations. Neuromuscul. Disord. 16, 571–582 [DOI] [PubMed] [Google Scholar]
- 19. Briñas L., Richard P., Quijano-Roy S., Gartioux C., Ledeuil C., Lacène E., Makri S., Ferreiro A., Maugenre S., Topaloglu H., Haliloglu G., Pénisson-Besnier I., Jeannet P. Y., Merlini L., Navarro C., Toutain A., Chaigne D., Desguerre I., de Die-Smulders C., Dunand M., Echenne B., Eymard B., Kuntzer T., Maincent K., Mayer M., Plessis G., Rivier F., Roelens F., Stojkovic T., Taratuto A. L., Lubieniecki F., Monges S., Tranchant C., Viollet L., Romero N. B., Estournet B., Guicheney P., Allamand V. (2010) Early onset collagen VI myopathies: Genetic and clinical correlations. Ann. Neurol. 68, 511–520 [DOI] [PubMed] [Google Scholar]
- 20. Lamandé S. R., Mörgelin M., Selan C., Jöbsis G. J., Baas F., Bateman J. F. (2002) Kinked collagen VI tetramers and reduced microfibril formation as a result of Bethlem myopathy and introduced triple helical glycine mutations. J. Biol. Chem. 277, 1949–1956 [DOI] [PubMed] [Google Scholar]
- 21. Old S. L., Johnson M. A. (1989) Methods of microphotometric assay of succinate dehydrogenase and cytochrome c oxidase activities for use on human skeletal muscle. Histochem. J. 21, 545–555 [DOI] [PubMed] [Google Scholar]
- 22. Schmidt T. G., Skerra A. (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 [DOI] [PubMed] [Google Scholar]
- 23. Wilson R., Freddi S., Bateman J. F. (2002) Collagen X chains harboring Schmid metaphyseal chondrodysplasia NC1 domain mutations are selectively retained and degraded in stably transfected cells. J. Biol. Chem. 277, 12516–12524 [DOI] [PubMed] [Google Scholar]
- 24. Kuntal B. K., Aparoy P., Reddanna P. (2010) EasyModeller: A graphical interface to MODELLER. BMC Res. Notes 3, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Irwin W. A., Bergamin N., Sabatelli P., Reggiani C., Megighian A., Merlini L., Braghetta P., Columbaro M., Volpin D., Bressan G. M., Bernardi P., Bonaldo P. (2003) Mitochondrial dysfunction and apoptosis in myopathic mice with collagen VI deficiency. Nat. Genet. 35, 367–371 [DOI] [PubMed] [Google Scholar]
- 26. Higuchi I., Shiraishi T., Hashiguchi T., Suehara M., Niiyama T., Nakagawa M., Arimura K., Maruyama I., Osame M. (2001) Frameshift mutation in the collagen VI gene causes Ullrich's disease. Ann. Neurol. 50, 261–265 [DOI] [PubMed] [Google Scholar]
- 27. Camacho Vanegas O., Bertini E., Zhang R. Z., Petrini S., Minosse C., Sabatelli P., Giusti B., Chu M. L., Pepe G. (2001) Ullrich scleroatonic muscular dystrophy is caused by recessive mutations in collagen type VI. Proc. Natl. Acad. Sci. U.S.A. 98, 7516–7521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brockington M., Brown S. C., Lampe A., Yuva Y., Feng L., Jimenez-Mallebrera C., Sewry C. A., Flanigan K. M., Bushby K., Muntoni F. (2004) Prenatal diagnosis of Ullrich congenital muscular dystrophy using haplotype analysis and collagen VI immunocytochemistry. Prenat. Diagn. 24, 440–444 [DOI] [PubMed] [Google Scholar]
- 29. Malandrini A., Scarpini C., Fabrizi G. M., Parrotta E., Salvadori C., Guazzi G. C. (1995) Early-onset benign limb-girdle myopathy with contractures and facial involvement affecting a father and daughter. J. Neurol. Sci. 132, 195–200 [DOI] [PubMed] [Google Scholar]
- 30. Jöbsis G. J., Keizers H., Vreijling J. P., de Visser M., Speer M. C., Wolterman R. A., Baas F., Bolhuis P. A. (1996) Type VI collagen mutations in Bethlem myopathy, an autosomal dominant myopathy with contractures. Nat. Genet. 14, 113–115 [DOI] [PubMed] [Google Scholar]
- 31. Zhang R. Z., Zou Y., Pan T. C., Markova D., Fertala A., Hu Y., Squarzoni S., Reed U. C., Marie S. K., Bönnemann C. G., Chu M. L. (2010) Recessive COL6A2 C-globular missense mutations in Ullrich congenital muscular dystrophy: role of the C2a splice variant. J. Biol. Chem. 285, 10005–10015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tooley L. D., Zamurs L. K., Beecher N., Baker N. L., Peat R. A., Adams N. E., Bateman J. F., North K. N., Baldock C., Lamandé S. R. (2010) Collagen VI microfibril formation is abolished by an {α}2(VI) von Willebrand factor type A domain mutation in a patient with Ullrich congenital muscular dystrophy. J. Biol. Chem. 285, 33567–33576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yorimitsu T., Klionsky D. J. (2007) Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy 3, 160–162 [DOI] [PubMed] [Google Scholar]
- 34. Meusser B., Hirsch C., Jarosch E., Sommer T. (2005) ERAD: the long road to destruction. Nature Cell Biology 7, 766–772 [DOI] [PubMed] [Google Scholar]
- 35. Ding W. X., Yin X. M. (2008) Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy 4, 141–150 [DOI] [PubMed] [Google Scholar]
- 36. Bateman J. F., Boot-Handford R. P., Lamandé S. R. (2009) Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 10, 173–183 [DOI] [PubMed] [Google Scholar]
- 37. Fitzgerald J., Lamandé S. R., Bateman J. F. (1999) Proteasomal degradation of unassembled mutant type I collagen pro-α1(I) chains. J. Biol. Chem. 274, 27392–27398 [DOI] [PubMed] [Google Scholar]
- 38. Lamandé S. R., Chessler S. D., Golub S. B., Byers P. H., Chan D., Cole W. G., Sillence D. O., Bateman J. F. (1995) Endoplasmic reticulum-mediated quality control of type I collagen production by cells from osteogenesis imperfecta patients with mutations in the pro α1 (I) chain carboxyl-terminal propeptide, which impair subunit assembly. J. Biol. Chem. 270, 8642–8649 [DOI] [PubMed] [Google Scholar]