

Abstract
Keratinocyte-associated C-type lectin (KACL) is a peculiar C-type lectin-like receptor (CTLR) due to its selective expression by human keratinocytes and cognate interaction with the genetically coupled CTLR NKp65. KACL and NKp65 are members of the CLEC2 and NKRP1 subfamilies of natural killer gene complex (NKC)-encoded CTLR, respectively. Most NKRP1 molecules are expressed on NK cells and T cells and act as receptors of CLEC2 glycoproteins with their genes being intermingled in a certain sub-region of the mammalian NKC. The reasons for the tight genetic linkage of these dedicated receptor/ligand pairs are unknown, as is the physiological expression of NKp65. Recently, we reported that the CTLR NKp65 and KACL interact with high affinity, resulting in activation of NKp65-expressing NK-92MI cells. Here, we address the molecular basis of this high-affinity interaction by analysing KACL mutants with KACL-specific monoclonal antibodies (mAb), soluble NKp65 (sNKp65) and NK-92MI-NKp65 cells. We find that none of the three N-linked carbohydrates of KACL glycoproteins significantly contributes to KACL surface expression and NKp65 interaction. However, KACL mutants with non-conservative amino acid substitutions of arginine 158 or isoleucine 161 abrogated binding of both KACL-specific mAb OMA1 and sNKp65, well in line with the blockade of NKp65–KACL interaction by OMA1. Accordingly, functional recognition of these KACL mutants by NK-92M-NKp65 cells was completely abolished. Arginine 158 and isoleucine 161 located at the membrane-distal surface of KACL were defined as residues, decisively determining functional KACL–NKp65 interaction that is independent of KACL glycosylation.
Keywords: epitope mapping, immune receptors, natural killer gene complex, skin
Introduction
Natural killer (NK) cells not only eradicate virally infected or malignantly transformed cells but also modulate immune responses through the production of cytokines and chemokines.1–4 These functions are governed by a plethora of germline-encoded activating and inhibitory NK receptors,5–7 which can be grouped based on their structure as members of the immunoglobulin-like or the C-type lectin-like receptor (CTLR) superfamily. Although most members of the immunoglobulin-like superfamily, e.g. the natural cytotoxicity receptor NKp46 or killer cell immunoglobulin-like receptors, are encoded in the leucocyte receptor complex on human chromosome 19, most CTLR, e.g. the activating receptor NKG2D, are encoded in the NK gene complex (NKC) on human chromosome 12.8,9 Natural killer receptors mostly recognize cellular ligands such as MHC class I and MHC class I-like molecules whose surface expression is altered upon cellular stress, viral infection or malignant transformation. Thereby, NK cells are able to sense and monitor the cellular state and contribute to an immune response against ‘harmful’ cells.10–13
The NKC-encoded CTLR can be further divided into killer cell lectin-like receptors, which usually are expressed on NK cells, and C-type lectin-like (CLEC) molecules, which are more broadly expressed.14 The human NKRP1 subfamily comprises the killer cell lectin-like receptors NKp65, NKp80 and NKR-P1A, which do not ligate MHC class I- or MHC class I-like molecules but recognize cognate CTLR of the CLEC2 family.15,16 Human CLEC2 family members are keratinocyte-associated C-type lectin (KACL), activation-induced C-type lectin (AICL), lectin-like transcript 1 (LLT1), CD69, and also the recently characterized non-NKC-encoded brain-associated C-type lectin (BACL). Whereas CD69 and BACL17 remain orphan receptors, KACL, AICL and LLT1 are ligated by the genetically linked NKRP1 receptors NKp65,18 NKp8019 and NKR-P1A,20,21 respectively. In mice, the NKRP1 receptor family is represented by the Nkrp1 proteins, which also form genetically linked receptor–ligand pairs with certain members of the CLEC2 family, the C-type lectin-related (Clr) proteins.22–24
The activating NK receptor NKp80 is expressed on all peripheral NK cells25,26 whereas its ligand AICL is expressed on monocytes, some malignant myeloid cell lines19 and monokine-activated NK cells.27 Upon ligation, NKp80 stimulates both cytotoxicity towards AICL-expressing cells and cytokine secretion. NKR-P1A was shown to function as an inhibitory receptor in NK cells upon encounter of its ligand LLT1,20,21,28 which is expressed on activated lymphocytes and antigen-presenting cells.28,29 In contrast to NKp80 and NKR-P1A, NKp65 is not expressed on peripheral NK cells, and the physiological expression of NKp65 remains to be explored.18 So far, substantial expression of NKp65 has only been described for the human NK cell line NK-92, where NKp65 stimulates interferon-γ secretion and mediates cytotoxicity towards KACL-expressing cells. The NKp65 ligand KACL is specifically expressed in the epidermis on the surface of keratinocytes, suggesting a role for the CTLR pair NKp65–KACL in skin-specific immunosurveillance.18,30
Immune-related CTLR of the NKC belong to the subgroup V of the CTLR superfamily14,31 and are type II transmembrane proteins with a highly variable N-terminal cytoplasmic domain, a transmembrane domain, a stalk region of variable length and a C-terminal C-type lectin-like ectodomain (CTLD). In contrast to ‘classical’ C-type lectins, e.g. the mannose-binding protein, NKC-encoded CTLR have lost the ability to bind calcium and the calcium-dependent interaction with carbohydrates. The CTLD is built up by two α-helices and two anti-parallel β-sheets that form a compact C-type lectin fold that is stabilized by two or three intramolecular disulphide bonds.9,31 Most NKC-encoded CTLR form homodimers that often are disulphide-linked through cysteines in the stalk region. Another conserved feature is a stretch of four hydrophobic amino acid residues, i.e. ‘WIGL’, forming the core of the CTLD.31
Structures of human CTLR are available, for example for CD69,32,33 for NKG2D in complex with MICA34 or ULBP3,35 and for CD94/NKG2A bound to HLA-E.36,37 The crystallized homodimer of CD69 revealed a membrane-distal flat surface that was suggested to serve as a potential ligand-binding site.
We set out to identify KACL residues important for NKp65 binding and to address the relevance of KACL glycosylation for NKp65 binding. All three putative N-linked glycosylation sites of KACL were mutated as well as selected residues of the predicted membrane-distal surface of KACL based on the CD69 structure. These KACL mutants were used to assess NKp65 interaction, and also to map the epitope of the previously reported KACL-specific monoclonal antibody (mAb) OMA1, which fully blocks NKp65 binding. We also report another KACL-specific mAb, OMA6 that does not block NKp65 binding. Our analysis reveals no role for N-linked carbohydrates in NKp65 binding and defines KACL residues that are necessary for NKp65 ligation. In the course of these studies, the complex structure of NKp65–KACL was recently reported by Mariuzza and colleagues identifying key residues that centrally contribute to NKp65–KACL binding, including residues defined in our study.38 Furthermore, we assessed the functional impact of KACL mutations on effector responses and demonstrate that mutation of certain single key residues fully abrogates functional responses mediated by the high-affinity interaction of KACL with NKp65.
Materials and methods
Cells and transfectants
Keratinocytes were isolated from human foreskin samples with approval of the local ethics committee as described previously.18 In brief, the cutaneous fat layer was removed and skin pieces were incubated for 16 hr at 4° in thermolysin (0·5 mg/ml; Sigma-Aldrich, St Louis, MO) solution. Subsequently, the epidermal layer was separated from the dermis and incubated for 20 min at 37° in trypsin (1× solution; Sigma) solution. The reaction was stopped by the addition of fetal calf serum (Biochrom, Berlin, Germany) and keratinocytes were filtered through a 100-μm cell strainer. 293T and COS-7 cells were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich), 293 cells in Iscove's modified Dulbecco's medium (PAA, Pasching, Austria). NK-92MI-NKp65 transductants were cultured in Iscove's modified Dulbecco's medium supplemented with 5 μg/ml puromycin (AppliChem, Darmstadt, Germany) as previously described.18 Cells were transfected using AppliFect (AppliChem). KACL cDNA was cloned into pIRES2-dsRED (ClonTech, Mountain View, CA) or RSV.5neo and mutated using a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) or by inverse vector PCR using Phusion Polymerase (NEB, Ipswich, UK) with mutation-containing primers followed by blunt-end ligation. Before transfection of 293 cells, the pIRES2-dsRED constructs were linearized with AflII (NEB). Stable transfectants were selected using 2 mg/ml G418 (AppliChem).
Recombinant proteins and antibodies
Soluble ectodomains of KACL, AICL, LLT1 and CD69 were purified from 293T supernatants and biotinylated using BirA ligase as previously described.19 The monoclonal KACL-specific antibody OMA1 was previously described.18 OMA6 was generated by standard hybridoma technology.39 In brief, BALB/c mice were immunized with P815–KACL and boosted with sKACL. Resulting hybridoma were screened by immunoblotting and flow cytometry.
Flow cytometry
Cells were washed with ice cold FACS buffer (PBS, 2% fetal bovine serum, 2 mm EDTA, 0·01% sodium azide) and then stained with relevant antibodies for 20 min on ice and washed again with FACS buffer. Flow cytometry analysis was performed with a FACS Canto II (BD Biosciences, Franklin Lakes, NJ) and data were analysed using flowjo (Tree Star, Ashland, OR). Unconjugated antibodies were stained with allophycocyanin (APC) -conjugated F(ab)2 fragments of goat anti-mouse IgG (Jackson ImmunoResearch, Newmarket, UK). Dead cells were excluded by DAPI stain. Ectodomains of NKp65 were biotinylated using BirA ligase and, before use, were tetramerized using APC-labelled or phycoerythrin-labelled streptavidin (Jackson ImmunoResearch).18 Streptavidin-coated microspheres (Bangs Laboratories, Inc., Indianapolis, IN) were loaded with 1 μg biotinylated ectodomains and stained as described above. Antibodies: anti-CD107a-APC (BD Biosciences), anti-FLAG (M2; Sigma-Aldrich), CD56-Peridinin chlorophyll protein (PerCP, Biolegend, San Diego, CA).
Immunofluorescence
Immunofluorescence was performed on cryosections of human foreskin. Eight-micrometre sections were fixed in 100% cold acetone for 10 min and dried at room temperature followed by rehydration in Tris-buffered saline (TBS) for 2 min before blocking in 3% BSA in TBS for 10 min. Sections were incubated in the presence of KACL antibodies OMA1, OMA6 or an IgG2a isotype control (all 5 μg/ml in 3% BSA/TBS) for 60 min at room temperature. After repeated washing in TBS for 2 min, secondary antibody staining was performed using AlexaFluor546-conjugated goat anti-mouse antibody (Molecular Probes, Eugene, OR, dilution 1 : 100 in 3% BSA/TBS) for 20 min at room temperature. For counterstaining, FITC-conjugated cytokeratin-14 antibody (Merck Millipore, Darmstadt, Germany; dilution 1 : 50 in 3% BSA/TBS) was employed after repeated washing. Slides were mounted in DAPI-containing Vectashield (Vector Laboratories, Burlingame, CA) and visualized with a DMI6000B with AFC microscope and a DFC3000G camera (Leica, Wetzlar, Germany).
Immunoblotting
Cells were lysed with ice-cold lysis buffer [1% Nonidet P40, 50 mm Tris–HCl pH 8, 150 mm NaCl (all from AppliChem), complete protease inhibitor (Roche, Mannheim, Germany)], then centrifuged and the supernatants containing cellular protein were separated by SDS–PAGE. For protein deglycosylation, cell lysates were treated with PNGase F (NEB) according to the manufacturer's protocol before SDS–PAGE. After blotting onto Roti-PVDF (Roth, Karlsruhe, Germany), the membrane was probed with KACL-specific mAb OMA6 or anti-FLAG (M2; Sigma-Aldrich) followed by detection with horseradish peroxidase-conjugated goat anti-mouse IgG (Jackson ImmunoResearch). Signals were generated with SuperSignal West Pico Chemiluminescent Substrate (ThermoScientific, Rockford, IL).
Degranulation and cytotoxicity assays
Degranulating NK-92MI-NKp65 cells were quantified by analysis of surface CD107a. After a 2-hr co-culture with 293–KACL transfectants in the presence of anti-CD107a-APC mAb and Golgi-Stop (both from BD Biosciences), NK-92MI-NKp65 cells were electronically gated using CD56-PerCP and assessed for CD107a staining in flow cytometry. For cytotoxicity assays, 293 cells were labelled with 50 μCi 51Cr for 2 hr at 37°. Subsequently, cells were washed and co-cultured for 4 hr at 37° with NK-92MI-NKp65 effector cells at different E : T ratios. Supernatants were mixed with an OptiPhase Supermix scintillation cocktail (PerkinElmer, Waltham, MA) and measured with a microbeta2 plate counter (PerkinElmer).
Statistical analysis, structural illustration and protein sequence analysis
Statistical analysis and data presentation were performed using prism 5 (GraphPad, San Diego, CA). Structural data from the KACL–NKp65 heterodimer (PDB ID: 4IOP) was retrieved from the protein data bank (http://www.rcsb.org) and represented using ucsf chimera 1.8.1 (http://www.cgl.ucsf.edu/chimera).40,41 Protein sequence alignment was performed using clustalx242 and the phylogenetic tree was visualized with treeview.43 Microscope images were illustrated with imagej 1.47v.44
Results
KACL-specific antibodies OMA1 and OMA6
We previously described the mAb OMA1 specifically binding to KACL, but not to the related CLEC2 family members LLT1 and AICL.18 Although OMA1 binds KACL both in flow cytometry and immunohistochemistry and blocks NKp65 interaction,18 it fails to detect denatured KACL, e.g. in immunoblotting. Therefore, we sought to generate another KACL-specific mAb not competing with NKp65 for KACL binding and suited for immunoblotting. Screening hybridoma raised from mice immunized with P815–KACL transfectants, resulted in isolation of mAb OMA6. OMA6 specifically binds to the recombinant soluble CTLD of KACL, but not to those of the other CLEC2 subfamily members AICL, LLT1 and CD69 (Fig.1a). OMA6, like OMA1, strongly binds the pro-monocytic cell line U937 and freshly isolated keratinocytes both endogenously expressing KACL (Fig.1b). However, while OMA1 efficiently blocked sNKp65 binding to both cell types, pre-incubation with OMA6 did not (Fig.1b). Immunoblots of lysates of KACL transfectants, U937 cells, and freshly isolated keratinocytes revealed that OMA6 binds denatured KACL when cell lysates were separated under non-reducing conditions (Fig.1c).
Figure 1.
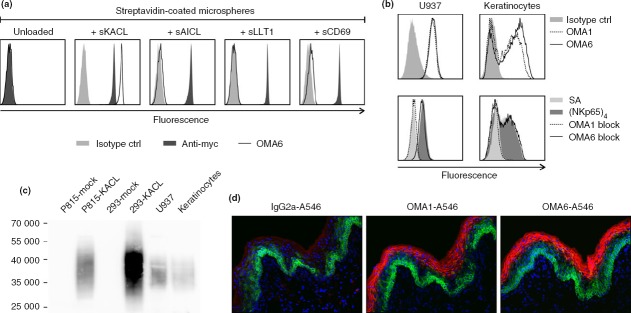
Characterization of keratinocyte-associated C-type lectin (KACL) -specific monoclonal antibodies (mAb) OMA1 and OMA6. (a) Streptavidin-coated microspheres loaded with biotinylated, c-myc-tagged soluble ectodomains of C-type lectin-like family 2 (CLEC2) members KACL, activation-induced C-type lectin (AICL), lectin-like transcript 1 (LLT1), and CD69, respectively, were stained with KACL-specific mAb OMA6 (open histograms). Control stainings with an isotype control (grey filled) or an anti-myc mAb 9E10 (black filled). (b) Flow cytometric analysis of U937 cells and keratinocytes with mAb OMA1 (stippled line), OMA6 (solid line), or an isotype control (grey filled) (upper panels), or with NKp65 tetramers (black filled), NKp65 tetramers after pre-incubation with mAb OMA1 (stippled) or OMA6 (solid) (lower panels). Control staining with allophycocyanin-conjugated streptavidin (grey filled). (c) Immunoblotting of lysates of P815 and 293 KACL transfectants, U937 and keratinocytes with mAb OMA6 after non-reducing SDS–PAGE. (d) KACL expression in human epidermis. Cryosections of human foreskin were stained with OMA1, OMA6 or an IgG2a isotype control, and counterstained for cytokeratin-14.
OMA1 and OMA6 were also evaluated in immunofluorescence. The epidermis of human skin was visualized by cytokeratin-14 that is strongly expressed in the proliferating stratum basale and expression decreases towards the more differentiated epidermal layers. In contrast, staining with OMA1 and OMA6 revealed that KACL expression was most pronounced in the more differentiated keratinocytes, whereas it remained undetectable in the proliferating basal layer and in the dermis (Fig.1d).
N-linked carbohydrates of KACL are dispensable for NKp65 binding
We previously described KACL as a non-disulphide-linked homodimeric glycoprotein expressed at the cellular surface of keratinocytes. Since the CTLD of KACL harbours three putative N-linked glycosylation sites at positions 78, 130 and 143,18 we sought to analyse the relevance of KACL glycosylation for NKp65 binding. According to the recently published crystal structure,38 asparagine 78 is located at the N-terminal end of the α1 helix, whereas asparagines 130 and 143 are part of loops L2 and L3, respectively (Fig.2a). For both Asn78 and Asn143, electron densities attributable to glycosylation were identified in the crystal structure, but there was no evidence for Asn143 glycosylation.
Figure 2.
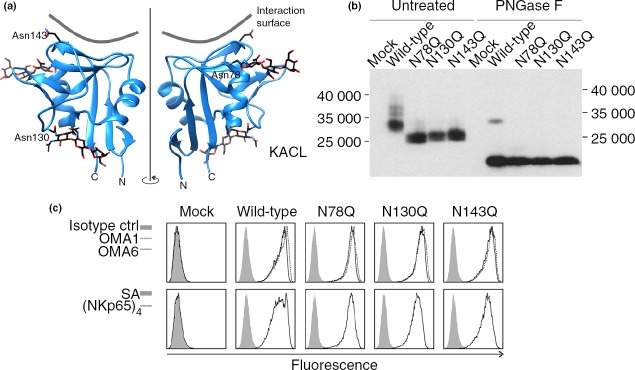
N-linked glycosylation of keratinocyte-associated C-type lectin (KACL) is dispensable for NKp65 interaction. (a) Localization of the three putative glycosylation sites Asn78, Asn130 and Asn143 in the KACL monomer structure (4IOP, PDB) depicted as a ribbon diagram in front (left) and back view (right). Carbohydrates linked to Asn78 and Asn130, as well as Asn143 are shown as sticks (carbon, black; oxygen, red; nitrogen, blue). Interacting surface of NKp65 is indicated as grey line. N-terminus (N) and C-terminus (C) are denoted below. (b) Each potential N-linked glycosylation site in KACL is glycosylated. KACL cDNA and various single mutants (Asn to Gln) thereof were transiently transfected into 293T cells. Cellular lysates were deglycosylated using PNGase F or left untreated, subsequently separated by SDS–PAGE, and FLAG-tagged KACL proteins detected by immunoblotting using monoclonal antibody (mAb) anti-FLAG M2. (c) N-linked glycosylation of KACL is irrelevant for binding to NKp65 or mAb OMA1 and OMA6. 293T cells were transiently transfected with various KACL cDNA in a bicistronic dsRed plasmid. The dsRed-positive cells were analysed by flow cytometry for binding of OMA1 (dashed line), OMA6 (solid line), and an isotype control (solid grey) (upper panels) and binding of sNKp65 tetramers (solid line) or allophycocyanin-conjugated streptavidin for control (solid grey) (lower panels).
To assess N-linked glycosylation, each of the three asparagines was individually mutated to glutamine and resulting KACL mutants were transiently transfected into 293T cells. To verify N-linked carbohydrate chains at the respective positions, cellular lysates were subjected to PNGase F digestion or left untreated and analysed by immunoblotting (Fig.2b). The predicted molecular mass of monomeric KACL protein is ∼20 kDa, whereas addition of three N-linked carbohydrates may result in a molecular mass of ∼32 kDa. When compared with non-mutated KACL glycoproteins, all three individual KACL mutants migrated at a smaller molecular mass of about 26 000, showing that all three asparagines of KACL are glycosylated. As expected, deglycosylation of all KACL molecules resulted in molecular masses of about 20 000.
To determine the influence of the N-linked carbohydrate chains on NKp65 binding, transiently transfected COS-7 cells were stained with sNKp65 tetramers and analysed by flow cytometry. Of note, neither sNKp65 nor the antibodies OMA1 and OMA6 showed an altered binding to any of the glycosylation mutants (Fig.2c).
Mutational analyses of KACL identify membrane-distal residues critical for OMA1 and NKp65 binding
The NKC-encoded human CTLR KACL, AICL, CD69 and LLT1 are grouped into the CLEC2 subfamily because of the sequence-relatedness of their CTLD (Fig.3a,b). Their CTLD exhibit shared sequence features such as a conserved ‘FLkRy’ motif in the α2 helix and a short L3 loop (preceding the β3 strand), distinguishing them from other NKC-encoded CTLR including NKRP1 receptors (Fig.3a,b). However, although the CTLD of AICL and CD69 are stabilized by three intramolecular disulphide bonds, LLT1 and KACL lack the third disulphide bond clamping strands β3 and β4 (Fig.3a).32,33 To pinpoint the binding site of NKp65 within the KACL CTLD, we specifically considered amino acids corresponding to such residues of the CD69 CTLD that have previously been suggested to constitute a putative ligand-binding site: about 15 solvent-exposed amino acids of the membrane-distal surface of the CD69 CTLD were proposed to form the binding site for a putative CD69 receptor (Fig.3c).32 When aligned with the KACL sequence, four residues were found to be conserved or conservatively substituted and therefore omitted from analysis. From the remainder, six KACL residues (M113, G144, H155, R158, F160, I161) were chosen for mutational analysis and substituted by the corresponding amino acids of CD69, except for methionine 113 and glycine 144, which were substituted by aspartate and glutamate, respectively, the corresponding residues in the AICL sequence. As evident from the localization of these six residues within the recently published KACL structure, their positioning in the KACL CTLD corresponds to the positioning of the respective residues in the CD69 CTLD (Fig.3c).
Figure 3.
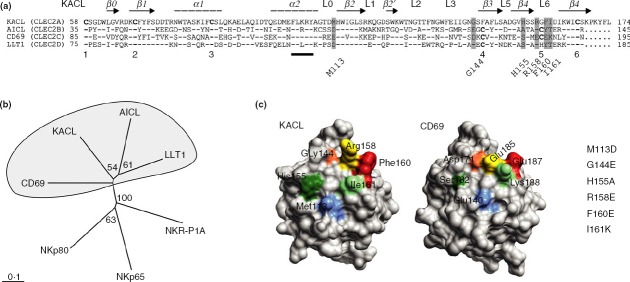
The C-type lectin-like domain (CTLD) of human C-type lectin-like family 2 (CLEC2) members. (a) Alignment of CTLD amino acid sequences of natural killer gene complex (NKC) -encoded human CLEC2 family members keratinocyte-associated C-type lectin (KACL; CLEC2A), activation-induced C-type lectin (AICL; CLEC2B), CD69 (CLEC2C), and lectin-like transcript 1 (LLT1; CLEC2D) using Gonnet series (gap opening 10; gap extension 0·2). Secondary structure elements deduced from the KACL structure are indicated above and analysed mutations are highlighted in grey. Cystein residues involved in disulphide bonds are numbered below and the ‘FLkRy’ motif is marked. Conserved amino acids in AICL, CD69 and LLT1 are depicted as dashes and missing acids as dots. (b) Phylogenetic tree of the CTLD sequences shown in (a) generated with clustalx2 using the bootstrap-neighbour-joining method (n = 100) and visualized with treeview. Node values are displayed. CTLD sequences of the corresponding receptors NKp80, NKp65 and NKR-P1A were also included. (c) Localization of mutations analysed in the CTLD structures of KACL (4IOP, PDB) and CD69 (1E8I, PDB), respectively. The molecular surface was visualized using ucsf chimera 1.8.1. Top view of the membrane-distal surface of the respective CTLD.
After transient transfection into COS-7 cells, surface expression of KACL mutants was monitored by flow cytometry. Strong surface expression of all seven KACL mutants was observed with mAb OMA6, indicating that the substitutions did not affect surface expression of the KACL mutants. However, although all KACL mutants were comparably stained by OMA6, binding of mAb OMA1 to KACL mutants M113D and F160E single mutants was reduced (Fig.4a). Further, there was no binding of OMA1 to the KACL mutant I161K and consequently also none to the double mutant R158E/I161K. Altogether, these data map the binding site of the NKp65-blocking antibody OMA1 to a non-contiguous epitope located at the membrane-distal surface of the KACL CTLD including the residues M113, F160 and I161.
Figure 4.
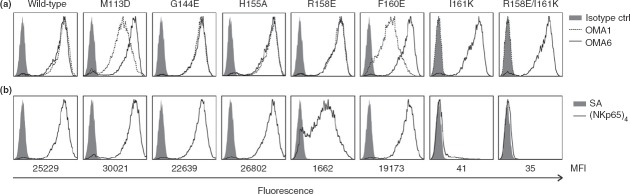
Mutational characterization of keratinocyte-associated C-type lectin (KACL) residues critical for binding of NKp65 and OMA1. (a, b) COS-7 cells were transiently transfected with KACL cDNA in a bicistronic dsRed plasmid and analysed in flow cytometry with gates set on dsRed-positive cells. (a) Stainings of dsRed-positive cells with monoclonal antibody (mAb) OMA1 (stippled line), OMA6 (solid line), and an isotype control (solid grey) or (b) with sNKp65 tetramers (solid line) or allophycocyanin-conjugated streptavidin for control (solid grey). Below, the median fluorescence intensity (MFI) is indicated.
To define residues involved in the KACL–NKp65 interaction, COS-7 cells expressing the KACL mutants were stained with APC-conjugated sNKp65 tetramers. While KACL mutants M113D, G144E, H155A and F160E did not show an altered sNKp65 binding compared with the KACL control, substitution of arginine 158 by glutamate resulted in a greatly impaired binding of sNKp65 tetramers, with staining intensities reduced by about 95% compared with KACL levels (Fig.4b). Substitution of isoleucine 161 by lysine even led to a complete abrogation of NKp65 binding that was also observed for the double mutant R158E/I161K.
Mutational disruption of functional KACL recognition by NKp65
We previously demonstrated that ligation of NKp65 on NK-92MI cells by KACL activates degranulation of NK-92MI cells and cytolysis of KACL-expressing target cells.18 Hence, we set out to address whether KACL amino acid residues identified as crucial for the binding of sNKp65 tetramers also impact on functional recognition of KACL by NK-92MI-NKp65 cells. With this aim, 293 cells were stably transfected with KACL, KACL R158E, KACL I161K, or KACL R158E/I161K. Flow cytometric analysis with OMA6 revealed similar cell surface expression levels of the various KACL mutants while staining with OMA1 and sNKp65 tetramers recapitulated the same differential binding pattern to the various KACL mutants as already observed for the corresponding COS-7 transfectants (Fig.5a).
Figure 5.

Mutational disruption of functional keratinocyte-associated C-type lectin (KACL) recognition by NKp65. (a) 293 cells stably transfected with cDNA of KACL or KACL mutants were analysed for binding of monoclonal antibodies (mAb) OMA1 (stippled line), OMA6 (solid line), or an isotype control (filled grey) (upper panels) or with sNKp65 tetramers (solid line) or allophycocyanin-conjugated streptavidin only (filled grey) (lower panels). (b) Surface CD107a on NK-92MI-NKp65 cells after co-culture with 293–KACL transfectants shown in (a). For control, CD107a expression on NK-92MI-NKp65 cells without co-culture (medium) and PMA/ionomycin treatment is shown. (c) Cytotoxicity of NK-92MI-NKp65 cells towards 293–KACL transfectants shown in (a) measured in a 4-hr chromium-release assay. All data are represented as mean ± standard deviation (SD). One-way analysis of variance (anova) (b) or two-way anova (c) was performed with Bonferroni post-test. ***P < 0·001; ****P < 0·0001.
To assess functional recognition of KACL mutants by NKp65, degranulation of NK-92MI-NKp65 cells was monitored after co-culture with 293–KACL transfectants. Mock-transfected 293 already exhibit a marked degranulation of NK-92MI-NKp65 cells presumably as the result of constitutive expression of ligands of NK activating receptors such as NKG2D39 and NCRs.45 As expected, degranulation was strongly increased in co-cultures with 293 cells expressing KACL. However, CD107a levels after co-culture with the KACL mutants were not significantly different from those observed for co-cultures with 293 mock transfectants indicating that these KACL mutants were functionally silenced. These results were further corroborated in cytotoxicity assays. Although cytolysis of 293 cells expressing KACL by NK-92MI-NKp65 cells was strongly enhanced compared with the control cells, lysis rates of 293 cells expressing the KACL mutants R158E and I161K did not differ from that of control cells, demonstrating that these KACL mutants are unable to trigger functional activation through NKp65.
Altogether, these data show that the impaired binding of sNKp65 tetramers to KACL mutants R158E and I161K correlate with their inability to trigger functional responses. The data also identify I161 as a key residue for the high-affinity interaction between NKp65 and KACL as its mutation completely abrogates NKp65 binding and functional activation.
Discussion
Natural killer cells recognize virally infected or malignantly transformed cells with a repertoire of germline-encoded NK receptors that ligate mostly MHC class I or MHC class I-like molecules. The NKC-encoded NKRP1 subfamily comprising NKp65, NKp80 and NKR-P1A, however, ligates genetically linked members of the CLEC2 family KACL, AICL and LLT1, respectively. These human NKRP1 receptors also do not associate with adapter molecules to relay signals into the cell, but rather bear signalling motifs in their cytoplasmic domain. Whereas AICL and LLT1 are expressed by haematopoietic cells, expression of KACL appears restricted to epidermal keratinocytes indicating a role in skin-specific immunosurveillance.
We previously presented OMA1 as a KACL-specific mAb not cross-reacting with other members of the CLEC2 family and efficiently blocking NKp65 binding. Here, we now present a second KACL-specific mAb, OMA6, that like OMA1 selectively binds to immobilized KACL ectodomains as well as to U937 cells and keratinocytes. However, in contrast to OMA1, OMA6 does not block NKp65 binding to KACL and can be used for immunoblotting.
In our efforts to characterize the molecular basis of NKp65–KACL interaction, we first addressed the contribution of N-linked carbohydrates to NKp65 binding by KACL. All three putative N-glycosylation sites identified in the KACL CTLD were shown to be glycosylated. For two out of these three sites, carbohydrate linkage was also seen in the recently published structure of KACL. According to this NKp65–KACL complex structure, only asparagine 143 is in the vicinity of the KACL–NKp65 interface, rendering it potentially relevant in ligand recognition. However, NKp65 interaction, and also OMA1 binding, was unaffected in a KACL mutant lacking glycosylation at position 143, showing that it is dispensable for NKp65–KACL interaction.
To identify residues determining KACL–NKp65 interaction, we took advantage of the CD69 structure and the putative ligand binding site as proposed in this study.32 Six positions were selected for mutational analysis based on their localization in the CD69 structure and non-conservation among human CLEC2 family members because these amino acid residues might be responsible for the non-redundant receptor specificity.
OMA1 binding turned out to be disturbed for KACL mutants M113D and F160E and fully abrogated for the I161K mutant. These amino acids are positioned in close proximity to each other, thereby forming a discontinuous epitope for OMA1. NKp65 binding was reduced for the KACL mutant R158E and completely absent for mutant I161K. The shared strict dependence of NKp65 and OMA1 binding on residue I161 explains the previously observed blockade of NKp65 recognition by OMA1.
Other CLEC2 family members carry polar or charged amino acids at this specific position; threonine in AICL and LLT1 and lysine for CD69. Therefore, this particular position may be considered as an interaction hot spot crucially determining the non-redundant receptor specificity of the various CLEC2 family members. In contrast to I161, R158 is conserved in CLEC2 family members AICL and LLT1 that may define this position as a key amino acid residue involved in receptor binding. Of note, this arginine is substituted in CD69 by the conversely charged glutamate for a yet unknown reason as a receptor of CD69 has not been identified. Li et al.38 had reported a decreased affinity for NKp65 to the KACL mutants S157A/R158A and F160A/I161A. From these results, the single contribution of one residue is difficult to determine but our additional data support a strong involvement of R158 and I161 in the KACL–NKp65 interaction. The structural data further imply similar binding modes of the CTLD in which the membrane-distal molecular surface makes contact with the respective ligand. In KACL, the NKp65 binding site involves the loops L0, L3, L5 and L6 as well as the β strands β3 and β4.38 The same protein elements were hypothesized to be a potential ligand-binding site in CD69.32
It will be interesting to address the contributions of these residues to NKp80–AICL and NKR-P1A–LLT1 interactions, once these respective complex structures become available to further decipher the molecular basis of the binding specificity of these structurally related genetically linked receptor–ligand pairs.
Acknowledgments
We thank Sandra Tafferner and Beate Pömmerl for excellent technical assistance. This work was supported by grants from the Deutsche Forschungsgemeinschaft (STE 828/5-1 and 5-2). Molecular graphics and analyses were performed with the ucsf chimera package. chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco.
Glossary
- AICL
activation-induced C-type lectin
- CLEC
C-type lectin-like
- Clr
C-type lectin-related
- CTLD
C-type lectin-like domain
- CTLR
C-type lectin-like receptor
- KACL
keratinocyte-associated C-type lectin
- LLT1
lectin-like transcript
- mAb
monoclonal antibody
- MHC
major histocompatibility complex
- NKC
natural killer gene complex
- NK
natural killer
- NKR
natural killer cell receptor
Author's contributions
BB designed and performed experiments, analysed the data and wrote the manuscript. CR performed experiments, JS designed and performed experiments, IV designed experiments. AS conceptualized research and wrote the manuscript.
Disclosures
AS holds a patent on NKp65. The other authors declare no conflict of interest.
References
- Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–9. doi: 10.1182/blood-2007-09-077438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Santo JP. Natural killer cells: diversity in search of a niche. Nat Immunol. 2008;9:473–5. doi: 10.1038/ni.f.201. [DOI] [PubMed] [Google Scholar]
- Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- Vivier E, Raulet DH, Moretta A, et al. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–9. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, Chiang SC, Darmanin S, et al. Molecular mechanisms of natural killer cell activation. J Innate Immun. 2011;3:216–26. doi: 10.1159/000325265. [DOI] [PubMed] [Google Scholar]
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502. doi: 10.1038/ni1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO, Sik KH, Liu D, Peterson ME, Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu Rev Immunol. 2013;31:227–58. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley J, Walter L, Trowsdale J. Comparative genomics of natural killer cell receptor gene clusters. PLoS Genet. 2005;1:129–39. doi: 10.1371/journal.pgen.0010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama WM, Plougastel BF. Immune functions encoded by the natural killer gene complex. Nat Rev Immunol. 2003;3:304–16. doi: 10.1038/nri1055. [DOI] [PubMed] [Google Scholar]
- Raulet DH, Vance RE. Self-tolerance of natural killer cells. Nat Rev Immunol. 2006;6:520–31. doi: 10.1038/nri1863. [DOI] [PubMed] [Google Scholar]
- Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. Regulation of ligands for the NKG2D activating receptor. Annu Rev Immunol. 2013;31:413–41. doi: 10.1146/annurev-immunol-032712-095951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Groh V, Wu J, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–9. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–43. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- Hao L, Klein J, Nei M. Heterogeneous but conserved natural killer receptor gene complexes in four major orders of mammals. Proc Natl Acad Sci USA. 2006;103:3192–7. doi: 10.1073/pnas.0511280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler I, Steinle A. Vis-a-vis in the NKC: genetically linked natural killer cell receptor/ligand pairs in the natural killer gene complex (NKC) J Innate Immun. 2011;3:227–35. doi: 10.1159/000324112. [DOI] [PubMed] [Google Scholar]
- Bartel Y, Bauer B, Steinle A. Modulation of NK cell function by genetically coupled C-type lectin-like receptor/ligand pairs encoded in the human natural killer gene complex. Front Immunol. 2013;4:362. doi: 10.3389/fimmu.2013.00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lysenko O, Schulte D, Mittelbronn M, Steinle A. BACL is a novel brain-associated, non-NKC-encoded mammalian C-type lectin-like receptor of the CLEC2 family. PLoS One. 2013;8:e65345. doi: 10.1371/journal.pone.0065345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreu J, Kuttruff S, Stejfova V, Dennehy KM, Schittek B, Steinle A. Interaction of C-type lectin-like receptors NKp65 and KACL facilitates dedicated immune recognition of human keratinocytes. Proc Natl Acad Sci USA. 2010;107:5100–5. doi: 10.1073/pnas.0913108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte S, Kuttruff S, Waldhauer I, Steinle A. Mutual activation of natural killer cells and monocytes mediated by NKp80–AICL interaction. Nat Immunol. 2006;7:1334–42. doi: 10.1038/ni1402. [DOI] [PubMed] [Google Scholar]
- Rosen DB, Bettadapura J, Alsharifi M, Mathew PA, Warren HS, Lanier LL. Cutting edge: lectin-like transcript-1 is a ligand for the inhibitory human NKR-P1A receptor. J Immunol. 2005;175:7796–9. doi: 10.4049/jimmunol.175.12.7796. [DOI] [PubMed] [Google Scholar]
- Aldemir H, Prod'homme V, Dumaurier MJ, et al. Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. J Immunol. 2005;175:7791–5. doi: 10.4049/jimmunol.175.12.7791. [DOI] [PubMed] [Google Scholar]
- Iizuka K, Naidenko OV, Plougastel BF, Fremont DH, Yokoyama WM. Genetically linked C-type lectin-related ligands for the NKRP1 family of natural killer cell receptors. Nat Immunol. 2003;4:801–7. doi: 10.1038/ni954. [DOI] [PubMed] [Google Scholar]
- Chen P, Belanger S, Aguilar OA, et al. Analysis of the mouse 129-strain Nkrp1–Clr gene cluster reveals conservation of genomic organization and functional receptor–ligand interactions despite significant allelic polymorphism. Immunogenetics. 2011;63:627–40. doi: 10.1007/s00251-011-0542-8. [DOI] [PubMed] [Google Scholar]
- Kveberg L, Dai KZ, Inngjerdingen M, Brooks CG, Fossum S, Vaage JT. Phylogenetic and functional conservation of the NKR-P1F and NKR-P1G receptors in rat and mouse. Immunogenetics. 2011;63:429–36. doi: 10.1007/s00251-011-0520-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roda-Navarro P, Arce I, Renedo M, Montgomery K, Kucherlapati R, Fernandez-Ruiz E. Human KLRF1, a novel member of the killer cell lectin-like receptor gene family: molecular characterization, genomic structure, physical mapping to the NK gene complex and expression analysis. Eur J Immunol. 2000;30:568–76. doi: 10.1002/1521-4141(200002)30:2<568::AID-IMMU568>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Vitale M, Falco M, Castriconi R, et al. Identification of NKp80, a novel triggering molecule expressed by human NK cells. Eur J Immunol. 2001;31:233–42. doi: 10.1002/1521-4141(200101)31:1<233::AID-IMMU233>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Klimosch SN, Bartel Y, Wiemann S, Steinle A. Genetically coupled receptor-ligand pair NKp80–AICL enables autonomous control of human NK cell responses. Blood. 2013;122:2380–9. doi: 10.1182/blood-2013-01-479790. [DOI] [PubMed] [Google Scholar]
- Rosen DB, Cao W, Avery DT, et al. Functional consequences of interactions between human NKR-P1A and its ligand LLT1 expressed on activated dendritic cells and B cells. J Immunol. 2008;180:6508–17. doi: 10.4049/jimmunol.180.10.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain C, Meier A, Jensen T, et al. Induction of lectin-like transcript 1 (LLT1) protein cell surface expression by pathogens and interferon-γ contributes to modulate immune responses. J Biol Chem. 2011;286:37964–75. doi: 10.1074/jbc.M111.285312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spreu J, Kienle EC, Schrage B, Steinle A. CLEC2A: a novel, alternatively spliced and skin-associated member of the NKC-encoded AICL-CD69-LLT1 family. Immunogenetics. 2007;59:903–12. doi: 10.1007/s00251-007-0263-1. [DOI] [PubMed] [Google Scholar]
- Zelensky AN, Gready JE. The C-type lectin-like domain superfamily. FEBS J. 2005;272:6179–217. doi: 10.1111/j.1742-4658.2005.05031.x. [DOI] [PubMed] [Google Scholar]
- Llera AS, Viedma F, Sanchez-Madrid F, Tormo J. Crystal structure of the C-type lectin-like domain from the human hematopoietic cell receptor CD69. J Biol Chem. 2001;276:7312–9. doi: 10.1074/jbc.M008573200. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Sawicki MW, Margulies DH, Mariuzza RA. Crystal structure of human CD69: a C-type lectin-like activation marker of hematopoietic cells. Biochemistry. 2000;39:14779–86. doi: 10.1021/bi0018180. [DOI] [PubMed] [Google Scholar]
- Li P, Morris DL, Willcox BE, Steinle A, Spies T, Strong RK. Complex structure of the activating immunoreceptor NKG2D and its MHC class I-like ligand MICA. Nat Immunol. 2001;2:443–51. doi: 10.1038/87757. [DOI] [PubMed] [Google Scholar]
- Radaev S, Rostro B, Brooks AG, Colonna M, Sun PD. Conformational plasticity revealed by the cocrystal structure of NKG2D and its class I MHC-like ligand ULBP3. Immunity. 2001;15:1039–49. doi: 10.1016/s1074-7613(01)00241-2. [DOI] [PubMed] [Google Scholar]
- Petrie EJ, Clements CS, Lin J, et al. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J Exp Med. 2008;205:725–35. doi: 10.1084/jem.20072525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser BK, Pizarro JC, Kerns J, Strong RK. Structural basis for NKG2A/CD94 recognition of HLA-E. Proc Natl Acad Sci USA. 2008;105:6696–701. doi: 10.1073/pnas.0802736105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang Q, Chen S, Brown PH, Mariuzza RA. Structure of NKp65 bound to its keratinocyte ligand reveals basis for genetically linked recognition in natural killer gene complex. Proc Natl Acad Sci USA. 2013;110:11505–10. doi: 10.1073/pnas.1303300110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte SA, Sinzger C, Lutz SZ, et al. Selective intracellular retention of virally induced NKG2D ligands by the human cytomegalovirus UL16 glycoprotein. Eur J Immunol. 2003;33:194–203. doi: 10.1002/immu.200390022. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38:305–20. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Page RD. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–8. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd A, Hoffmann SC, Jarahian M, Momburg F, Watzl C. Expression analysis of the ligands for the natural killer cell receptors NKp30 and NKp44. PLoS One. 2007;2:e1339. doi: 10.1371/journal.pone.0001339. [DOI] [PMC free article] [PubMed] [Google Scholar]