

Abstract
The cytoplasmic [PSI+] element of budding yeast represents the prion conformation of translation release factor eRF-3 (Sup35). Prions are transmissible agents caused by self-seeded highly ordered aggregates (amyloids). Much interest lies in understanding how prions are developed and transmitted. However, the cellular mechanism involved in the prion clearance is unknown. Recently we have reported that excess misfolded multi-transmembrane protein, Dip5ΔC-v82, eliminates yeast prion [PSI+]. In this study, we showed that the prion loss was caused by enlargement of prion amyloids, unsuitable for transmission, and its efficiency was affected by the cellular balance between the chaperone Hsp70-Ssa1 and Sgt2, a small cochaperone known as a regulator of chaperone targeting to different types of aggregation-prone proteins. The present findings suggest that Sgt2 is titrated by excess Dip5ΔC-v82, and the shortage of Sgt2 led to non-productive binding of Ssa1 on [PSI+] amyloids. Clearance of prion [PSI+] by the imbalance between Ssa1 and Sgt2 might provide a novel array to regulate the release factor function in yeast.
Keywords: eRF-3, yeast prion, [PSI+], misfolded multi-trans membranes, prion clearance, Hsp104, Ssa1, GET pathway, Sgt2, HSR
Introduction
Prions are transmissible agents caused by the self-propagating conformational change of proteins.1 According to the “protein only” hypothesis,1 the prion protein is the sole agent responsible for causing numerous infectious diseases including scrapie (sheep), bovine spongiform encephalopathy (cow), chronic wasting disease (deer, elk) as well as kuru and Creutzfeldt-Jakob disease (humans). In Saccharomyces cerevisiae, prions have also been characterized as non-Mendelian inheritable elements, notably [PSI+], [URE3] and [RNQ+].2-4 Molecular and genetic studies of these yeast prions have greatly facilitated the elucidation of the molecular basis for prion conversion and propagation.
The yeast prion [PSI+]2 is the amyloid-like structure of the eRF3 polypeptide release factor, Sup35, which is essential for terminating protein synthesis at stop codons (reviewed by ref. 5). [PSI+] cells are marked by an altered catalytic protein conformation of Sup35 whereby the Sup35 protein is converted from a soluble, active state to an aggregated, inactive state. When Sup35 is in the [PSI+] state, ribosomes exhibit an increased rate of stop codon readthrough, causing a non-Mendelian trait easily detected by nonsense suppression.6 The biological (or functional) significance of [PSI+] and other yeast prions is not well understood. It is speculated that heritable prion-encoded information has been harnessed during evolution to confer selective advantages.7
Several chaperone proteins are involved in prion maintenance and propagation. Of these, the best characterized is the Hsp100-family protein Hsp104, which facilitates the propagation of yeast prions by breaking apart amyloid filaments to generate prion seeds, which are transmissible to daughter cells.8,9 It is known that propagation of yeast prions depends on the balance between Hsp104 and the Hsp70-family protein Ssa1.10-12 The prion [PSI+] can be eliminated by excess Hsp104, and this effect is reversed by excess Ssa1.8,10 Recently, Chernoff and coworkers have found that the actions of Hsp104 and Ssa1 on [PSI+] are modulated by the small cochaperone Sgt2,13 which has previously been implicated in the guided entry of tail-anchored (TA) proteins (GET) trafficking pathway.14,15 Sgt2 is known to interact with aggregation-prone proteins, such as heat-shock proteins (Hsps), cytosolic GET proteins, and tail-anchored proteins.16,17 They demonstrated that Sgt2 overexpression reverses the curing inhibition effect in the presence of excess Hsp104 and Ssa1.13
We have conducted genome-wide screens for prion-eliminating factors or mutants using a multi-copy expression system in yeast.18-23 One of the newly found anti-prion agents was an excess of multi-transmembrane (MTM) mutant protein, Dip5ΔC-v82.24 Dip5 is an 11-spanning MTM protein, and Dip5ΔC-v82 is truncated at the 8th transmembrane domain and fused to a vector-coded polypeptide v82. The genetic mutational studies indicated that the abnormal accumulation of Dip5ΔC-v82 in the endoplasmic reticulum (ER) compartment triggered prion clearance, independently of the unfolded protein response (UPR), through the GET pathway.24 In this study, we pursued genetic mechanistic analysis of this phenomenon and found that the cellular balance between Ssa1 and Sgt2 plays an essential role for the [PSI+] clearance by excess Dip5ΔC-v82.
Results
Genetic evidence for the involvement of Sgt2 and Ssa1 in [PSI+] clearance by excess Dip5ΔC-v82
We have reported previously that overexpression of Dip5ΔC-v82 from pRS413GPDp in [PSI+] cells is prone to eliminate the yeast prion [PSI+], changing colony color from white to red in the ade1–14 strain (Fig. 1A, left).24 The clearance of [PSI+] by excess Dip5ΔC-v82 is disabled by the get3Δ deletion, implicating the involvement of the GET pathway in the [PSI+] clearance phenomenon.24 In this regard, it warrants mentioning that a deletion of the GET2 gene, as well as a deletion of any other GET genes, impairs [PSI+] clearance by excess Hsp104.13 Importantly, this clearance is modulated by Sgt2, which is known to bind prion amyloids and other aggregation-prone proteins.13 Collectively, these findings are interpreted as indicating that Sgt2 might preferentially bind excess Dip5ΔC-v82, resulting in the shortage of Sgt2.

Figure 1. Protective effects of excess Sgt2 and ssa1Δ deletion on [PSI+] from clearance by excess Dip5ΔC-v82. (A) [PSI+] clearance by excess Dip5ΔC-v82. Plasmids pRS413GPDp-Dip5ΔC-v82 (shown as [Dip5ΔC-v82]) and pRS415GPDp-Sgt2 (shown as [Sgt2]) were transformed in NPK265 [PSI+] cells. Transformants were incubated on SC-His-Leu plate for 3 d, and selected colonies were re-grown on YPD for 4 d. Empty vector was used as a negative control. (B) Excess Sgt2 attenuates [PSI+] clearance by excess Dip5ΔC-v82. The frequency of appearance of red colonies from NPK265 [PSI+] cells transformed by pRS413GPDp-Dip5ΔC-v82 with or without pRS415GPDp-Sgt2 was monitored. The frequency is shown as the mean and standard deviation from 3 independent experiments. (C) The ssa1Δ deletion protects [PSI+] from clearance by excess Dip5ΔC-v82. Wild-type (NPK265) and ssa1Δ deletion (NPK608) [PSI+] cells were transformed singly or doubly with plasmids pRS413GPDp-Dip5ΔC-v82, pRS415GPDp-Ssa1 (shown as [Ssa1]) and pRS415GPDp-Sgt2, and transformants were monitored by color as shown in (A).
This possibility was examined by overexpression of Sgt2. As expected, the frequency of [PSI+] clearance by excess Dip5ΔC-v82 is decreased upon overexpression of Sgt2 from pRS415GPDp (Fig. 1A, right and B). This finding suggests that the [PSI+] clearance is, at least in part, caused by the shortage of Sgt2 under overexpression of Dip5ΔC-v82. This is consistent with the previous report that Sgt2 interacts with several types of aggregation-prone proteins, i.e., not only prion amyloids, Hsps and cytosolic GET proteins but also TA proteins, which include MTM proteins in a broad sense.13
The profound effect on [PSI+] clearance by excess Dip5ΔC-v82 was observed with Ssa1. First, the ssa1Δ deletion completely protects [PSI+] from clearance by excess Dip5ΔC-v82, which is reversed by Ssa1 expression from pRS415GPDp (Fig. 1C). Second, in the wild-type SGT2 strain, [PSI+] clearance by excess Dip5ΔC-v82 was enhanced by overexpression of Ssa1 from pRS415GPDp (Fig. 2A, left). In the sgt2Δ deletion strain, however, [PSI+] clearance by excess Dip5ΔC-v82 was blocked and reversed by excess Ssa1 (Fig. 2A, right). Under the sgt2Δ deletion condition, Ssa1 was expressed from three different promoters of weak (CYC1), moderate (ADH), and strong (GPD) expression to examine the dosage-dependency of the clearance frequency. As shown in Figure 2B, [PSI+] clearance by excess Dip5ΔC-v82 was reversed in proportion to the promoter strength. These findings suggest that, in the absence of Sgt2, Ssa1 binds [PSI+] amyloids to generate a non-productive form. On the other hand, excess Sgt2 could not reverse [PSI+] clearance by excess Dip5ΔC-v82 in the ssa1Δ deletion strain (Fig. 1C, right). This implies that Sgt2 does not function as a co-chaperone in the absence of Ssa1.

Figure 2. Ssa1 reverses [PSI+] clearance by excess Dip5ΔC-v82 in the absence of Sgt2. (A) Wild-type (NPK265, left half) and sgt2Δ deletion (NA120, right half) [PSI+] strains were transformed with the indicated plasmids, transformants were monitored by colony color. (B) Ssa1 dosage effects on [PSI+] clearance by excess Dip5ΔC-v82. Ssa1 expression plasmids from weak (CYC1), moderate (ADH), and strong (GPD) promoters in pRS413 derivatives were transformed into the sgt2Δ deletion (NA120) [PSI+] strain and transformants were monitored by colony color.
Enlargement of [PSI+] amyloids by excess Dip5ΔC-v82
We have shown previously that the average size of the [PSI+] amyloids slightly increased after expression of Dip5ΔC-v82 when monitored by semi-denaturing detergent-agarose gel electrophoresis and this increase in the amyloid size was not observed in the get3Δ strain.24 Hence, we examined the dynamics of [PSI+] amyloids by fluorescence correlation spectroscopy (FCS).20,26 FCS is a technique to determine the diffusion coefficients of fluorescence molecules by calculating the autocorrelation function from fluorescence intensity fluctuation detected in a microscopic volume of detection under 10−15 L (1 femtoliter), thereby providing an estimation of the size of aggregates. For these experiments, we used strains with GFP integrated in the endogenous SUP35 ORF.25 Dip5ΔC-v82 was expressed from the GAL1 promoter in [PSI+] and [psi-] strains.
Using this FCS technique, the size change of Sup35-GFP amyloid and the process of loss-of-[PSI+] were analyzed in single living cells that express Dip5ΔC-v82. Single cell analysis showed that cells can be categorized into three diffusional types in terms of mother and daughter cell states 72 h after expression of Dip5ΔC-v82. The first type shows slow intracellular diffusion of Sup35-GFP in both mother and daughter cells, as would be expected for the [PSI+] state. The second type shows slow diffusion in mother cells and fast diffusion in daughter cells. The third type shows fast diffusion in both mother and daughter cells, as would be expected for the [psi-] state. Figure 3 shows the second type of FCS measurement on a representative mother and daughter cell pair with Dip5ΔC-v82 expression (Fig. 3B represents the normalized data of Figure 3A). Strikingly, the mother cell had freely diffusing Sup35 aggregates with higher than average fluorescent intensity and the high-broad peaks directly reflects the right-shifted correlation function in Figure 3A(red), which had a much larger diffusional component than [PSI+] cells without expression of Dip5ΔC-v82 as a control (Fig. 3B). In contrast, the daughter cell only had stationary fluctuation of fluorescent intensity without high-peaks of fluorescence and had fast diffusion similar to that of [psi-] cells (Fig. 3B). This single cell analysis likely shows that, with expression of Dip5ΔC-v82, the size of the enlarged amyloids observed in mother cells are directly related to the physical size limitation of amyloids to be efficiently transmitted from mother to daughter cells, accounting for the [PSI+] elimination that occurred at this time point.25 A similar enlargement of [PSI+] amyloids was observed upon expression of N-terminal non-prion domain deletion Rnq1Δ100, missense mutations of Rnq1, or Lsm4.20,23
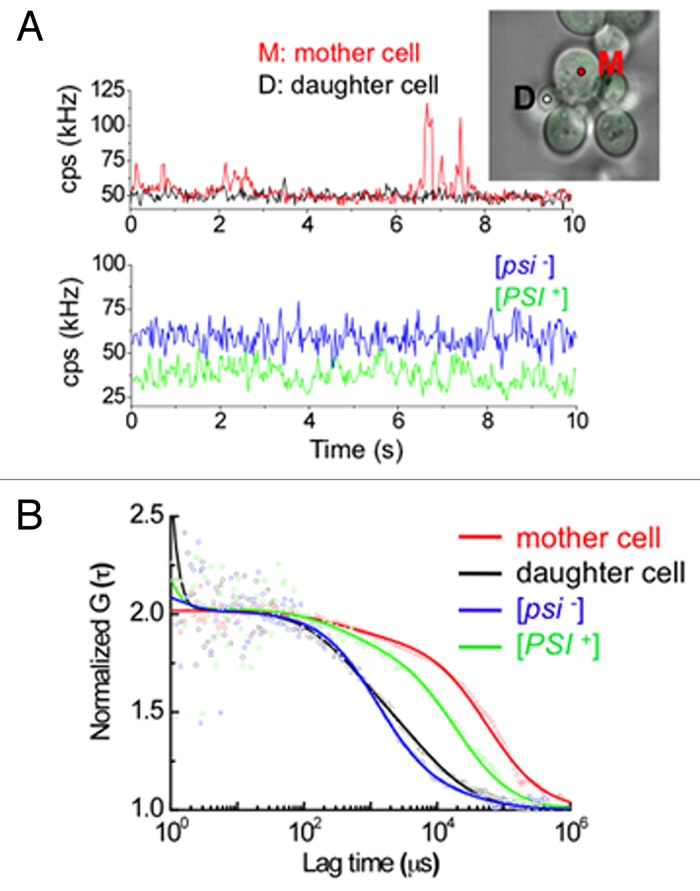
Figure 3. Diffusional properties of [PSI+] aggregates upon expression of Dip5ΔC-v82. (A) Traces of average fluorescence intensities (counts per second; cps) for Sup35-GFP in a single [PSI+] cell pair of mother (M, red) and daughter (D, black) 72 h after Dip5ΔC-v82 induction and traces of fluorescence intensities for Sup35-GFP in a single [psi-] cell (blue) and a single [PSI+] cell (green) without Dip5ΔC-v82. Insert: The fluorescent image shows the merge of confocal and light microscopic images of the mother and daughter cell pair used for FCS measurement. The white and red circles show the positions of the FCS measurement. (B) Normalized fluorescence autocorrelation functions of Sup35-GFP in a single [PSI+] cell pair of mother (red) and daughter (black) cells, 72 h after Dip5ΔC-v82 induction, and in a single [psi-] cell (blue) and a single [PSI+] cell (green) without Dip5ΔC-v82 induction. Each autocorrelation functions were respectively calculated from the fluorescence intensity fluctuations shown in (A). Solid lines depict the fitting of the functions by a two-component model.
Discussion
Propagation of yeast prions depends on the proper balance between several chaperone and cochaperone proteins. The prion clearance by excess Dip5ΔC-v82 is likely caused by the imbalance between these proteins. An excess of Dip5ΔC-v82 triggers the stress response HSR, which induces Hsps including Hsp104 and Ssa1, and most likely the shortage of the cochaperone Sgt2, which preferentially binds aggregation-prone proteins such as Dip5ΔC-v82.24 According to the model proposed by Chernoff and coworkers, Sgt2 increases access of Ssa1 to prion polymers, helps proper targeting of Hsp104 to polymer-bound Ssa1, and therefore results in normal prion fragmentation and propagation.13 We assume that, in the shortage of Sgt2, Ssa1 binds prion amyloids in a non-productive manner, which might prevent proper targeting of Hsp104 (Fig. 4, middle). This is conceivable since the role of Sgt2 is to coordinate chaperone interactions with various types of aggregates.28,29 Additionally, we found that the prion is stably maintained in the Sgt2 deletion strain (Fig. 2A). This is interpreted as indicating that Ssa1 binds efficiently the aggregates in the Sgt2 deletion strain, allowing Hsp104 to access and disaggregate them. On the other hand, excess of Sgt2 might lead to prion curing, probably masking the binding site for Ssa1 and thereby preventing the access of Hsp104 to the aggregates (Fig. 4, left). Several lines of observations are consistent with this “Ssa1-coated non-productive amyloid” hypothesis. First, [PSI+] was partially protected from clearance by excess Dip5ΔC-v82 with an excess of Sgt2 (Fig. 1B). Second, the frequency of [PSI+] clearance by excess Dip5ΔC-v82 was increased in proportion to the level of Ssa1 expression (Fig. 2B). Third, the size of [PSI+] amyloids increased by excess Dip5ΔC-v82 (Fig. 3). Fourth, it is known that excess Ssa1 increases the average size of prion polymers.11

Figure 4. Model for the effects of Sgt2 and Ssa1 on [PSI+] clearance by excess Dip5ΔC-v82. Chernoff and coworkers have proposed that in the wild-type strain Sgt2 increases access of Ssa1 to Sup35 prion polymers, helps proper targeting of Hsp104 to polymer-bound Ssa1, and therefore results in normal prion fragmentation and propagation.13 However, excess Dip5ΔC-v82 triggers HSR, which induces Hsp104 (hexagons) and Ssa1 (blue circles), and the shortage of Sgt2 (closed triangles) due to the titration onto Dip5ΔC-v82. Under these conditions, Ssa1 binds Sup35 prion polymers in a non-productive manner, which prevents proper entry of Hsp104, resulting in enlarged prion polymers unsuitable for proper transmission (middle). On the other hand, excess Sgt2 binds to the aggregates, allowing the entry of Ssa1 and Hsp104 onto the aggregates, leading to the prion propagation (left). In the sgt2Δ strain, Ssa1 doesn’t bind to the aggregates efficiently compared with wild type strain. However, Hsp104 might be able to bind to the aggregates, less efficiently but sufficiently to generate prion seeds for transmission (right).
The clearance of [PSI+] by excess Dip5ΔC-v82 is disabled by the get3Δ deletion24 as well as by get3Δ and get4Δ/get5Δ deletions (unpublished). In the GET pathway, Get5 and Get4 appear to regulate the handoff of substrates from Sgt2 to the downstream chaperone Get3, which functions in the ER integration of TA proteins.17,30,31 Therefore, in view of the fact that the prion clearance ability of Dip5ΔC-v82 is nullified by mutations of components in the GET complex pathway, it is tempting to speculate that the GET-pathway dependent accumulation of Dip5ΔC-v82 in the ER compartment is prerequisite for the clearance of yeast prions. One might speculate that, in the get deletion strains, excess Dip5ΔC-v82 might preferentially form cytoplasmic aggregates prior to access to Sgt2, and this in turn increases the fraction of free Sgt2 available for access to [PSI+] amyloids for normal prion propagation. Although further analysis of the mechanism underlying the yeast prion clearance of propagation is required, the take-home message from this study is that excess Ssa1 is able to eliminate yeast prions in the presence of Dip5ΔC-v82, and that the [PSI+] clearance by the imbalance between Ssa1 and Sgt2 might provide a novel means to regulate the release factor function in yeast.
Materials and Methods
Strains, plasmids and culture manipulations
S. cerevisiae strains used in this study are: NPK265 ([PSI+] [PIN+] MATa ade1–14 leu2Δ0 ura3–197 his3Δ200 trp1–289), NA120 ([PSI+] MATa ade1–14 leu2Δ0 ura3–197 his3Δ200 trp1–289 sgt2::KanMX), NPK608 (MATa ade1–14 leu2–3,112 ura3–52 his3Δ200 trp1–289 ssa1::KanMX) and ND21 ([PSI+] [pin-] MATa ade1–14 leu2 ura3 his3 trp1 sup35::SUP35-GFP) and ND20 ([psi-] [pin-] isogenic with ND21) (ND20 and ND21 are derivatives of G74-D694 [psi-]).25
Expression plasmids used in this study were constructed from pRS400 series vectors (Stratagene) carrying the CYC1, ADH and GPD promoters at the SacI-BamHI site and the CYC terminator at the XhoI-KpnI site. The Sgt2 and Ssa1 sequences were amplified by PCR using the following primers and cloned into the BamHI-XhoI site: P1 (CGCGGATCCATGTCAGCATCAAAAGAAG, all the sequence shown here are from 5′ to 3′) and P2 (CGCCTCGAGCTATTGCTTGTTCTCATTG) for wild-type Sgt2; P3 (CGCGGATCCATGTCAAAAGCTGTCGG) and P4 (CGCCTCGAGTTAATCAACTTCTTCAAC) for Ssa1.
The yeast media used were YPD, synthetic complete medium containing glucose (SC), Ssuc and Sgal.24
Fluorescence correlation spectroscopy
All of the FCS measurements were performed at 25°C on LSM510 confocal microscope combined with a ConfoCor 2 (Zeiss), as described in our previous studies.25-27 The fluorescence autocorrelation functions (FAF; G (τ)), from which the average diffusion time (τi), which is inversely proportional to the diffusion coefficient and the absolute number of fluorescent proteins in the detection volume are calculated, are obtained as follows;
| (Eq. 1) |
where I (t+τ) is the fluorescence intensity obtained by the single photon counting method in a detection volume at a delay time τ (brackets denote ensemble averages). The curve fitting for the multi-component model is given by:
| (Eq. | 2) |
where yi and τi are the fraction and the diffusion time of the component i, respectively, N is the total number of fluorescent molecules in the detection volume defined by the beam waist w0 and the axial radius z0, s is the structure parameter representing the ratio of w0 and z0. Structure parameter was determined with a standard Rh6G solution.27 The GFP fluorescence in living cells was excited by 488nm laser line with a minimal total power for enough signals to noise by adjusting the acousto-optical tunable filter. Five or ten sequential measurements of 10 s were performed in a single cell. The effect of photobleaching on FCS analysis was minimized by lowering the excitation intensity and by selecting cells with low fluorescence.
Glossary
Abbreviations:
- RF
release factor
- ER
endoplasmic reticulum
- MTM
multi-transmembrane
- UPR
unfolded protein response
- GET
guided entry of tail-anchored protein
- TA
tail-anchored
- HSR
heat shock response
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank H Taguchi and K Ito for the gift of strains and/or helpful discussion. This work was supported in part by grants from The Ministry of Education, Sports, Culture, Science and Technology of Japan (MEXT) to Y Nakamura and H Kurahashi.
References
- 1.Prusiner SB. . Novel proteinaceous infectious particles cause scrapie. Science 1982; 216:136 - 44; http://dx.doi.org/ 10.1126/science.6801762; PMID: 6801762 [DOI] [PubMed] [Google Scholar]
- 2.Cox BS. . ψ, a cytoplasmic suppressor of super-suppressor in yeast. Heredity 1965; 20:505 - 21; http://dx.doi.org/ 10.1038/hdy.1965.65 [DOI] [Google Scholar]
- 3.Wickner RB. . [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae.. Science 1994; 264:566 - 9; http://dx.doi.org/ 10.1126/science.7909170; PMID: 7909170 [DOI] [PubMed] [Google Scholar]
- 4.Sondheimer N, Lindquist S. . Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell 2000; 5:163 - 72; http://dx.doi.org/ 10.1016/S1097-2765(00)80412-8; PMID: 10678178 [DOI] [PubMed] [Google Scholar]
- 5.Ehrenberg M, Hauryliuk V, Crist CG, Nakamura Y. Translation termination, prion [PSI+], and ribosome recycling. In: Mathews MB, Sonenberg N, Hershey JWB, eds. Translational control in biology and medicine. New York: Cold Spring Harbor Laboratory Press 2007. [Google Scholar]
- 6.Liebman SW, Sherman F. . Extrachromosomal psi+ determinant suppresses nonsense mutations in yeast. J Bacteriol 1979; 139:1068 - 71; PMID: 225301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shorter J, Lindquist S. . Prions as adaptive conduits of memory and inheritance. Nat Rev Genet 2005; 6:435 - 50; http://dx.doi.org/ 10.1038/nrg1616; PMID: 15931169 [DOI] [PubMed] [Google Scholar]
- 8.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. . Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. [PSI+] Science 1995; 268:880 - 4; http://dx.doi.org/ 10.1126/science.7754373; PMID: 7754373 [DOI] [PubMed] [Google Scholar]
- 9.Shorter J, Lindquist S. . Hsp104 catalyzes formation and elimination of self-replicating Sup35 prion conformers. Science 2004; 304:1793 - 7; http://dx.doi.org/ 10.1126/science.1098007; PMID: 15155912 [DOI] [PubMed] [Google Scholar]
- 10.Newnam GP, Wegrzyn RD, Lindquist SL, Chernoff YO. . Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol Cell Biol 1999; 19:1325 - 33; PMID: 9891066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Allen KD, Wegrzyn RD, Chernova TA, Müller S, Newnam GP, Winslett PA, Wittich KB, Wilkinson KD, Chernoff YO. . Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI+]. [PSI+] Genetics 2005; 169:1227 - 42; http://dx.doi.org/ 10.1534/genetics.104.037168; PMID: 15545639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winkler J, Tyedmers J, Bukau B, Mogk A. . Chaperone networks in protein disaggregation and prion propagation. J Struct Biol 2012; 179:152 - 60; http://dx.doi.org/ 10.1016/j.jsb.2012.05.002; PMID: 22580344 [DOI] [PubMed] [Google Scholar]
- 13.Kiktev DA, Patterson JC, Müller S, Bariar B, Pan T, Chernoff YO. . Regulation of chaperone effects on a yeast prion by cochaperone Sgt2. Mol Cell Biol 2012; 32:4960 - 70; http://dx.doi.org/ 10.1128/MCB.00875-12; PMID: 23045389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, et al. . Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell 2005; 123:507 - 19; http://dx.doi.org/ 10.1016/j.cell.2005.08.031; PMID: 16269340 [DOI] [PubMed] [Google Scholar]
- 15.Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS. . The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 2008; 134:634 - 45; http://dx.doi.org/ 10.1016/j.cell.2008.06.025; PMID: 18724936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Angeletti PC, Walker D, Panganiban AT. . Small glutamine-rich protein/viral protein U-binding protein is a novel cochaperone that affects heat shock protein 70 activity. Cell Stress Chaperones 2002; 7:258 - 68; http://dx.doi.org/; PMID: 12482202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang F, Brown EC, Mak G, Zhuang J, Denic V. . A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol Cell 2010; 40:159 - 71; http://dx.doi.org/ 10.1016/j.molcel.2010.08.038; PMID: 20850366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurahashi H, Ishiwata M, Shibata S, Nakamura Y. . A regulatory role of the Rnq1 nonprion domain for prion propagation and polyglutamine aggregates. Mol Cell Biol 2008; 28:3313 - 23; http://dx.doi.org/ 10.1128/MCB.01900-07; PMID: 18332119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kurahashi H, Shibata S, Ishiwata M, Nakamura Y. . Selfish prion of Rnq1 mutant in yeast. Genes Cells 2009; 14:659 - 68; http://dx.doi.org/ 10.1111/j.1365-2443.2009.01297.x; PMID: 19371377 [DOI] [PubMed] [Google Scholar]
- 20.Kurahashi H, Pack CG, Shibata S, Oishi K, Sako Y, Nakamura Y. . [PSI(+)] aggregate enlargement in rnq1 nonprion domain mutants, leading to a loss of prion in yeast. Genes Cells 2011; 16:576 - 89; http://dx.doi.org/ 10.1111/j.1365-2443.2011.01511.x; PMID: 21453425 [DOI] [PubMed] [Google Scholar]
- 21.Shibata S, Kurahashi H, Nakamura Y. . Localization of prion-destabilizing mutations in the N-terminal non-prion domain of Rnq1 in Saccharomyces cerevisiae.. Prion 2009; 3:250 - 8; http://dx.doi.org/ 10.4161/pri.3.4.10388; PMID: 20009538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishiwata M, Kurahashi H, Nakamura Y. . A G-protein γ subunit mimic is a general antagonist of prion propagation in Saccharomyces cerevisiae.. Proc Natl Acad Sci U S A 2009; 106:791 - 6; http://dx.doi.org/ 10.1073/pnas.0808383106; PMID: 19129493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oishi K, Kurahashi H, Pack CG, Sako Y, Nakamura Y. Q/N-rich proteins: Lsm4 amyloid causes clearance of yeast prions. Microbiologyopen 2013; doi: http://dx.doi.org/ 10.1002/mbo3.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arai C, Kurahashi H, Ishiwata M, Oishi K, Nakamura Y. . Clearance of yeast prions by misfolded multi-transmembrane proteins. Biochimie 2013; 95:1223 - 32; http://dx.doi.org/ 10.1016/j.biochi.2013.01.009; PMID: 23384482 [DOI] [PubMed] [Google Scholar]
- 25.Kawai-Noma S, Pack CG, Tsuji T, Kinjo M, Taguchi H. . Single mother-daughter pair analysis to clarify the diffusion properties of yeast prion Sup35 in guanidine-HCl-treated [PSI] cells. Genes Cells 2009; 14:1045 - 54; http://dx.doi.org/ 10.1111/j.1365-2443.2009.01333.x; PMID: 19674118 [DOI] [PubMed] [Google Scholar]
- 26.Kawai-Noma S, Ayano S, Pack CG, Kinjo M, Yoshida M, Yasuda K, Taguchi H. . Dynamics of yeast prion aggregates in single living cells. Genes Cells 2006; 11:1085 - 96; http://dx.doi.org/ 10.1111/j.1365-2443.2006.01004.x; PMID: 16923127 [DOI] [PubMed] [Google Scholar]
- 27.Pack C, Saito K, Tamura M, Kinjo M. . Microenvironment and effect of energy depletion in the nucleus analyzed by mobility of multiple oligomeric EGFPs. Biophys J 2006; 91:3921 - 36; http://dx.doi.org/ 10.1529/biophysj.105.079467; PMID: 16950841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, et al. . High-quality binary protein interaction map of the yeast interactome network. Science 2008; 322:104 - 10; http://dx.doi.org/ 10.1126/science.1158684; PMID: 18719252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, Sherman MY. . Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J 2009; 23:451 - 63; http://dx.doi.org/ 10.1096/fj.08-117614; PMID: 18854435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mateja A, Szlachcic A, Downing ME, Dobosz M, Mariappan M, Hegde RS, Keenan RJ. . The structural basis of tail-anchored membrane protein recognition by Get3. Nature 2009; 461:361 - 6; http://dx.doi.org/ 10.1038/nature08319; PMID: 19675567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang F, Whynot A, Tung M, Denic V. . The mechanism of tail-anchored protein insertion into the ER membrane. Mol Cell 2011; 43:738 - 50; http://dx.doi.org/ 10.1016/j.molcel.2011.07.020; PMID: 21835666 [DOI] [PMC free article] [PubMed] [Google Scholar]