

Abstract
A3 adenosine receptor activation has been previously demonstrated to result in both neuroprotective and neurodegenerative effects, depending upon specific pathophysiological conditions. This dual effect may depend on receptor regulation mechanisms that are able to change receptor availability and/or function. In the present study, we investigated desensitization, internalization, and down-regulation of native A3 adenosine receptors in human astrocytoma cells after exposure to the agonist 2-chloro-N6-(3-iodobenzyl)-N-methyl-5'-carbamoyladenosine (Cl-IBMECA). Cl-IBMECA induced a concentration-dependent inhibition of adenylyl cyclase activity with an EC50 value of 2.9 ± 0.1 nM. The effect was suggested to be mediated by A3 adenosine receptor subtype by the use of selective adenosine receptor antagonists. Cell treatment with pertussis toxin abolished Cl-IBMECA-mediated inhibition of adenylyl cyclase activity, evidencing an A3 receptor coupling to inhibitory G protein. Short-term exposure to the agonist Cl-IBMECA (100 nM) caused rapid receptor desensitization, within 15 min. Agonist-induced desensitization was accompanied by receptor internalization: A3 adenosine receptor internalized with rapid kinetics, within 30 min, after cell exposure to 100 nM Cl-IB-MECA. The localization of A3 adenosine receptors on the plasma membrane and in intracellular compartments was directly revealed by immunogold electron microscopy. After desensitization, the removal of agonist led to the restoration of A3 adenosine receptor functioning through receptor recycling to the cell surface within 120 min. Prolonged agonist exposure (1–24 h) resulted in a marked down-regulation of A3 adenosine receptors that reached 21.9 ± 2.88% of control value after 24 h. After down-regulation, the recovery of receptor functioning was slow (24 h) and associated with the restoration of receptor levels close to control values. In conclusion, our results demonstrated that A3 receptors, in astrocytoma cells, are regulated after short- and long-term agonist exposure.
Actions of adenosine are mediated by four G protein-coupled membrane receptors (GPCRs): A1, A2A, A2B, and A3 receptors (Fredholm et al., 1994). Although expressed at very low levels in mammalian brain, the A3 adenosine receptor (AR) subtype has been implicated in behavioral depression (Jacobson et al., 1993) and modulation of ischemic cerebral damage (for review, see von Lubitz, 1999). Through the development of selective A3 AR agonists (e.g., Cl-IBMECA) and antagonists (e.g., MRS 1191 and MRS 1220) (Kim et al., 1994, 1996; Jacobson et al., 1997), the putative pathophysiological roles of this receptor have became clear. It has been demonstrated that A3 AR agonists profoundly affect cell survival, by promoting cell protection or cell death, depending upon the cell type and/or agonist concentration (for reviews, see Jacobson, 1998; Jacobson et al., 1999). In human astrocytoma cells (ADF cells), exposure to nanomolar Cl-IBMECA concentrations increased resistance to apoptosis, by a mechanism involving signaling to the small G proteins Rho, cytoskeletal rearrangement, and intracellular redistribution of the antiapoptotic protein Bcl-XL (Abbracchio et al., 1997, 2001). The cytoprotective effect induced by nanomolar Cl-IBMECA concentrations is specifically mediated by activation of the A3 AR, as shown by the ability of selective A3 AR antagonists to fully prevent this response (Abbracchio and Burnstock, 1998) and by the recent demonstration that despite low levels of expression in brain-derived tissues (Jacobson et al., 1993; Fredholm et al., 1994), ADF cells indeed express the A3 AR protein to significant levels (Abbracchio et al., 2001). In contrast, Cl-IBMECA induced cell death in ADF cells when used at concentrations in the high micromolar range (Abbracchio and Burnstock, 1998). In the same way, micromolar concentrations of Cl-IBMECA markedly impaired cell cycle progression in CHO cells transfected with the human A3 receptor (CHO-A3R) (Brambilla et al., 2000). Although interpretation of the latter results is still debated (effects induced by micromolar Cl-IBMECA concentrations were prevented by A3 AR antagonists in CHO-A3R, but not in human ADF cells; see Brambilla et al., 2000), the differential effects induced by different A3 AR agonist concentrations on cell survival may depend upon agonist-induced A3 AR desensitization and down-regulation.
All A3 AR-induced effects are initiated by interaction of agonist-occupied receptors with members of the Gi family of guanine nucleotide-binding regulatory proteins (G proteins) (Ali et al., 1990; Palmer et al., 1995b). Like many other GPCRs, intracellular signals initiated by agonist-occupied A3 ARs are subject to several regulation processes, including homologous desensitization and internalization (short-time response), as well as down-regulation (long-time response) as demonstrated in studies on CHO cells (Ali et al., 1990; Palmer et al., 1995a, 1997; Palmer and Stiles, 2000; Trincavelli et al., 2000). Because desensitization is also influenced by receptor density and transfected receptors are often overexpressed in engineered cellular systems, to define its exact pathophysiological significance, it is important that the information obtained in such experimental systems is also confirmed in native cells. At present, no data are available on the mechanisms involved in the regulation of A3 AR in native systems, because of low expression levels of this receptor subtype, coexpression with other receptor subtypes, and lack of studies carried out with specific and selective agonists/antagonists. In the present study, we investigated, for the first time, the desensitization, internalization, and down-regulation of A3 ARs in human ADF cells after treatment of cells with the selective agonist Cl-IBMECA.
Materials and Methods
Materials
N6-(4-Amino-3-[125I]iodobenzyl)adenosine-5'-N-methyluronamide ([125I]AB-MECA) and [α-32P]ATP were from PerkinElmer Life Sciences (Köln, Germany). Cl-IBMECA was supplied by National Institutes of Health (Bethesda, MD). Cell culture media and fetal calf serum were from EuroClone (Pero, Italy) and Roche Applied Science (Mannheim, Germany), respectively. Electrophoresis reagents were from Bio-Rad (Hercules, CA). MRS 1191 and MRS 1220 antagonists, DPCPX, and pertussis toxin were from Sigma-Aldrich (St. Louis, MO). All other chemicals were supplied from standard commercial sources. Human A3 AR antibody and human A3 AR control peptide were supplied by Alpha Diagnostic (San Antonio, TX) and gold-conjugate secondary antibodies (ImmunoGold reagents) were from Aurion (Wageningen, The Netherlands). CHO-A1 and CHO-A3 cells were kindly supplied by K. N. Klotz (University of Würzburg, Würzburg, Germany).
Cell Culture
Human astrocytoma ADF cells were grown adherently at 37°C in humidified atmosphere as described previously (Abbracchio et al., 1997) and maintained in RPMI 1640 medium containing 10% fetal calf serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, and 1% nonessential amino acids. Cells were split two or three times per week at ratio between 1:5 and 1:20 and used at subconfluence for adenylyl cyclase activity, internalization, and down-regulation experiments. Cells had viability >95%, as assessed by the exclusion of trypan blue. CHO cells were grown as described previously (Trincavelli et al., 2000).
Antibody Neutralization by Preabsorption
To evaluate anti-A3 AR antibody-nonspecific binding, a blocking peptide corresponding to the peptide antigen (Alpha Diagnostic) was used. Primary antibody binding was neutralized by preabsorption with blocking peptide. A3 AR antibody was incubated overnight at 4°C with a 5-fold (by weight) excess of blocking peptide in a small volume of phosphate-buffered saline (PBS). In parallel, the same amount of A3 AR antibody was incubated overnight at 4°C in PBS alone. The signals obtained in experiments with neutralized and non-neutralized antibody were then compared. The final working dilutions of treated and nontreated antibody were 1:20 in immunogold electron microscopy and 1:1000 in immunoblot assay, respectively.
Desensitization/Resensitization Studies
The coupling of A3 ARs to Gi proteins in ADF cells was assessed by evaluating the ability of the agonist Cl-IBMECA (1 nM–1 μM) to inhibit adenylyl cyclase activity stimulated by 10 μM forskolin. Adenylyl cyclase assays were performed as described by Olah et al. (1994), except for the use of 20 μM Ro201724 as phosphodiesterase inhibitor. Cl-IBMECA-mediated inhibition of adenylyl cyclase activity was also assayed in the presence of 100 nM DPCPX or 1 nM to 10 μM MRS 1191 and 1 nM to 10 μM MRS 1220. A3 AR coupling to inhibitory G protein was evaluated using 300 ng/ml pertussis toxin (Suh et al., 2001).
Desensitization treatments were carried out by incubating cells with the agonist Cl-IBMECA (100 nM) for 1 to 60 min at 37°C. To evaluate receptor resensitization, cells were desensitized for short (30-min) and long (24-h) time periods and then reincubated at 37°C in agonist-free medium for various times. After desensitization/resensitization treatments, medium was removed and cell monolayer was washed quickly three times with 5 ml of warm culture medium before cyclase assay. In each experiment, an appropriate number of flasks was used so that cells could be harvested before desensitization, after desensitization, and during the resensitization period. After cell treatment, membranes were then prepared and immediately tested for adenylyl cyclase activity.
Receptor Internalization/Recycling
Radioligand Binding Studies
A3 AR internalization was quantified by evaluating the changes of receptor surface density after treatment of the cells with agonist at 37°C for different times (Edwardson and Szekeres, 1999). Cells were incubated with 100 nM Cl-IBMECA at 37°C for 5 to 90 min. At the end of incubation, cells were placed on ice and rapidly washed three times with 120 mM NaCl, 5 mM KCl, 2 mM CaCl2, 50 mM Tris, and 1 mM EDTA, pH 3.5 (acid T1 buffer) to remove agonist. Then, cells were incubated with 0.5 nM [125I]AB-MECA at 4°C to prevent receptor cycling, in T1 buffer at pH 8.12. The assay was performed in the absence or in the presence of 100 nM Cl-IBMECA for nonspecific binding determination.
In receptor recycling studies, ADF cells were incubated with 100 nM Cl-IBMECA for 30 min at 37°C, rapidly acid-washed for 1 min, and then rewarmed at 37°C in the culture medium, in the absence of the A3 AR agonist. Then, [125I]AB-MECA binding was carried out at 4°C to evaluate the receptor surface binding activity (Trincavelli et al., 2000).
Immunogold Electron Microscopy
ADF cells, grown to subconfluence on 10-mm2 plates, were treated for 30 min at 4°C, for 10 to 60 min at 37°C with 100 nM Cl-IBMECA, or for 30 min at 37°C with 100 nM Cl-IBMECA in the presence of 100 nM MRS 1220, washed with PBS, and fixed for1h at 4°C with 4% formaldehyde and 1% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2. Cells were scraped, pelleted, and postfixed with 0.5% osmium tetroxide in the same buffer. Samples were then dehydrated with ethanol, infiltered with Unicryl (British Biocell International, Cardiff, UK) and allowed to polymerize for 3 days at 4°C under UV light. Thin sections were placed on formvar carbon-coated nickel grids and incubated for 1 h on a drop of 5% fish gelatin in PBS at room temperature. After washing in PBS, grids were floated on a drop of rabbit polyclonal antibodies raised against human A3 AR and diluted 1:20 in PBS containing 0.1% gelatin and 0.5% bovine serum albumin. Incubation of the primary antibody was prolonged overnight in a humid chamber set at 4°C. After rinses in a solution containing 0.1% gelatin, 0.5% bovine serum albumin, and 0.05% Tween 20 in PBS, grids were incubated for 1 h at room temperature with gold-conjugated goat anti-rabbit IgG, diluted 1:10 with 1% fish gelatin in PBS. Sections were finally fixed with 2% glutaraldehyde for 15 min at room temperature, washed with distilled water, and stained with uranyl acetate and lead citrate. Controls were run using antigenically unrelated primary antibodies. Ultrathin sections were examined using a 100 SX electron microscope (JOEL, Tokyo, Japan).
Down-Regulation and Receptor Recovery Assays
Down-regulation assays were performed by assessing changes in the total levels of A3 ARs as determined by immunoblotting. Cells were incubated with 100 nM Cl-IBMECA at various time intervals (1–24 h) at 37°C. To evaluate receptor recovery, cells were treated with agonist for 24 h and then reincubated at 37°C in agonist-free medium (1–24 h). After incubations, cells were washed extensively with PBS buffer and solubilized by scraping into 1 ml of lysis buffer radioimmunoprecipitation assay (9.1 mM Na2H2PO4, 1.7 mM Na2HPO4, 150 mM NaCl, pH 7.4, 0.5% sodium deoxycholate, 1% Nonidet P-40, and 0.1% SDS, containing protease inhibitors). Cell lysis was carried out for 60 min at 4°C. After centrifugation at 15,000g for 30 min, soluble fraction was assayed for protein content using protein kit (Bio-Rad) with bovine serum albumin as standard.
SDS-PAGE and Immunoblotting
Equivalent amounts of protein (typically 100 μg/sample) were resolved by SDS-PAGE using 12% (w/v) polyacrylamide gels. The appropriate amounts of cell lysate were prepared for electrophoresis by boiling for 5 min before loading for SDS-PAGE. Resolved proteins were transferred to nitrocellulose and anti-human A3 AR primary antibody was used for immunoblotting at concentrations of 1 μg/ml overnight at 4°C. After extensive washing with Tris-buffered saline (10 mM Tris-HCl and 150 mM NaCl, pH 8), containing 0.05% Tween 20, nitrocellulose membrane was incubated for 120 min at room temperature with horseradish peroxidase goat anti-rabbit-conjugated secondary antibody diluted to 1:2000 in Blotto A (Tris-buffered saline, 0.05% Tween 20, and 5% low-fat dried milk). After a second series of washes, reactive proteins were visualized by the enhanced chemiluminescence protocol ECL (Amersham Biosciences, Piscataway, NJ). The same membranes were stripped and treated with primary antibody against actin (1:500) for 2 h at room temperature and then with horseradish peroxidase goat secondary antibody (1:15,000). Immunoreactive bands were visualized by chemiluminescence and quantified by densitometric scanning of films exposed in the linear range using an image analysis system (GS-670; Bio-Rad). The optical density of each sample was corrected by the optical density of the corresponding actin bands.
In all experiments (desensitization, internalization, and down-regulation) agonist/antagonist cell treatments were performed in the presence of 1 U/ml adenosine deaminase to remove endogenous adenosine.
Data Analysis
Data analysis was performed with the nonlinear multipurpose curve-fitting computer program GraphPad Prism (GraphPad Software, San Diego, CA). Significant differences between measurements were calculated using GraphPad InStat.
Results
A3 AR-Mediated Effects on Adenylyl Cyclase in Human Astrocytoma Cells
The functional coupling of A3 AR to inhibitory G proteins in ADF cells was assessed by evaluating the ability of the A3 AR agonist Cl-IBMECA to inhibit forskolin-stimulated adenylyl cyclase activity. Results showed that Cl-IBMECA (1 nM–10 μM) induced a concentration-dependent inhibition of forskolin-stimulated adenylyl cyclase activity with an EC50 value of 2.9 ± 0.1 nM (Fig. 1). The dose-response effect of this agonist on adenylyl cyclase suggested the presence of more than one AR subtype in these cells, probably represented by the A1 AR and A3 AR (Jacobson et al., 1997). To shed light on this issue, Cl-IBMECA inhibition curve on adenylyl cyclase was also carried out in the presence of the A1 AR antagonist DPCPX at 100 nM concentration, able to selectively block the A1 AR subtype (Gessi et al., 2001). Under these experimental conditions, Cl-IBMECA inhibited forskolin-stimulated adenylyl cyclase activity (Fig. 2) with an EC50 value of 1.8 ± 0.12 nM. This value was comparable with the EC50 value of Cl-IBMECA on human A3 AR in CHO-transfected cells (Jacobson et al., 1997). The ability of the selective A3 AR antagonists MRS 1191 and MRS 1220 to reduce 100 nM and 1 μM Cl-IBMECA-mediated adenylyl cyclase inhibition was also evaluated. MRS 1191 and MRS 1220 completely antagonized cAMP inhibition mediated by 100 nM Cl-IBMECA, with an EC50 value of 304 ± 20 and 3.8 ± 0.1 nM, respectively (Fig. 3A). These results suggested that the effect evoked by 100 nM Cl-IBMECA was primarily mediated by the A3 AR subtype. On the contrary, the two selective A3 AR antagonists did not completely antagonize the inhibitory effects induced by higher agonist concentration (1 μM) (Fig. 3B), indicating that at micromolar concentration, Cl-IBMECA may also activate other coexpressed adenosine receptor subtypes (i.e., A1 AR). On this basis, to investigate the homologous A3 AR regulation mechanisms we selected an agonist concentration of 100 nM, which selectively activated the A3 AR subtype in our experimental conditions.
Fig. 1.
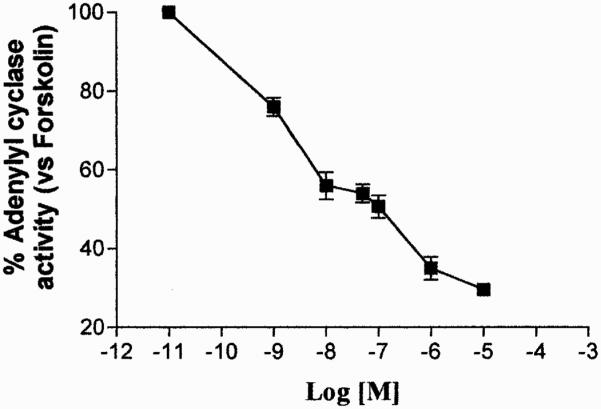
Inhibition curve of forskolin-stimulated adenylyl cyclase activity by Cl-IBMECA in ADF cells. Data (means of three experiments ± S.E.M.) are given as percentage of forskolin stimulated adenylyl cyclase activity set to 100%.
Fig. 2.
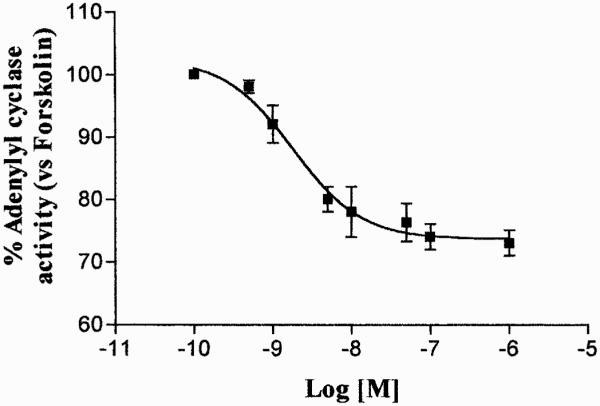
Inhibition curve of forskolin-stimulated adenylyl cyclase activity by Cl-IBMECA in the presence of 100 nM DPCPX. Data (means of three experiments ± S.E.M.) are given as percentage of forskolin stimulated adenylyl cyclase activity set to 100%.
Fig. 3.

Effect of the A3 AR antagonists MRS 1220 and MRS 1191 on Cl-IBMECA (A, 100 nM; B, 1 μM) inhibition of forskolin-stimulated adenylyl cyclase activity in ADF cells. Cell membranes were incubated with the agonist in the presence of the antagonist concentrations reported on the x-axis (1 nM–10 μM); adenylyl cyclase activity is expressed as percentage of basal value (100%). Data represent means ± S.E.M. from three separate experiments. The EC50 values for MRS 1220 and MRS 1191 were 3.8 ± 0.1 and 304 ± 20 nM, respectively.
Finally, we performed adenylyl cyclase experiments in cultures pre-exposed to pertussis toxin, which selectively uncouples Gi/Go proteins from GPCRs by catalyzing the ADP-ribosylation of the αi/α0 subunits (Simon et al., 1991). As shown in Table 1a, although forskolin-stimulated adenylyl cyclase activity was unaffected, Cl-IBMECA-mediated inhibitory effect on cAMP production was almost completely prevented by a 12-h treatment of cells with 300 ng/ml pertussis toxin. These data indicate that, in these cells, A3 ARs are coupled to adenylyl cyclase through pertussis toxin-sensitive inhibitory G proteins.
TABLE 1.
Effects of pertussis toxin on Cl-IBMECA-induced inhibition of cyclic AMP production in human ADF cells
| cAMP Production |
||
|---|---|---|
| Vehicle | Pertussis Toxin-Treated | |
| pmol/min/mg | ||
| Basal | 61.8 ± 3.4 | 73.8 ± 6.3 |
| Forskolin | 182 ± 11.2 | 227 ± 14.3 |
| Cl-IBMECA | 138 ± 9.8* | 212 ± 15.2** |
, p < 0.05 with respect to corresponding value of forskolin alone.
, not statistically different from corresponding value of forskolin-stimulation of adenylyl cyclase alone.
Confluent cells were pretreated with either 300 ng/ml pertussis toxin or vehicle for 12 h, and the effect of 100 nM Cl-IBMECA on forskolin-stimulated cAMP production was measured in membrane cell preparations as described under Materials and Methods. Data are presented as mean ± S.E.M. of three independent experiments.
Desensitization and Internalization of A3 AR after Short-Term Agonist Exposure
The functional desensitization of native A3 ARs in ADF cells was determined by analysis of Cl-IBMECA-mediated inhibition of forskolin-stimulated adenylyl cyclase activity in isolated membranes before and after cell treatment with the agonist (100 nM) for 1 to 60 min at 37°C. In control membranes the agonist (100 nM) inhibited forskolin-stimulated adenylyl cyclase activity by 33.3 ± 0.6%. Upon agonist exposure, A3 AR underwent desensitization with rapid kinetics (t1/2 of 3.23 ± 0.22 min−1) (Fig. 4). The concomitant presence of the A3 AR antagonist MRS 1220 (100 nM) completely prevented agonist-mediated desensitization, demonstrating that loss of function is mediated by a specific A3 AR regulation mechanism (Fig. 4). Short-term desensitization did not result in any changes in the efficacy of forskolin to stimulate adenylyl cyclase activity, suggesting that G proteins were not affected by the desensitization process (data not shown). Therefore, the observed desensitization process is specifically due to the impairment of A3 AR ability to effectively interact with Gi protein.
Fig. 4.
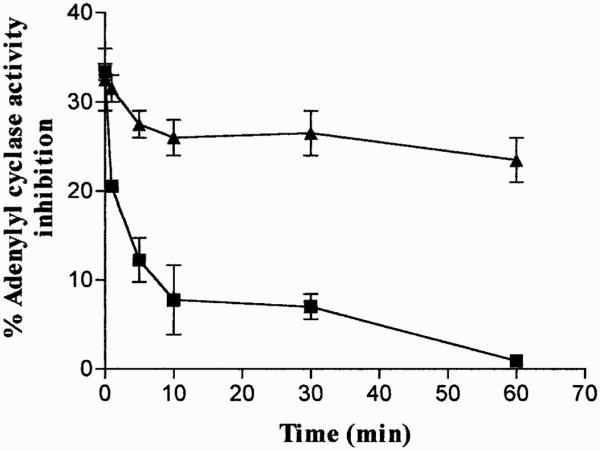
A3 AR desensitization kinetics. ADF cell monolayers were treated with Cl-IBMECA (100 nM) for 1 to 60 min at 37°C in the absence (f) or the presence (Œ) of MRS 1220 (100 nM). At each time point, cells were washed to remove agonist/antagonist and membranes were prepared for adenylyl cyclase activity. Desensitization of A3 AR responsiveness was evaluated as the ability of the agonist Cl-IBMECA (100 nM) to inhibit forskolin-stimulated adenylyl cyclase activity compared with untreated control cells (33.3 ± 0.6% inhibition). Data are mean ± S.E.M. of three separate experiments performed with similar results.
Resensitization of A3 ARs was also investigated and data obtained showed that, after removal of the agonist, A3 AR responsiveness resensitized to 100% of control values within 120 min (Fig. 5).
Fig. 5.
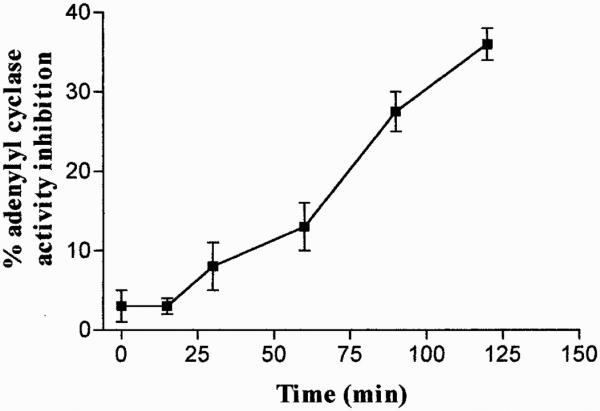
Resensitization of A3 AR signal in ADF cells. After desensitization with 100 nM Cl-IBMECA for 30 min at 37°C, agonist removal, and incubation in agonist-free medium for distinct time intervals, cells were harvested and assessed for adenylyl cyclase activity. Data are means ± S.E.M. values of three separate experiments and are expressed as percentage of forskolin-stimulated adenylyl cyclase inhibition.
To correlate the agonist-induced loss of A3 AR function with receptor internalization, we evaluated the changes in the number of A3 AR remaining on the cell surface after cell treatment with 100 nM Cl-IBMECA. Specific binding of [125I]AB-MECA to A3 AR was determined in the presence of 100 nM Cl-IBMECA. The association kinetic of [125I]AB-MECA binding to the receptor cell surface was rapid and reached equilibrium after 90-min incubation (Fig. 6). To measure receptor internalization in response to stimulation, cells were treated with agonist for 5 to 60 min at 37°C, to allow receptor cycling between the plasma membrane and intracellular compartment, and then the number of cell surface receptors was quantified by using [125I]AB-MECA. After 30-min agonist treatment, 85% of the total cell surface receptors were internalized (Fig. 7). The half-time of internalization was estimated to be 11.79 ± 0.97 min and the rate constant of receptor internalization was 0.0588 ± 0.0033 min−1.
Fig. 6.
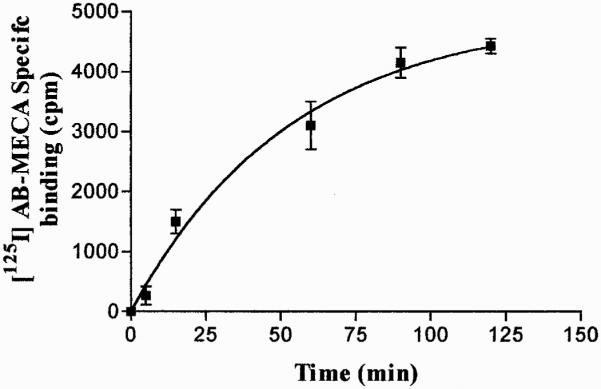
[125I]AB-MECA association kinetics to surface human A3 ARs. Cells were exposed to 0.5 nM [125I]AB-MECA at 4°C in the presence or in the absence of 100 nM Cl-IBMECA for nonspecific binding determination. The amount of [125I]AB-MECA associated with the receptor was determined at each time. Data are means ± S.E.M. values of three determinations. Specific binding at equilibrium (120 min) was 4425 ± 125 cpm.
Fig. 7.
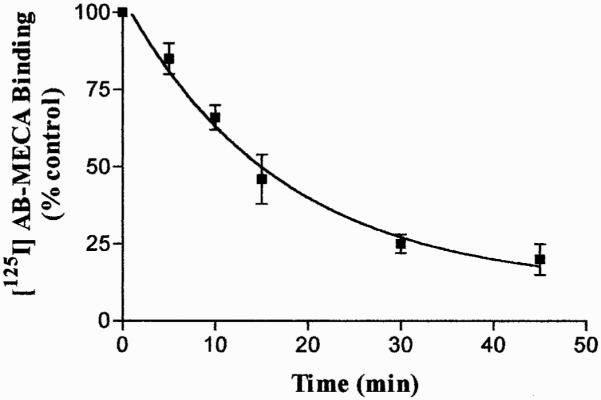
Time course of Cl-IBMECA-induced loss of cell surface [125I]AB-MECA binding sites. Cells were incubated with 100 nM Cl-IBMECA at 37°C for different incubation times (5–60 min). After incubation cells were washed to remove agonist and A3 ARs on the cell surface was evaluated by measuring the [125I]AB-MECA binding at 4°C for 120 min. Data, expressed as percentage of total binding versus control untreated cells (100%), represented the mean ± S.E.M. (bars) values of three different determinations.
A direct visualization of surface and internalized receptors was achieved by sequential labeling of the receptors with human A3 AR antibody followed by gold-conjugated goat anti-rabbit secondary antibody. To evaluate nonspecific antibody binding, grids were incubated with both neutralized and non-neutralized antibody (Fig. 8, A and B). No signal was detected in the presence of blocking peptide, demonstrating that the majority of gold particles really indicate A3 AR (Fig. 8B). Incubation with Cl-IBMECA for 30 min at 4°C did not result in receptor internalization: gold particles were distributed on the cell surface usually in small aggregates and rarely as single particles (Fig. 9A). After incubation with the agonist at 37°C for 10 min, receptors were found predominantly in large uncoated pits and vesicles (Fig. 9, B and C). In addition smaller coated pits and vesicles not containing any gold particles were observed in the same area, showing that sample preparation was also adequate to identify coated structures. After 30 min of exposure to the agonist, receptors were localized in the deeper cytoplasm in endosome-like structures (Fig. 9D). In addition, when cells were incubated for 30 min with agonist in the presence of an excess of the A3 AR-selective antagonist MRS 1220 (100 nM), gold particles were visible only on the cell surface, suggesting that internalization occurred by a receptor-mediated pathway (Fig. 9F).
Fig. 8.

A, labeling of human ADF cells with A3 AR polyclonal antibody revealed by immunogold electron microscopy. Clusters of gold particles are visible on the cell surface, including cytoplasmic projections (arrow). B, labeling of human ADF cells with neutralized A3 AR antibody preadsorbed with blocking peptide. No gold particles are visible on the cell surface. Scale bars, 0.25 μm.
Fig. 9.

Localization of A3 ARs in human astrocytoma cells by immunogold electron microscopy. Scale bars, 0.25 μm. A, preincubation of cells with 100 nM Cl-IBMECA at 4°C for 30 min. Small clusters and single particles of gold (arrow) are visible on the plasma membrane. B, preincubation of the cells with 100 nM Cl-IBMECA at 37°C for 10 min. Smooth-surfaced pit with a cluster of gold particles (arrow). C, preincubation of the cells with 100 nM Cl-IBMECA at 37°C for 10 min. Uncoated vesicle containing gold particles in the cortical cytoplasm. D, preincubation of the cells with 100 nM Cl-IBMECA at 37°C for 30 min. Gold particles are visible in vesicular endosomes. E, preincubation of cells with 100 nM Cl-IBMECA at 37°C for 60 min. Arrows point to gold particles presumably recycled on the plasma membrane. F, preincubation of cells with 100 nM Cl-IBMECA plus 100 nM MRS 1220 at 37°C for 30 min. Gold particles are localized only on the cell surface.
After 60 min of agonist incubation, gold particles were visible in small vesicles both under and on the plasma membrane, suggesting recycling of A3 AR to the cell surface (Fig. 9E). Receptor recycling was also demonstrated by radioligand binding studies inducing one round of receptor internalization and measuring the re-expression kinetic of [125I]AB-MECA binding sites on the cell surface (Fig. 10). The results showed that internalized receptors are quantitatively cycled back to the plasma membrane within 120 min with a rate constant of 0.0056 ± 0.00023 min−1.
Fig. 10.
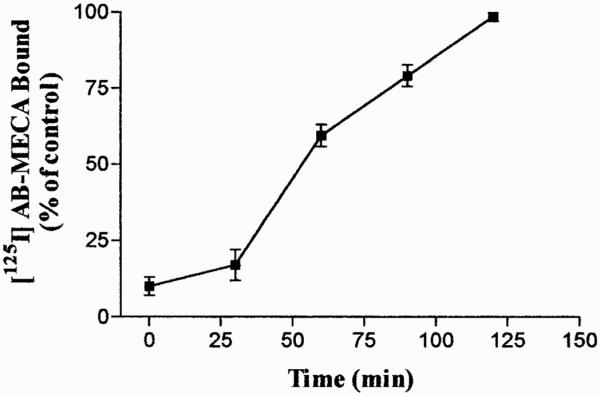
Surface re-expression of [125I]AB-MECA receptor binding after one round of internalization. Cells were incubated with 100 nM unlabeled Cl-IBMECA for 30 min at 37°C to allow A3 AR internalization. Upon removal of the unlabeled agonist, cells were maintained at 37°C for different time periods. The recovery of A3 ARs on the cell surface was evaluated by measuring the [125I]AB-MECA binding at 4°C for 120 min. Data are mean ± S.E.M. (bars) values of three different experiments.
Down-Regulation of A3 AR after Long-Term Agonist Exposure
To analyze the possible changes of the total A3 AR population after long-term Cl-IBMECA exposure, we performed immunoblotting experiments. As a first step, to assess the specificity of the anti-A3 AR antibody used in the present study, CHO cells transfected with either the human A3 AR or A1 AR were used as positive and negative controls, respectively. A specific immunoreactive band at a molecular mass of 36 kDa corresponding to the A3 AR was detected in CHO-A3 AR and ADF cells, but not in CHO-A1 AR cells (Fig. 11A). To further confirm antibody specificity, immunoblotting experiments performed in parallel by using neutralized primary antibody revealed no signals at the molecular mass corresponding to the A3 AR protein (Fig. 11B).
Fig. 11.

Immunoblot analysis of the A3 AR in CHO A1, CHO A3, and ADF cells. Cells were lysed and equal amounts of protein (100 μg) were electrophoresed in SDS-containing buffer. Immunoblotting was performed using A3 AR antibody (A) or A3 AR neutralized antibody preadsorbed with blocking peptide (B). No immunoreactive bands were detected in CHO A1 cells (used as a negative control) and upon utilization of neutralized antibody.
Cells exposed for 1 to 24 h to A3 AR agonist showed a time-dependent reduction of A3 AR, as detected by immunoblot analysis. After 1 h of agonist exposure, it was already possible to detect a reduction in A3 AR protein levels (69.2 ± 2.8% versus control set to 100%); such a reduction increased over time and reached its maximum value after 24 h (21.9 ± 2.88% versus control) (Fig. 12). These results demonstrated that, in human ADF cells, A3 AR is down-regulated in response to long-term agonist stimulation. Therefore, under conditions of agonist treatment resulting in a profound reduction of A3 AR levels (e.g., 24-h agonist treatment), the signaling capacity of the A3 AR was maintained in a desensitized state.
Fig. 12.

Down-regulation of A3 AR in ADF cells. Cells were grown in the absence (control, C) or presence of 100 nM Cl-IBMECA for the indicated time periods. Cells were then lysed and equal amounts of protein (100 μg) were electrophoresed in SDS-containing buffer. Immunoblotting was performed using A3 AR antibody as described under Materials and Methods. A, representative immunoblot. Arrow indicates the 36-kDa electrophoretic band corresponding to A3 AR. B, semiquantitative analysis of the receptor performed by image densitometry and expressed as percentage of the receptor amount found in control untreated cells (100%). Data are mean ± S.E.M. from three separate experiments. **, p < 0.01; ***, p < 0.001.
As shown in Fig. 13A, long-term exposure to the A3 AR agonist did not result in a significantly greater reduction of A3 AR functioning with respect to that observed after 30 min. However, removal of Cl-IBMECA after 24 h of agonist exposure resulted in a recovery of A3 AR responsiveness with a slow kinetics that occurred over a period of several hours. After induction of A3 AR down-regulation, agonist removal, and maintenance of cells in agonist-free medium for 1 to 24 h, the time-dependent recovery of A3 AR responsiveness (evaluated as cyclase inhibition) was paralleled by the recovery of receptor protein levels (evaluated by immunoblot analysis) (Fig. 13B), suggesting that the two phenomena are related to each other.
Fig. 13.

Recovery of A3 AR function after long-term desensitization. ADF cells were exposed to 100 nM Cl-IBMECA for 24 h before extensive washing in PBS buffer, addition of agonist-free medium, and further incubation at 37°C for indicated times. A, recovery of A3 AR responsiveness assessed by adenylyl cyclase assay. Data are means ± S.E.M. values of three separate experiments, expressed as percentage of forskolin-stimulated adenylyl cyclase activity inhibition. B, time course of A3 AR recovery, as evaluated by immunoblotting: B', representative immunoblot. B", corresponding densitometric analysis of immunoreactive bands. Data are expressed as percentage of total A3 AR levels (untreated cells), set to 100%, and represent means ± S.E.M. from three separate experiments.
Discussion
In the present article, we describe for the first time A3 AR regulation mechanisms in human ADF cells that natively express this receptor subtype. Stimulation of GPCRs by their agonists results in activation of heterotrimeric G proteins, leading to the regulation of a variety of signal transduction events in the cells. One of the major mechanisms modulating the cellular response to agonists is regulation of GPCRs themselves (Bohm et al., 1997). The phenomenon whereby receptor signaling responses plateau and then diminish despite the continuous presence of agonist is termed desensitization and is induced by the uncoupling of GPCR from its associated G protein (Bunemann and Hosey, 1999). Moreover, the responsiveness of target cells is also critically dependent on the subcellular distribution of the receptors, and agonist-induced endocytosis is important in the desensitization and resensitization of signaling (for reviews, see Ferguson, 2001; von Zastrow, 2001). Down-regulation is caused by long-term receptor exposure to agonists for hours to days and controls total receptor levels either through intracellular protein degradative pathways and/or by modulating mRNA transcription and translation; recovery of receptor responsiveness is similarly slow, because long-term agonist exposure may result in a distinct phosphorylation pattern or in a particular receptor conformation that exposes lysosomal targeting sequences, leading the receptor away from the recycling pathway (Tsao et al., 2001; Tsao and von Zastrow, 2001).
So far, no studies have been focused on the pharmacological characterization of the regulation of the A3 AR in native cell systems. The receptor protein has been identified by radioligand binding only in relatively low number of cells in both human and rodent (Ji et al., 1994; Olah et al., 1994; Gessi et al., 2001, 2002; Merighi et al., 2001), which is surprising given the many actions ascribed to the A3 AR. Nevertheless, the gene transcript for the A3 AR has been detected by polymerase chain reaction in many central tissues (Salvatore et al., 1993). Pharmacological studies, with Cl-IBMECA, a relatively selective agonist at the rat and human A3 receptor subtype (Kim et al., 1994), demonstrated an important role of this receptor in the control of neuronal cell death in brain slices or astroglial cells maintained in vitro (Abbracchio et al., 1997; Appel et al., 2001). In particular, in ADF cells, we have demonstrated the presence of the A3 AR and its involvement in the control of cytoskeletal rearrangement (Abbracchio et al., 2001) and of the intracellular distribution of the antiapoptotic protein Bcl-XL (Abbracchio et al., 1997), events that may be at the basis of modulation of cell survival by this receptor.
Several studies have described agonist-mediated A3 AR desensitization and internalization in transfected cell lines (Palmer et al., 1997; Trincavelli et al., 2000) and also demonstrated that the agonist-occupied A3 AR is a substrate for the G protein-coupled receptor kinase family of kinases (Palmer et al., 1995). Phosphorylation results in a decreased number of receptors in the high-affinity conformation and in decreased potency of agonists to inhibit adenylyl cyclase after a 10-min agonist exposure. Over a similar time course, the receptor is internalized in an agonist-dependent and reversible manner (Trincavelli et al., 2000).
The evaluation of A3 AR regulation in native systems has several advantages and disadvantages with respect to transfected cell lines. On one side, there are major problems due to the possible coexpression of different receptor subtypes and the difficulty to selectively label the A3 AR subtype. However, the study of A3 AR regulation mechanisms in native systems that also coexpress other adenosine receptor subtypes represents an important means to evaluate the cross talk between different receptor subtypes in the control of intracellular response. Moreover, evaluation of A3 AR regulatory mechanisms in native systems provides a clue to assess the pathophysiological significance of agonist-induced desensitization. To address the issue of A3 AR regulation in human astrocytoma cells, we first demonstrated the presence of A3 AR functionally coupled to Gi proteins. In these cells, Cl-IB-MECA indeed inhibited adenylyl cyclase activity with an EC50 value of 1.8 ± 0.12 nM, a value comparable with that reported for A3 AR in human transfected cell lines (Jacobson et al., 1997). The inhibitory effect exerted by nanomolar concentrations of this agonist on forskolin-stimulated adenylyl cyclase activity was completely blocked by the selective A3 AR antagonists MRS 1191 and MRS 1220, confirming a specific A3 AR-mediated effect. In contrast, these antagonists only partially reversed Cl-IBMECA inhibition of adenylyl cyclase induced by micromolar agonist concentrations, in agreement with previous studies demonstrating that the effects elicited by higher concentrations of N-methyl-5'-carbamoyladenosine analogs in the central nervous system may be due to the activation of A1 adenosine receptors, the predominant adenosine receptor subtype in the brain. Based on these data, and on the immunoblotting demonstration that ADF cells also coexpress the A1 AR subtype (data not shown), we selected a Cl-IBMECA concentration (100 nM) that selectively activates this receptor subtype to study A3 AR desensitization/internalization. Short-time exposure of ADF cells to 100 nM Cl-IBMECA induced a rapid reduction of A3 AR responsiveness as demonstrated by the loss of the ability of this agonist to inhibit adenylyl cyclase in isolated membranes. Desensitization kinetics was comparable with that obtained in CHO cells transfected with human A3 AR (Trincavelli et al., 2000). This effect is not accompanied by any decrease in adenylyl cyclase stimulation by forskolin, suggesting that both Gi protein and the adenylyl cyclase catalytic subunit were unaffected by the desensitization process. These data are consistent with the “homologous nature” of A3 AR desensitization, and support a model whereby functional A3 AR desensitization selectively diminishes the number of its signaling-competent receptor/G protein complexes. In contrast, Palmer et al. (1997) demonstrated that long-term exposure of the human A3 AR to agonists, enhances the stimulation of adenylyl cyclase activity by GTP/forskolin through an up-regulation of Gs proteins. These inconsistencies may be due to differences in short- and long-term receptor activation, suggesting different regulatory mechanisms in acute and chronic receptor overstimulation.
Using the selective A3 AR antagonists MRS 1191 and MRS 1220 we demonstrated that the reduction of agonist responsiveness induced by Cl-IBMECA is exclusively due to A3 AR subtype desensitization. These results are also supported by the known differences in desensitization properties between the A1 and A3 AR subtypes (Palmer et al., 1996), whereas A3 AR undergoes a rapid desensitization (as also supported by the present results), signaling via the A1 AR does not easily subside upon sustained agonist exposure. It takes about 12 h to down-regulate A1 AR receptor number and to attenuate its ability to inhibit adenylyl cyclase (Ciruela et al., 1997; Gao et al., 1999). These desensitization pattern differences have been attributed to a different sensitivity of the two receptor subtypes to phosphorylation by G protein-coupled receptor kinase proteins (Ferguson et al., 2000).
A3 AR endocytosis was demonstrated by radioligand binding and electron microscopic immunogold techniques. Data obtained demonstrated that, in ADF cells, A3 AR internalized with rapid kinetics, which reached equilibrium within 30 min. The availability of a polyclonal anti-human A3 AR antibody allowed us to determine the intracellular trafficking of A3 AR in response to receptor activation. Our data show that after treatment with the agonist at 4°C, human A3 AR is mainly located on the plasma membranes. After agonist treatment at 37°C for 10 to 30 min, a reorganization of receptor distribution, including endocytosis and formation of endocytotic vesicles, was observed. On the basis of the present ultrastructural evidence, we have been able to demonstrate that, in ADF cells, A3 AR is internalized by means of an endocytotic pathway that involves non–clathrin-coated vesicles.
As demonstrated for other GPCRs, A3 AR internalization induced by agonist binding is followed by surface re-expression of receptors and recovery of receptor responsiveness. Recycling of receptors to the cell surface after agonist-triggered internalization followed an exponential time course; within 120 min 100% of receptors reappeared on the cell surface. Moreover, ultrastructural analysis showed that after 60 min of incubation with the agonist, gold particles were visible under and on the plasma membranes, confirming the recycling of A3 AR. The removal of the agonist, after the desensitization period, induced the progressive reappearance of receptor responsiveness within 120 min. Resensitization kinetic seemed comparable with that obtained in CHO cells stably transfected with the human A3 AR (Trincavelli et al., 2000).
Finally, we have investigated the A3 AR regulatory mechanisms induced by long-term agonist exposure. Long-term treatment of ADF cells with the agonist for up to 24 h did not produce any further reduction of A3 AR function, compared with that observed in short-time (e.g., 30-min) agonist treatment. However, this second phase of A3 AR regulation was associated with a reduction in the levels of immunoreactive A3 AR. Moreover, at variance from the quick reversal of receptor desensitization observed after short-term agonist treatment, complete recovery from the desensitization produced by long-term agonist exposure required a period of several hours, further suggesting the involvement of different desensitization mechanisms at 30 min and 24 h. In addition, in the latter case, the recovery of A3 AR responsiveness occurred with a kinetics comparable with that required for the recovery of total A3 AR immunoreactivity, suggesting the involvement of “ex novo” protein synthesis. Reverse transcription-polymerase chain reaction studies are in progress to evaluate the regulation of A3 AR transcript levels by long-term cell exposure to agonist. These data could also clarify whether down-regulation occurs by intracellular degradative mechanisms or can be attributed to alterations of A3 AR gene transcription. We cannot rule out that a desensitization-induced conformational change of A3 AR is responsible for the reduction of the receptor immunoreactivity with the antibody. However, this seems very unlikely, because cells were solubilized and electrophoresed under denaturing conditions.
In conclusion, we have demonstrated for the first time that multiple, temporally distinct, and sequential processes are associated with regulation of A3 AR responsiveness in ADF cells. Short-term agonist exposure causes a rapid impairment of the receptor/G protein interaction, leading to reduced A3 AR inhibition of adenylyl cyclase activity. This is associated with receptor sequestration into an intracellular endosomal compartment. Long-term treatment leads to receptor down-regulation, recovery from which takes several hours. In previous studies, we have demonstrated that a 24- or 48-h exposure to Cl-IBMECA results in cytoprotection, as shown by a significant reduction of spontaneous apoptotic cell death (Abbracchio and Burnstock, 1998). We now speculate that desensitization/down-regulation of the A3 AR under such agonist exposure conditions is indeed at the basis of cytoprotection. This implies a causative role for this receptor subtype in induction of cell death, as nevertheless suggested by data obtained in different experimental models (Shneyvais et al., 1998; Appel et al., 2001; Kim et al., 2002).
Acknowledgments
This work was supported by a grant from the Italian Ministero dell’Università; e della Ricerca Scientifica (Cofinanziamento di ricerche di interesse nazionale, 2001 on “Recettori purinergici e neuroprotezione”).
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- AR
adenosine receptor
- Cl-IBMECA
2-chloro-N6-(3-iodobenzyl)-N-methyl-5'-carbamoyladenosine
- MRS 1191
3-ethyl, 5-benzyl 2-methyl-6-phenyl-4-phenylethynyl-1,4-(±)-dihydropyridine-3,5-dicarboxylate
- MRS 1220
9-chloro-2-(2-furyl)-5-phenylacetamino[1,2,4]triazolo[1,5-c]quinazoline
- CHO
Chinese hamster ovary
- CHO-A3R
Chinese hamster ovary cells transfected with the human A3 receptor
- [125I]AB-MECA
N6-(4-amino-3-[125I]iodobenzyl)-N-methyl-5'-carbamoyladenosine
- R0201724
4-[(3-butoxy-4-methoxyphenyl)methyl]-2-imidazolidinone
- DPCPX
1,3-dipropyl-8-cyclopentylxanthine
- PBS
phosphate-buffered saline
- R0201724
4-[(3-butoxy-4-methoxyphenyl)methyl]-2-imidazolidinone
References
- Abbracchio MP, Burnstock G. Purinergic signalling: pathophysiological roles. Jpn J Pharmacol. 1998;78:113–145. doi: 10.1254/jjp.78.113. [DOI] [PubMed] [Google Scholar]
- Abbracchio MP, Camurri A, Ceruti S, Cattabeni F, Falzano L, Giammarioli AM, Jacobson KA, Trincavelli L, Martini C, Malorni W, et al. The A3 adenosine receptor induces cytoskeleton rearrangement in human astrocytoma cells via a specific action on Rho proteins. Ann NY Acad Sci. 2001;939:63–73. doi: 10.1111/j.1749-6632.2001.tb03613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbracchio MP, Rainaldi G, Giammarioli AM, Ceruti S, Brambilla R, Cattabeni F, Barbieri D, Franceschi C, Jacobson KA, Malorni W. The A3 adenosine receptor mediates cell spreading, reorganization of actin cytoskeleton and distribution of Bcl-XL: studies in human astroglioma cells. Biochem Biophys Res Commun. 1997;241:297–304. doi: 10.1006/bbrc.1997.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali H, Cunha-Melo JR, Saul WF, Beaven MA. Activation of phospholipase C via adenosine receptors provides synergistic signals for secretion in antigen-stimulated RBL-2H3 cells. Evidence for a novel adenosine receptor. J Biol Chem. 1990;265:745–753. [PubMed] [Google Scholar]
- Appel E, Kazimirsky G, Ashkenazi E, Kim SG, Jacobson KA, Brodie C. Roles of BCL-2 and caspase 3 in the adenosine A3 receptor-induced apoptosis. J Mol Neurosci. 2001;17:285–292. doi: 10.1385/JMN:17:3:285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm SK, Grady EF, Bunnett NW. Regulatory mechanisms that modulate signalling by G-protein coupled receptors. Biochem J. 1997;322:1–18. doi: 10.1042/bj3220001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Cattabeni F, Ceruti S, Barbieri D, Franceschi C, Kim YC, Jacobson KA, Klotz KN, Lohse MJ, Abbracchio MP. Activation of the A3 adenosine receptor affects cell cycle progression and cell growth. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;361:225–234. doi: 10.1007/s002109900186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunemann M, Hosey MM. G-protein coupled receptor kinases as modulators of G-protein signalling. J Physiol (Lond) 1999;517:5–23. doi: 10.1111/j.1469-7793.1999.0005z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciruela F, Saura C, Canela EI, Mallol J, Lluis C, Franco R. Ligand-induced phosphorylation, clustering and desensitization of A1 adenosine receptors. Mol Pharmacol. 1997;52:788–797. doi: 10.1124/mol.52.5.788. [DOI] [PubMed] [Google Scholar]
- Edwardson JM, Szekeres PG. Endocytosis and recycling of muscarinic receptors. Life Sci. 1999;64:487–494. doi: 10.1016/s0024-3205(98)00592-x. [DOI] [PubMed] [Google Scholar]
- Ferguson G, Watterson KR, Palmer TM. Subtype-specific kinetics of inhibitory adenosine receptor internalization are determined by sensitivity to phosphorylation by G protein-coupled receptor kinases. Mol Pharmacol. 2000;57:546–552. [PubMed] [Google Scholar]
- Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacol Rev. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Robeva AS, Linden J. Purification of A1 adenosine receptor-G-protein complexes: effects of receptor down-regulation and phosphorylation on coupling. Biochem J. 1999;338:729–736. [PMC free article] [PubMed] [Google Scholar]
- Gessi S, Varani K, Merighi S, Morelli A, Ferrari D, Leung E, Baraldi PG, Spalluto G, Borea PA. Pharmacological and biochemical characterization of A3 adenosine receptors in Jurkat T cells. Br J Pharmacol. 2001;134:116–126. doi: 10.1038/sj.bjp.0704254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessi S, Varani K, Merighi S, Cattabriga E, Iannotta V, Leung E, Baraldi PG, Borea PA. A(3) adenosine receptors in human neutrophils and promyelocytic HL60 cells: a pharmacological and biochemical study. Mol Pharmacol. 2002;61:415–424. doi: 10.1124/mol.61.2.415. [DOI] [PubMed] [Google Scholar]
- Jacobson KA. Adenosine A3 receptors: novel ligands and paradoxical effects. Trends Pharmacol Sci. 1998;19:184–191. doi: 10.1016/s0165-6147(98)01203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Hoffmann C, Cattabeni F, Abbracchio MP. Adenosine-induced cell death: evidence for receptor-mediated signalling. Apoptosis. 1999;4:197–211. doi: 10.1023/a:1009666707307. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Nikodijevic O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JWA. Role for central A3-adenosine receptors. Mediation of behavioral depressant effects. FEBS Lett. 1993;336:57–60. doi: 10.1016/0014-5793(93)81608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Pharmacological characterization of novel A3 adenosine receptor-selective antagonists. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji XD, Gallo-Rodriguez C, Jacobson KA. A selective agonist affinity label for A3 adenosine receptors. Biochem Biophys Res Commun. 1994;203:570–576. doi: 10.1006/bbrc.1994.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HO, Ji XD, Melman N, Olah ME, Stiles GL, Jacobson KA. Selective ligands for rat A3 adenosine receptors: structure-activity relationships of 1,3-dialkylxanthine 7-riboside derivatives. J Med Chem. 1994;37:4020–4030. doi: 10.1021/jm00049a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Ji XD, Jacobson KA. Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J Med Chem. 1996;39:4142–4148. doi: 10.1021/jm960482i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Ravi G, Hoffmann C, Jung YJ, Kim M, Chen A, Jacobson KA. p53-independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative, Cl-IB-MECA. Biochem Pharmacol. 2002;63:871–880. doi: 10.1016/s0006-2952(02)00839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi S, Varani K, Gessi S, Cattabriga E, Iannotta V, Ulouglu C, Leung E, Borea PA. Pharmacological and biochemical characterization of adenosine receptors in the human malignant melanoma A375 cell line. Br J Pharmacol. 2001;134:1215–1226. doi: 10.1038/sj.bjp.0704352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. 125I-4-aminobenzyl-5'-N-methylcarboxamidoadenosine, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- Palmer TM, Benovic JL, Stiles GL. Agonist-dependent phosphorylation and desensitization of the rat A3 adenosine receptors. J Biol Chem. 1995a;270:29607–29613. doi: 10.1074/jbc.270.49.29607. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Benovic JL, Stiles GL. Molecular basis for subtype-specific desensitization of inhibitory adenosine receptors. Analysis of a chimeric A1-A3 adenosine receptor. J Biol Chem. 1996;271:15272–15278. doi: 10.1074/jbc.271.25.15272. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Gettys TW, Stiles GL. Differential interaction with and regulation of multiple G-proteins by the rat A3 adenosine receptors. J Biol Chem. 1995b;270:16895–16902. doi: 10.1074/jbc.270.28.16895. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Harris CA, Coote J, Stiles GL. Induction of multiple effects on adenylyl cyclase regulation by chronic activation of the human A3 adenosine receptor. Mol Pharmacol. 1997;52:632–640. doi: 10.1124/mol.52.4.632. [DOI] [PubMed] [Google Scholar]
- Palmer TM, Stiles GL. Identification of threonine residues controlling the agonist-dependent phosphorylation and desensitization of the rat A(3) adenosine receptor. Mol Pharmacol. 2000;57:539–545. [PubMed] [Google Scholar]
- Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc Natl Acad Sci USA. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Kim TD, Lee JU, Seong JK, Kim KT. Pharmacological characterization of adenosine receptors in PGT-f3 mouse pineal gland tumour cells. Br J Pharmacol. 2001;134:132–142. doi: 10.1038/sj.bjp.0704218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MI, Strathmann MP, Gautam N. Diversity of G proteins in signal transduction. Science (Wash DC) 1991;252:802–808. doi: 10.1126/science.1902986. [DOI] [PubMed] [Google Scholar]
- Shneyvais V, Nawrath H, Jacobson KA, Shainberg A. Induction of apoptosis in cardiac myocytes by an A3 adenosine receptor agonist. Exp Cell Res. 1998;243:383–397. doi: 10.1006/excr.1998.4134. [DOI] [PubMed] [Google Scholar]
- Trincavelli ML, Tuscano D, Cecchetti P, Falleni A, Benzi L, Klotz K-N, Gremigni V, Cattabeni F, Lucacchini A, Martini C. Agonist-induced internalization and recycling of the human A3 adenosine receptors: role in receptor desensitization and resensitization. J Neurochem. 2000;75:1492–1501. doi: 10.1046/j.1471-4159.2000.0751493.x. [DOI] [PubMed] [Google Scholar]
- Tsao P, Cao T, von Zastrow M. Role of endocytosis in mediating down-regulation of G-protein-coupled receptors. Trends Pharmacol Sci. 2001;22:91–96. doi: 10.1016/s0165-6147(00)01620-5. [DOI] [PubMed] [Google Scholar]
- Tsao PI, von Zastrow M. Diversity and specificity in the regulated endocytic membrane trafficking of G-protein-coupled receptors. Pharmacol Ther. 2001;89:139–147. doi: 10.1016/s0163-7258(00)00107-8. [DOI] [PubMed] [Google Scholar]
- von Lubitz DK. Adenosine and cerebral ischemia: therapeutic future or death of a brave concept? Eur J Pharmacol. 1999;371:85–102. doi: 10.1016/s0014-2999(99)00135-1. [DOI] [PubMed] [Google Scholar]
- von Zastrow M. Role of endocytosis in signalling and regulation of G-protein-coupled receptors. Biochem Soc Trans. 2001;29:500–504. doi: 10.1042/bst0290500. [DOI] [PubMed] [Google Scholar]