

Abstract
Mitogen‐activated protein kinases (MAPK) are critical mediators of cellular responses to pathogens and are activated in response to infection, but investigation is difficult in multi‐cell hosts due to developmental lethality of mutations. Mycobacterium marinum (Mm) is an established model for tuberculosis, a disease afflicting nearly one‐third of the world's population. We found that Mm‐infected Caenorhabditis elegans display >80% mortality, but nonpathogenic M. smegmatis cause <15% mortality. C. elegans display pathological changes when infected with Mm, whereas Mm mutants produce lower mortality, suggesting that C. elegans is a promising virulence model for detailed genetic analysis. C. elegans MAPK mutants are hypersusceptible to mycobacterial infection; however, the C. elegans TOL‐like, TGF‐β and insulin‐like pathway genes do not play important roles in susceptibility. We show that pathogenic mycobacteria inhibit MAPK‐mediated protection through the MAPK phosphatase gene and demonstrate that C. elegans provide a genetically tractable pathogenicity model of both the host and pathogen.
Keywords: C. elegans, innate immunity, Mycobacterium
Introduction
The innate immune response acts as a first line of defense in protecting the host against pathogens and activates the adaptive response by producing specific cytokines and chemokines (Iwasaki and Medzhitov 2010). Pattern recognition receptors (PRRs) recognize conserved structures not present in eukaryotic cells and provide partial protection until the adaptive immune system takes over (Janeway 1992). Several classes of PRRs can trigger innate immunity, including C‐type lectin receptors (CLRs), tol‐like receptors (TLRs), and NOD‐like receptors (NLRs) (Kleinnijenhuis et al. 2011; Kingeter and Lin 2012). All identified PRRs activate the p38 mitogen‐activated protein kinase (MAPK) pathway in response to pathogen‐associated molecular patterns (PAMPs) (Kawai and Akira 2010), suggesting that p38 MAPK is a key mediator of immunity (Koul et al. 2004). Mycobacteria are some of the most important pathogens worldwide, encompassing the causative agents of leprosy and tuberculosis. Tuberculosis (TB) causes ~1.4 million deaths annually, latently infecting a third of the world's population and multidrug‐resistant (MDR‐TB) strains are now prevalent in all continents (Glaziou et al. 2013). While many interventions focus on modulating the adaptive immune system, the innate immune response is also critical for host protection (Pan et al. 2005).
The innate immune response is initially activated by macrophages at the site of mycobacterial infection (Roach and Schorey 2002; Kleinnijenhuis et al. 2011). The T‐cell‐mediated adaptive response is activated shortly thereafter and can reduce the bacterial load (Stenger and Modlin 1999). Pathogenic mycobacteria activate all three major MAPK pathways, but there is little direct evidence for a role in pathogenesis due to the detrimental effects of mutations during development in mice. When mycobacteria infect macrophages, ERK and p38 MAPK are activated by phosphorylation, inducing an inflammatory response (Schorey and Cooper 2003). Mycobacterial lipoarabinomannan (LAM) acts as a PAMP that activates the innate immune response through MAPK (Strohmeier and Fenton 1999). LAM on the surface of pathogenic mycobacteria is mannose‐capped (Man‐LAM); whereas, nonpathogenic mycobacteria have arabinosylated LAM (Ara‐LAM). Ara‐LAM induces cytokines and inflammatory mediators, whereas Man‐LAM promotes dephosphorylation that suppresses MAPK activation and, thereby, inflammatory cytokines (Roach et al. 1993; Knutson et al. 1998; Juffermans et al. 2000). Despite the differences in activation of MAPK and the inflammatory response, the mechanisms for suppression of MAPK and their significance during disease remain elusive.
Genetically tractable virulence models are valuable for pathogenesis studies, since they allow rapid analysis of complex interactions often difficult to study in mammals. Several virulence models have been used for study of mycobacteria, including tissue culture cells, mice, guinea pigs, and rabbits (Bishai et al. 1999; Jain et al. 2007), but can be costly and difficult to genetically manipulate. C. elegans is a valuable model for study of host responses because it has a well‐characterized genome and an extensive array of molecular tools (Ashrafi et al. 2003; Irazoqui et al. 2010). The innate immune response and host–pathogen interactions can be analyzed in great detail using C. elegans (Garsin et al. 2001; Couillault and Ewbank 2002; Irazoqui et al. 2010). C. elegans use reactive oxygen species (ROS) and lysosomes to defend against invading pathogens, similar to mammalian macrophages and neutrophils (Mallo et al. 2002; Chavez et al. 2007). The C. elegans p38 isoforms of MAPKs, designated pmk‐1‐3, are localized to the intestine and are important in the response to pathogenic bacteria (Coleman and Mylonakis 2009; Shivers et al. 2009). Many such responses are mediated by PMK‐1, the best characterized of the p38 isoforms (Aballay et al. 2003; Kim et al. 2004; Jebamercy et al. 2013), making it potentially important for a host response to pathogenic mycobacteria. Mycobacterium marinum (Mm) is commonly used as a model for M. tuberculosis due to its rapid growth rate and ease of use (Shiloh and Champion 2010).
We combined the model organisms Mm and C. elegans to analyze the role of MAPK in mycobacterial pathogenesis. We found that infection of C. elegans with Mm causes higher morbidity and mortality than the nonpathogen M. smegmatis (Ms). Mm‐infected C. elegans undergo irreversible pathological changes that lead to nematode death. Mortality of C. elegans correlates well with virulence of several Mm mutants, suggesting that the nematode serves as a useful virulence model. We found that C. elegans protection is controlled by the p38 MAPK pathway and Mm can hijack MAPK phosphatase to interfere with protection. These studies demonstrate that C. elegans has particular value for study of MAPK, which can be difficult to study in mammals and has provided insight into the mechanisms that lead to mycobacterial pathogenesis.
Materials and Methods
Bacterial strains and growth conditions
A wild‐type clinical isolate of Mm strain M, wild‐type Ms (mc2155), and E. coli (OP50) were used. Two constitutively expressing tdTomato fluorescent mycobacterial strains (ψmm91 and ψms23) were derived by transforming Mm and Ms, respectively, with pJDC60 (pFJS8ΔGFP::tdTomato expressed by L5 promoter, KanR). M. marinum cultures were grown at 32°C standing in T25 tissue culture flasks. E. coli and M. smegmatis cultures were grown at 37°C shaking in sterile disposable glass test tubes. M. marinum and M. smegmatis were grown in Middlebrook 7H9 media (Difco, Sparks, MD) supplemented with 0.5% glycerol, 10% albumin‐dextrose complex (ADC), and 0.25% Tween 80 (M‐ADC‐TW), whereas E. coli was grown in Miller's Luria Broth (NPI, Mt. Prospect, IL).
C. elegans strains
The C. elegans strains were obtained from the Caenorhabditis Genetics Center (CGC), University of Minnesota. The wild‐type strain was N2 and mutants included: TP12 [kaIs12(col‐19::GFP)], GR1307 [daf‐16(mgDf50)], NU3 [dbl‐1(nk3)], KU25 [pmk‐1(km25)], EU31 [skn‐1(zu135)], IG10 [tol‐1(nr2033)], and JT366 [vhp‐1(sa366)]. Nematodes were grown and maintained on nematode growth media (NGM) plates using standard methods at 19°–21°C (Brenner 1974). Synchronous cohorts of C. elegans were obtained by lysing gravid nematodes using an alkaline bleach solution (Emmons et al. 1979). After removing the bleach solution and washing the embryos, they were stored in M9 buffer overnight to obtain L1 larvae. These L1 larvae were transferred on NGM plates seeded with OP50 E. coli for growth of age‐synchronized nematodes.
C. elegans infection
E. coli, Ms and Mm grown to stationary phase were seeded on tissue culture dishes with NGM agar. Three‐day‐old age‐synchronized adult C. elegans were infected for 4, 24, or 48 h with each bacterial strain. Volumes of 70 μL for E. coli, M. smegmatis, or M. marinum were seeded on small tissue culture dishes (35 × 10 mm, Falcon), with NGM agar by spreading the bacterial cultures to cover over 3/4th of the infection plates. Infection plates were placed at room temperature overnight to allow bacterial cultures to grow, equilibrate/stabilize and be ready for infection the next day. Three‐day‐old age‐synchronized adult C. elegans were washed with ddH2O to remove residual E. coli on their surface and transferred onto plates seeded with E. coli, M. smegmatis, or M. marinum for infection. Cohorts of C. elegans were exposed for a period of 4, 24, or 48 h to each individual bacterial strain. After infection, nematodes were transferred onto NGM plates seeded with E. coli and assessed for pathology and mortality. After the period of infection, the nematodes were transferred onto small NGM recovery plates with E. coli seeded in the center of each plate. A total of 20 nematodes were incubated per small recovery NGM plate and cohorts of 60 nematodes were used for each bacterial infection. Nematodes were counted daily and transferred onto fresh E. coli seeded plates every other day until experimental nematodes stopped laying eggs. Then they were transferred every 3–4 days to avoid overgrowth of seeded E. coli until the nematodes reached senescence and died. The nematodes were considered dead if they were unresponsive to touch with a wire pick. Bacterial infection experiments were repeated at least three times with 20, 40, or 60 nematodes, unless stated otherwise.
Survival and morphological characterization of C. elegans
C. elegans L1 larvae incubated on E. coli seeded NGM plates after synchronization were considered 0 days old. They were grown for 3 days at room temperature before subjection to bacterial infection. On day 4, they were recovered on to fresh E. coli seeded NGM plates and followed up for survival and changes in morphology. Number of nematodes that died due to bagging of the adult nematode (where embryos hatch within the adult and cause the death of the adult) on day 5 and 6 were counted. Nematodes that lost their dark pigmentation after bacterial infection were characterized as depigmented. Nematodes that were less than 2/3rd the length of a healthy nematode were characterized as having a shortened length. On day 6, depigmented and shorter nematodes were counted. Nematodes that died prior to 15 days were considered to have a shortened lifespan.
Imaging of C. elegans for morphological differences and fluorescent mycobacteria
TP12 [kaIs12(col‐19::GFP)] C. elegans expressing cuticular eGFP‐tagged col‐19, a member of the collagen superfamily, were infected with ψms23 (Ms::tdTomato) or ψmm91 (Mm::tdTomato). Exposed C. elegans were mounted on a 3% agar pad with a thickness of two labeling tapes. 15 μL of 5 mmol/L levamisole was added to immobilize the nematodes and keep the agar pad moist for imaging. 25‐mm round cover slips with a thickness of 0.17 mm (#1.5) were placed on the agar pad to keep C. elegans in place and for imaging. Using a confocal microscope (Nikon A1R+), exposed nematodes were imaged under differential interface contrast (DIC) and fluorescent filters (FITC and TRITC) using a 10× objective. The nematodes were imaged at 4, 12, 18, and 24 h during infection, and 6 and 24 h post infection. The 60 nematodes for each bacterial infection and time point were imaged. DIC images were used to identify patterns of morphological change in cohorts of nematodes infected with different bacteria. Morphological changes in M. smegmatis and M. marinum exposed nematodes were compared with that of E. coli exposed nematodes. Fluorescent imaging using FITC and TRITC filters was used to determine localization during colonization of M. marinum and M. smegmatis within C. elegans. The 60 nematodes imaged for each time point were randomly divided into three groups and mycobacterial fluorescent signal determined. Three segments of each nematode were imaged (upper, middle, and lower) and fluorescent bacteria were quantified within each defined area imaged.
CFU assay for bacterial load and colonization of C. elegans
Nematodes were transferred and colony forming units (CFU) determined by homogenization and plating dilutions. At 4, 12, and 24 h during infection and 6 h post infection, a total of 30 C. elegans infected with wild‐type M. marinum or M. smegmatis were transferred to 500 μL of M9 buffer with 0.05% Tween‐20 in 1.5 mL microcentrifuge tubes. After briefly vortexing, they were spun down at 800 rpm for 15 sec and the supernatant removed. These nematodes were washed three times with 500 μL of 1× PBS with 0.05% Tween‐20 and resuspend in 600 μL of 1× PBS with 0.05% Tween‐20 and 100 μL plated for levels of bacteria in serial dilutions to determine background. The nematodes remaining in 500 μL of 1× PBS with 0.05% Tween‐20 were homogenized with a hand held motorized pestle for a total of 45 sec. After the nematodes were broken up, releasing the bacteria, they were vortexed on high and plated for CFU in serial dilutions. A quantity of 20 μL spot plating was carried out in triplicate for each of the dilutions. M. smegmatis was grown at 37°C and M. marinum was grown at 32°C overnight.
Macrophage infection mutant (MIM) M. marinum infection
Nematodes were infected with M. marinum mutants that were constructed previously and are known to impact infection of mammalian macrophages. Similar methods were used to those described for wild‐type M. marinum. Mm MIMs, mimA‐K, nrp, ppe24, sdhD, pks12, fadD29, fadD30, and ppe53, were constructed by insertion of kanamycin resistance within the coding region of each gene (Mehta et al. 2006). Three genes were complemented with their respective Mtb gene orthologs (mimA::Rv0246, mimG::Rv3242c, mimI::Rv1502), as described previously (Mehta et al. 2006). We used these mutants and exposed 3 day old adult N2 nematodes for a period of 24 h. MIM cultures were grown until stationary phase of growth (OD >1.2) and 70 μL of each were seeded on small NGM plates, the day prior to infection. Triplicates of 20 N2 worms (total of 60) were infected with each strain of M. marinum. They were then allowed to recover and mortality rates assessed 2 days post‐infection (day 6). To confirm the attenuation was due to a specific gene in M. marinum, three strains complemented with their M. tuberculosis orthologs were also used to expose C. elegans and mortality rates were compared, 2 days post infection (day 6).
RNAi knock‐down and confirmation
After bleaching was used to age‐synchronize C. elegans, N2 L1 larvae were inoculated on small NGM plates seeded with E. coli strain HT115 (DE3) with RNAi constructs cloned in the pL4440‐DEST vector and selected with ampicillin resistance (100 μg/mL). E. coli strains producing dsRNA corresponding to the C. elegans pmk‐1, tol‐1, dbl‐1, daf‐16, skn‐1, and vhp‐1 genes were used (Table S2). A quantity of 50 μL of E. coli carrying the RNAi plasmid was added to the plates on day one for 24 h. Adult N2 knock‐down animals were then infected with wild‐type M. smegmatis or M. marinum on day three and susceptibility to infection with mycobacteria assessed.
For confirmation, RNA from 3 day old N2 nematodes grown on small NGM plates seeded with E. coli strain HT115 (DE3) expressing individual RNAi constructs for pmk‐1, tol‐1, dbl‐1, daf‐16, skn‐1, and vhp‐1 was isolated after dissolving the nematodes in Trizol. cDNA was produced using random primers and specific qPCR run for each target gene (Table S3). Cdc‐42 was used as a housekeeping gene control. Age‐synchronized 3 day old nematodes from each RNAi knock‐down mutant strain used in this study were subjected to RT‐PCR and qPCR confirmation. RNA levels were normalized to housekeeping genes pmp‐3 and cdc‐42, and compared against N2‐infected with E. coli expressing the empty RNAi vector.
High‐resolution confocal imaging of C. elegans
Using a confocal microscope (Nikon A1R+) with spectral capability, 25 emission channels with a resolution of 6.0 nm were use to detect the wavelengths of 500–640 nm. This wavelength range was used to detect excitation of GFP (500–520 nm) and tdTomato (560–620 nm). Using a 40× oil immersion objective, TP12 nematodes infected with E. coli (OP50), ψms23 (M. smegmatis::tdTomato), or ψmm91 (M. marinum::tdTomato) were imaged during (4 and 24 h) and post infection (30 h). The nematodes were immobilized with 15 μL of levamisole (5 mmol/L) and the head, mid‐gut region, and lower‐gut regions were imaged. About 15 nematodes for each time point for infection with ψms23 and ψmm91 and about 5 nematodes were imaged for each time point for infection with E. coli. Representative images are presented to show differences in bacterial colonization and morphological changes in nematodes after infection.
Transmission electron microscopy imaging of C. elegans
C. elegans infected with bacteria were fixed, embedded, sectioned, and imaged using previously described methods with slight modifications (Hall et al. 2012; Schultz et al. 2014) at 4 and 24 h during infection and 6 h postinfection. Nematodes from Ms or Mm were embedded and longitudinal sections were obtained using a microtome. C. elegans from each group (E. coli, M. smegmatis and M. marinum) were obtained and washed twice with 500 μL of 1 × M9 buffer. The nematodes were then immersed in fixative with 2.5% glutaraldehyde, 2% paraformaldehyde and 0.1% (w/v) malachite green in a working buffer (0.1 HEPES, pH 7.4, containing 2 mmol/L MgCl2). Using a PELCO BioWave® microwave, the nematodes in the fixative solution were microwaved under vacuum at 100 W for 10 min and allowed to stand at room temperature for 3 min. This process was repeated and the specimens were placed at room temperature for 1 h. A second step of microwaving at 500 W for 10 sec, stand for 20 sec and microwaved for 10 sec. The nematodes were washed three times with 500 μL of working buffer and microwaved for 1 min after each wash. To improve contrast, the nematodes were postfixed in 1% (w/v) osmium tetroxide with 1.5% (w/v) potassium ferricyanide in working buffer. They were microwaved at 100 W for 2 min, followed by letting them stand for 2 min. This was repeated four times. The specimens were dehydrated by rinsing the samples with 50% acetone, 70% acetone, 90% acetone, and three times with 100% acetone. After each rinse, the specimens were microwaved at 150 W for 1 min. The specimens were then infiltrated with resin, Quetol 651‐modified Spurr low viscosity epoxy resin, by 1:1 acetone:resin followed by 100% resin three times. They were microwaved at 200 W for 4 min between each infiltration step. The specimens were transferred into embedding tubes and the resin was allowed to polymerize overnight at 60°C. Longitudinal sections were obtained from the embedded specimens using a microtome and poststained with 2% (w/v) aqueous uranyl acetate. A JEOL 1200EX transmission electron microscope at an accelerated voltage of 100 kV was used to image these specimens.
C. elegans mutant confirmation
Mutant confirmation was accomplished using PCR with specific primers. Age‐synchronized N2 L1 larvae were incubated on plates seeded with E. coli producing each of the individual dsRNA for the target gene (Table S2). Three‐day‐old adult nematodes were obtained and RNA was extracted after dissolving the nematodes in Trizol. cDNA was produced using random primers and PCR was run for each target gene (Table S3). Cdc‐42 was used as a housekeeping gene control. Age‐synchronized 3‐day‐old nematodes from each mutant strain used in this study were also subjected to PCR for confirmation.
Statistical analyses
All experiments were repeated at least twice with consistent results. Parametric two‐tailed unpaired t‐test statistical analyzes were used to compare the means of different bacterial infection groups at distinct time points using GraphPad Prism version 5 software (La Jolla, CA, USA). Means and standard deviations or standard errors are presented. Means, standard deviations, and standard errors were calculated using GraphPad Prism 5 software and Microsoft Excel. Nonparametric log‐rank statistics were used to determine the difference in survival for groups of C. elegans after infection, using an online application for survival analysis of lifespan assays found on http://sbi.postech.ac.kr/oasis (released on May 2009; last accessed on April 20th 2014) (Yang et al. 2011).
Results
Pathogenic mycobacteria cause mortality in C. elegans
We infected C. elegans with M. smegmatis (Ms), a nonpathogenic mycobacteria, and Mm. C. elegans use E. coli as their natural food, which is harmless for these nematodes and necessary for their survival. We found that Mm causes higher rates of mortality in C. elegans than Ms or E. coli. In fact, Ms does not display significantly different rates of mortality as compared with E. coli where the nematodes die at the normal age of 18–20 days. When infected with Mm for 24 h, C. elegans display >80% mortality at 2 d post infection; whereas, Ms produce <15% mortality (Fig. 1). The majority of nematode deaths in the Mm‐infected group occur due to an event described as bagging, where the gravid adult nematode is unable to lay eggs and viviparity (egg hatching) occurs within the worm, resulting in the death of the adult, but survival of progeny. Consistent with these observations, bagging can occur as a C. elegans response to bacterial infection (Mosser et al. 2011). Even the <20% of nematodes that survive >24 h after Mm infection display pathology (Fig. S1). Longer infections with Mm increase the rate and number of mortalities, but do not impact mortality from Ms, suggesting that the mechanisms involved are specific to the pathogenic species.
Figure 1.
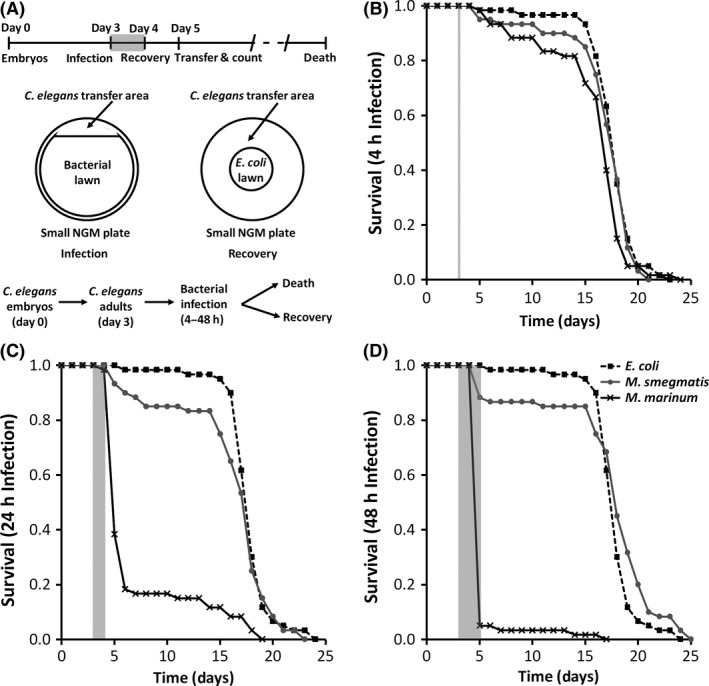
Pathogenic mycobacteria cause C. elegans mortality. (A) Procedure for infecting C. elegans. Adult nematodes were grown on NGM plates seeded with E. coli and infected for (B) 4, (C) 24 or (D) 48 h on day 3 as indicated by the gray shaded regions on each graph. During recovery on NGM plates seeded with E. coli, nematode survival was determined. Data shown are survival for 20 nematodes in each group. Key in (D) is for (B–D). Log‐rank analysis was used to compare survival of each infection group. (B) Mm versus. Ms P = 0.0567; Mm versus. E. coli P = 0.0095; Ms versus E. coli P = 0.3776. (C) Mm versus E. coli P < 0.0001; Ms versus E. coli P = 0.2207; Mm versus Ms P < 0.0001. (D) Mm versus Ms P < 0.0001; Mm versus E. coli P < 0.0001; Ms versus E. coli P = 0.0750.
Mm cause irreversible pathological changes in C. elegans
C. elegans were imaged by light microscopy during infection for 24 h and for 24 h postinfection. Nematodes infected with mycobacteria displayed reduced pigmentation and increased retention of embryos as compared with those infected with E. coli (Fig. 2). E. coli‐infected nematodes did not lose pigmentation or display pathology and lay eggs (Fig. 2A–D, S2). In the case of Ms, nematodes initially lose pigmentation and retain embryos, but regain pigmentation and lay eggs within 6 h post infection (Fig. 2E–H). In contrast to Ms, nematodes infected with Mm undergo irreversible pathological changes and are unable to lay eggs, leading to mortality (Fig. 2I–L). Greater than 80% of Mm‐infected nematodes died via bagging by 24 h post infection (Fig. S1). While both species of mycobacteria induce pathological changes, the immune response most likely clears Ms, preventing permanent damage, but cannot clear Mm.
Figure 2.
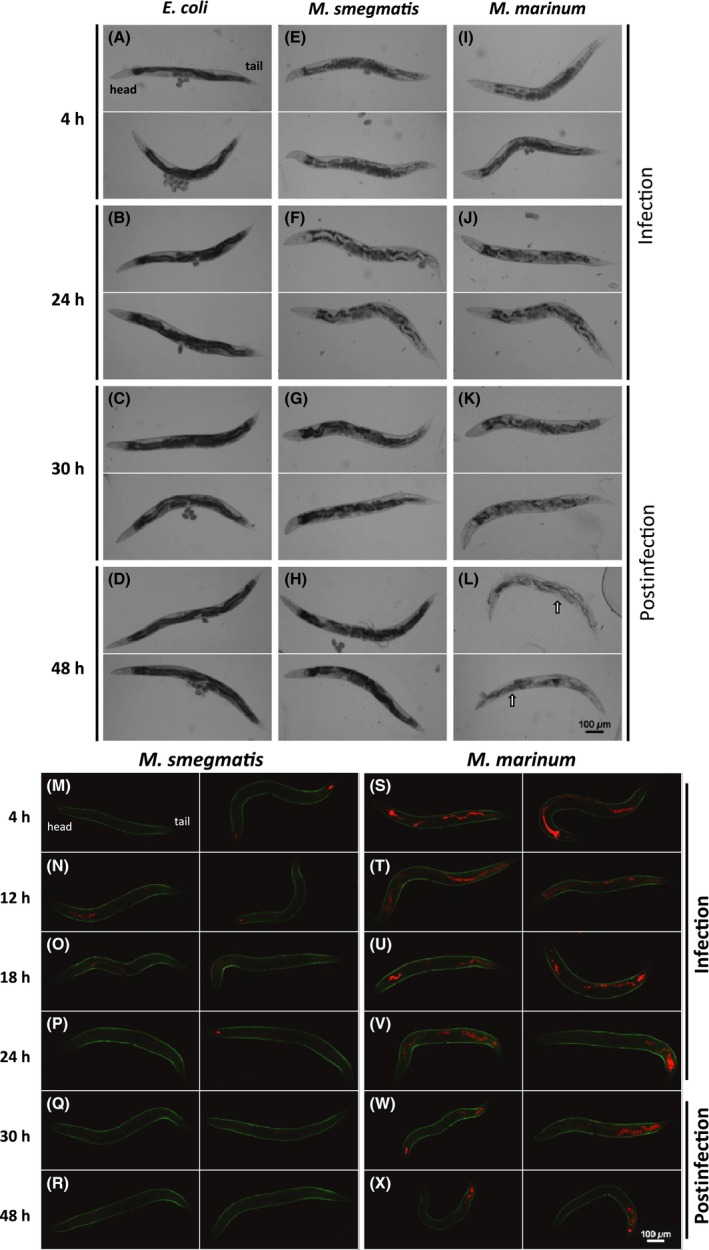
M. marinum persists in C. elegans and causes irreversible pathology. Morphology of C. elegans by light microscopy (A–L) and GFP fluorescent C. elegans by confocal microscopy (M–X) infected for 24 h and recovered on E. coli 48 h postinfection. (A–D) E. coli (OP50) infected nematodes are able to lay eggs and retain their dark pigmentation. (E–H) M. smegmatis (tdTomato)‐infected nematodes undergo reversible retention of eggs and loss of pigmentation during infection, but regain pigmentation and the ability to lay eggs postinfection. (I–L) M. marinum (tdTomato)‐infected nematodes experience irreversible retention of eggs and loss of pigmentation during infection. (L) The arrows indicate “bagging” morphological changes that occur when embryos are hatched in utero.
Mm colonize C. elegans more extensively than Ms
We infected green fluorescent C. elegans with red fluorescent Mm (ψms91) and Ms (ψms23), and found that they behave similarly to their nonfluorescent counterparts (Fig. S3 and S4). These observations allowed use of fluorescent markers to localize the bacteria during infection. Mm and Ms are found within C. elegans 4 h during infection (Fig. 2 M and S). At 4 h, >60% of the nematodes are colonized by Mm, with >10 bacteria/nematode (Fig. 3C). In contrast, <15% of the nematodes display >10 intact Ms per nematode. Apparently, C. elegans can digest Ms in a manner similar to E. coli, but Mm persists. Quantitative analyses by microscopy confirm that Ms is present in lower quantities than Mm at all time points (Fig. 3). The majority of mycobacteria are within the pharyngeal and lower gut regions. Even 6 h after the 24 h infection period, Ms was barely observable within nematodes, while Mm persists (Figs. 2 and 3). We found that >50% of C. elegans carried Mm, while <5% still had Ms at 24 h post infection. We confirmed observations from fluorescent microscopy by plating for colony‐forming units (CFU). We consistently observed >2‐fold higher levels of Mm CFU in C. elegans as compared with Ms (Fig. 3D). We confirmed that the fluorescent labels in the bacteria do not impact these observations using unlabeled organisms in similar experiments and obtained comparable results (Fig. S5). There is a decrease in Ms within nematodes by 6 h post infection; whereas, Mm persists beyond 24 h post infection.
Figure 3.
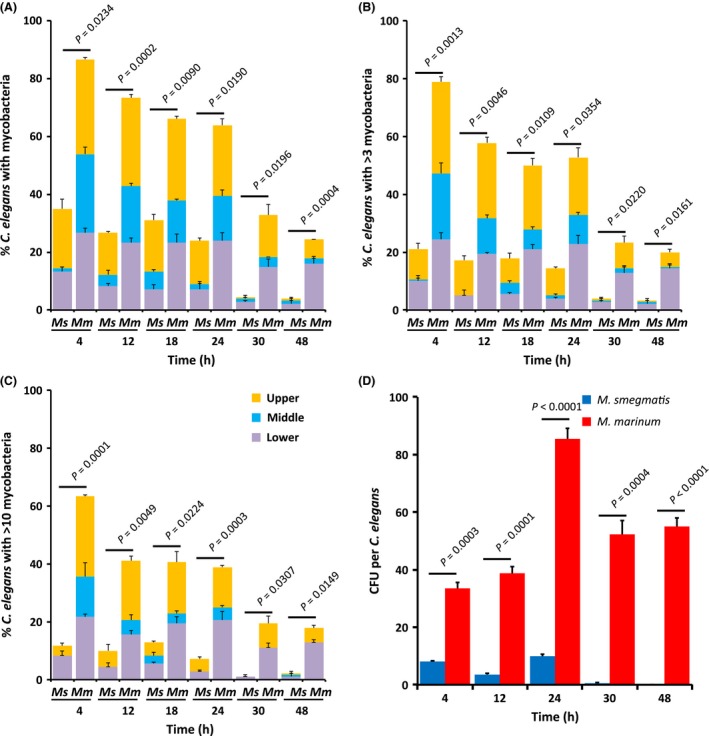
M. marinum colonize C. elegans. C. elegans were infected with either M. smegmatis (Ms) or M. marinum (Mm) for 24 h and quantified during and postinfection. 60 nematodes were randomly split into three groups of 20 at each time point. The number of bacteria in the upper, middle, and lower third of each nematode was determined. P ‐values at each time point are provided (unpaired t‐test). (A–C) Quantification of bacteria in the upper, middle, and lower regions of nematodes. Data are presented for means and standard deviations. Key in (C) is for (A–C). (A) C. elegans with at least one, (B) 4 or more, or (C) 11 or more cluster(s) of mycobacteria in the upper, middle, or lower segment. (D) Colony‐forming units (CFU) of mycobacteria per nematode recovered from 20 to 30°C. elegans at each time point.
Mm colonize the C. elegans intestine
In order to gain insight into the mechanisms involved, we imaged infected C. elegans using high‐resolution confocal and electron microscopy. We imaged the head, mid‐gut, and lower‐gut regions of nematodes where accumulations of mycobacteria were observed (Figs. 3 and 4). Bacteria are found in the head from 4 to 24 h during infection, but by 6 h post infection Ms is cleared and Mm is retained. Ms is rarely found in the lower gut, though some individual bacteria are observed. In contrast, Mm is present in the lower gut throughout and after the infection period. Observations by electron microscopy correlated well with those by confocal, with numerous Mm debris and bacteria present in the intestine; whereas, little Ms debris remains (Fig. 3 S–X). Interestingly, the villi of the Mm‐infected nematodes were less well organized and had greater variability in density, suggesting that Mm disrupts the actin core of villi, known to be composed of ACT‐5 (MacQueen et al. 2005).
Figure 4.
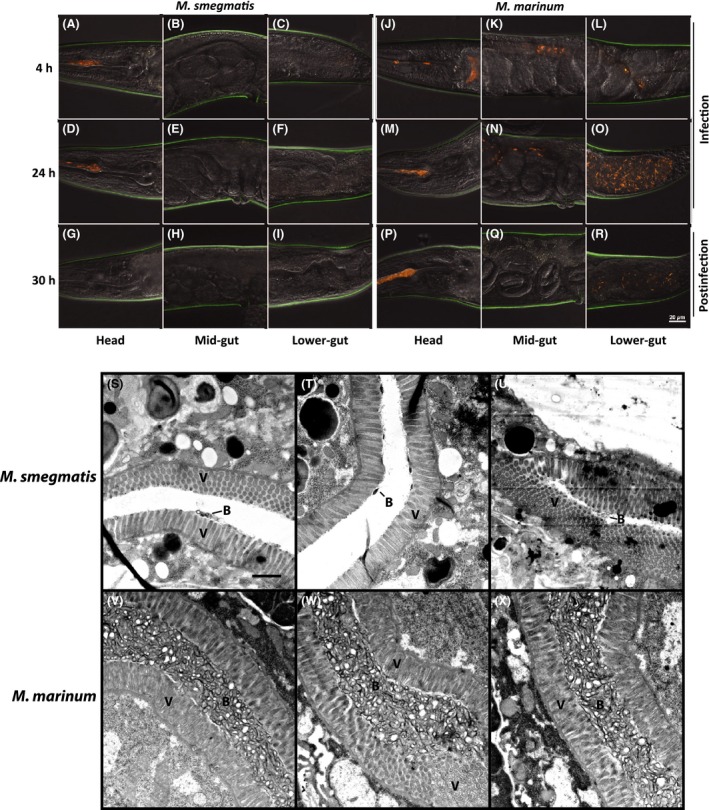
Confocal and transmission electron microscopy of infected C. elegans. C. elegans infected with (A–C, G–I, M–O, S–U) M. smegmatis (tdTomato) or (D–F, J–L, P–R, V–X) M. marinum (tdTomato) were imaged at (A–F) 4 and (G–L) 24 h during infection and (M–X) 6 h postinfection (30 h). (A–R) The head, mid‐gut, and lower‐gut of 13–15 nematodes each at 4, 24, and 30 h were imaged using a confocal microscope and (S–X) at 30 h using transmission electron microscopy. A spectral filter for excitation wavelengths of 500–640 nm was used for confocal microscopy. The scale bar in R (20 μm) applies to panels A–R. The scale bar in S (1 μm) applies to panels S–X. V indicates nematode villi and B indicate mycobacterial debris and mycobacteria.
C. elegans is a novel virulence model for Mm
Mm mutants that are defective for infection and growth within mammalian macrophages (Mehta et al. 2006) were screened for pathogenesis in C. elegans (Fig. 5). A total of eight (44%) of the Mm mutants were attenuated in C. elegans (P < 0.05). These observations suggest that there are parallels between the Mm genes involved in mammalian macrophage infection and those required for colonization of C. elegans. Three of these mutants, mimA, mimG, and mimI were complemented with their respective gene, restoring wild‐type levels of C. elegans killing, confirming that these genes are specifically involved in C. elegans pathogenesis (Fig. S6). Our results suggest that C. elegans is a genetically tractable host for study of Mm pathogenesis and can be used to analyze virulence mechanisms in a manner that complements existing mammalian models.
Figure 5.
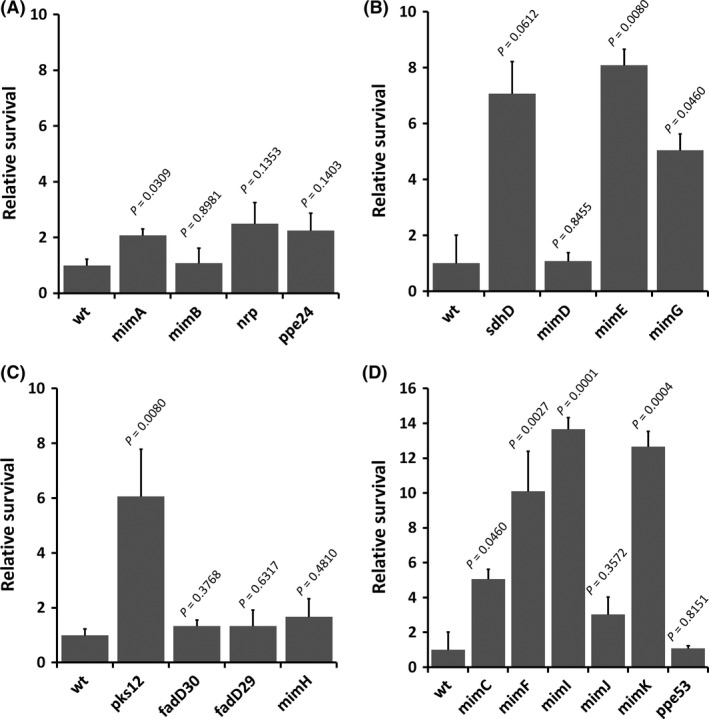
C. elegans can be used to measure virulence of M. marinum. (A–D) Relative survival of C. elegans infected with M. marinum wild type (wt) and macrophage infection mutants (MIMs), 2 d postinfection (day 6). Three trials of 20 nematodes (n = 60) were infected with each of the MIM strains and survival was assessed on day 6. All survival calculations are relative to survival after infection with wild type, e.g. relative survival = number of nematodes surviving with strain/number of nematodes surviving with wild type. Data are presented for means and standard deviations. P values are provided comparing relative survival with each MIM to infection with wild type (unpaired t‐test).
p38 MAPK plays an important role in protection
The p38 MAPK pathway is triggered during the initial response of mammalian macrophages to mycobacterial infection (Roach and Schorey 2002), but its role in the multi‐cellular response in whole animals has been difficult to determine due to the essential nature of this gene during mammalian development. The p38 MAPK can play a role in protection of C. elegans from other pathogens, but mycobacteria have not been examined (Aballay et al. 2003; Jebamercy et al. 2013). We used E. coli expressing RNAi to knock‐down p38 MAPK (pmk‐1) expression as well as other relevant genes in C. elegans. We confirmed that all RNAi knock‐downs display decreased expression of the targeted gene by qPCR. There was >10‐fold decrease in expression for all genes, other than vhp‐1 (MAPK phosphatase), which displayed a >4‐fold decrease. C. elegans with reduced MAPK expression are more susceptible to both Mm and Ms (Fig. 6A, S7). We confirmed that this phenotype was not due to off‐target effects of RNAi using a specific C. elegans p38 MAPK (pmk‐1) mutant. The p38 MAPK mutant displays a similar increase in mortality, with 100% mortality during the first 24 h (Fig. 6B, S7). The C. elegans MAPK mutant not only displayed an increase in mortality, but also increased pathology (Fig. S8), demonstrating that p38 MAPK is an important pathway for protection from mycobacteria.
Figure 6.
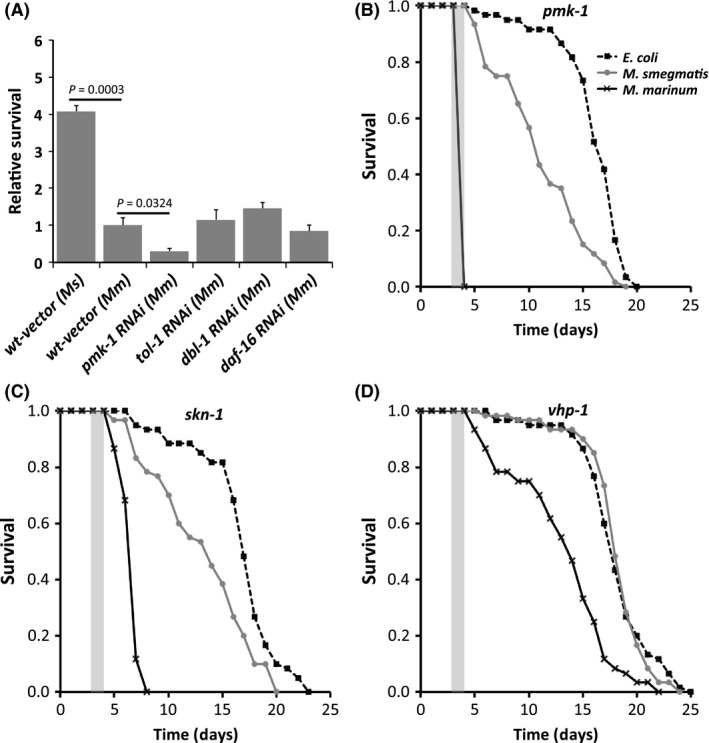
Pathogenic mycobacteria block C. elegans MAPK‐mediated protection using MAPK phosphatase. (A) Relative survival of RNAi knock‐down of p38 MAPK (pmk‐1) TLR (tol‐1), TGF‐ β (dbl‐1) and insulin‐like receptor (daf‐16) at 2 d postinfection as compared with vector alone wild type. Relative survival = number of RNAi knock‐down nematodes surviving/number of wild‐type surviving. Data are means and standard deviations. (B) Survival curve for MAPK (pmk‐1) mutant after 24 h of infection. Mm versus Ms P < 0.0001; Mm versus E. coli P < 0.0001; Ms versus E. coli P < 0.0001. Key in (B) is for (B‐D). (C) Survival curve for the C. elegans the downstream regulator of MAPK (skn‐1) mutant after 24 h infection. Mm versus Ms P < 0.0001; Mm versus E. coli P < 0.0001; Ms versus E. coli P < 0.0001. (D) Survival curve for the C. elegans MAPK phosphatase (vhp‐1) mutant after 24 h infection. Mm versus Ms P < 0.0001; Mm versus E. coli P < 0.0001; Ms versus E. coli P = 0.9173. Survival curve P values from log‐rank analysis.
MAPK is the dominant pathway involved in protection from mycobacteria
Several alternative signaling pathways are important in defense of C. elegans from other pathogenic bacteria, including TLR, TGF‐β, and insulin‐like receptor pathways (Evans et al. 2008; Tenor and Aballay 2008; Singh and Aballay 2009). We used RNAi to decrease expression of tol‐1 (TLR), dbl‐1 (TGF‐β pathway), and daf‐16 (insulin‐like pathway) genes in C. elegans and evaluated mycobacterial susceptibility. None of these signaling pathways had an impact on susceptibility (Fig. 6A). We confirmed these results using C. elegans mutants in tol‐1, dbl‐1, and daf‐16 (Fig. S9 and also found no impact on susceptibility. In fact, C. elegans mutants in other signaling pathways display a trend toward increased resistance to mycobacterial infection. As expected, the C. elegans tol‐1, dbl‐1, and daf‐16 mutants display similar pathology to wild‐type infected with Ms and Mm (Fig. S10, S11 and S12). These observations suggest that the C. elegans model for mycobacterial infection has the advantage that it can be used to focus analysis of host protection and susceptibility mechanisms on the p38 MAPK pathway.
MAPK mediates protection through SKN‐1
One of the primary downstream regulators that C. elegans p38 MAPK controls is SKN‐1 (Papp et al. 2012). The skn‐1 gene, analogous to the essential mammalian nrf1 and nrf2 cap’n’collar subfamily of basic leucine zipper transcription factors, is activated during the C. elegans response to oxidative stress and bacterial infection (Papp et al. 2012). We infected C. elegans skn‐1 RNAi knock‐down nematodes and found no difference compared with wild type in susceptibility as measured by survival (Fig. S13). However, we found that Mm‐infected skn‐1 RNAi knock‐down C. elegans display increased depigmentation and shortened length, suggesting that the role of skn‐1 is only partially observed in knock‐down strains. We investigated the possibility that a complete skn‐1 knockout would have a more obvious phenotype using a skn‐1 mutant. The skn‐1 mutant displays more rapid mortality as compared with wild type after infection with Mm (Fig. 6C). The Mm‐infected skn‐1 mutant displays depigmentation, shortening, and bagging (Fig. S13), but less bagging at 48 h than the pmk‐1 mutant (P < 0.0001), demonstrating that SKN‐1 is a downstream regulator for MAPK‐mediated protection against mycobacteria in C. elegans.
Hijacking the host mapk phosphatase
MAPK phosphatase (vhp‐1) acts as an inhibitor of the p38 MAPK pathway and can modulate the innate immune response (Kim et al. 2004). We evaluated the role of MAPK phosphatase in the susceptibility of C. elegans to mycobacterial infection using RNAi. We found that the C. elegans vhp‐1 knock‐down strain displayed increased survival after infection with Mm (Fig. S15). Similarly, a vhp‐1 mutant displayed improved survival postinfection by Mm (Fig. 6D). The vhp‐1 mutant also displays a reduction in bagging (<5%) as compared with wild type (>80%) and the pmk‐1 mutant (100%) (Fig. S16), demonstrating that the p38 MAPK pathway is involved in combating mycobacterial infection and that the presence of vhp‐1 increases susceptibility to mycobacteria in C. elegans.
Discussion
The C. elegans model provides an extremely simple and effective system for pathogenesis studies, particularly mechanisms for avoiding ROS and lysosomal defenses (Mallo et al. 2002; Chavez et al. 2007). Research has focused on the adaptive immune response to pathogenic mycobacteria, but the innate immune response and roles of MAPK are not fully understood (Philips and Ernst 2012). Part of the reason that C. elegans has not been used to study Mm pathogenesis is because another study did not observe mortality (Couillault and Ewbank 2002), but conditions were not optimized. We used age‐synchronized, 3‐day‐old gravid adult nematodes and all infections were carried out at ~20°C. Use of adult gravid nematodes is important, since bagging and depigmentation are only observed in adults. The pathological processes of depigmentation and bagging are consistent with observations with other pathogens (Mosser et al. 2011). Pseudomonas aeruginosa was one of the first human pathogens studied in C. elegans, where they also cause increased mortality (Mahajan‐Miklos et al. 1999). S. pyogenes, E. faecium, and P. aeruginosa cause mortality via toxin‐mediated killing (Jansen et al. 2002; Moy et al. 2004). E. faecalis, S. marcescens, and S. enterica attach to the gut epithelium of C. elegans (Kurz et al. 2003; Maadani et al. 2007; Sem and Rhen 2012). The actinomycete Streptomyces albireticuli and the fungus Drechmeria coniospora, natural pathogens of C. elegans, invade the gut epithelium (Jansson et al. 1984; Park et al. 2002). Interestingly, Microbacterium nematophilum forms an adhesive biofilm on the C. elegans cuticle (Hodgkin et al. 2000). Despite the diversity of bacterial interactions observed, the mechanisms of susceptibility to mycobacteria are relatively unique.
Mm cause mortality in C. elegans through inhibition of the nematode's ability to lay eggs. Inhibition of egg laying causes infected adult nematodes to produce a bag of nematodes (bagging), which is protection for the progeny (Mosser et al. 2011). Most likely the bagging phenotype is the result of stress induced within the worm as a result of attempts to defend against mycobacterial invasion of the intestinal epithelium, involving production of reactive oxygen species. Increased oxidative stress causes loss of pigmentation in adult nematodes and lower survival. Future studies evaluating the mechanisms of Mm pathogenesis in the absence of bagging would be an important next step using this host–pathogen model. These studies could be accomplished with C. elegans mutants that do not produce progeny, CF512: fer‐15;fem‐1, and thereby, would not die due to bagging (Troemel et al. 2006). The identification of several Mm mutants that impact mortality due to bagging suggests that specific Mm pathogenic mechanisms are involved and separation from early mortality due to bagging might offer one way to gain further discriminatory power for subtle virulence defects. Interestingly, we also observed differences in time to death and percent mortality when worms are exposed to more M. marinum, as shown in Figure 1. These observations suggest that additional discriminatory power for subtle virulence defects might be accomplished by variation in the exposure time to M. marinum mutants in different virulence pathways of interest.
On the surface, one might consider the absence of a clear macrophage lineage of cells in C. elegans (Engelmann and Pujol 2010) to be a disadvantage for study of intracellular pathogens, including Mm. However, other intracellular pathogens, including Legionella pneumophila (Komura et al. 2010), have been successfully examined using C. elegans as a host that allowed analysis of specific virulence mechanisms playing an important role within macrophages. There are specialized cells that continuously endocytose fluid from the lumen of the worm intestine called coelomocytes, but these have not been shown to phagocytose large particles (Fares and Greenwald 2001). Interestingly, when ROS and lysozymes are produced in worms, these bactericidal host defense molecules are delivered to the lumen of the worm intestine (Mallo et al. 2002; Chavez et al. 2007), rather than to an endosomal or sorting compartment, as would occur within macrophages or single‐cell phagocytes, such as amoebae (Cirillo 1999; Yan et al. 2004). These observations, combined with those within this study, suggest the interesting concept that, in the case of C. elegans, the lumen of the worm intestine might serve a similar role to the sorting compartment where food or energy sources are sorted from toxic or pathogenic particles and pathogenic particles are attacked with ROS, lysozyme, and potentially other innate immune effectors to defend the worm. Thus, the entire digestive tract of the worm serves as an analogous system to that of a phagocyte. Using this analogy, the worm phagocytoses bacteria by eating them and the lumen of the worm intestine serves as an endosome, with which ROS and lysosomal vesicles fuse, resulting in bacterial killing and degradation for nonpathogenic species, but pathogens resist or inhibit these innate immune mechanisms.
Activation of MAPK differs between pathogenic and nonpathogenic mycobacteria infection in C. elegans, consistent with observations in mammalian tissue culture models (Roach and Schorey 2002; Schorey and Cooper 2003). The p38 MAPK plays a role in protection, but MAPK phosphatase (vhp‐1) plays a role in susceptibility. C. elegans lacking p38 MAPK, display 100% mortality, which is reduced in the absence of vhp‐1. Ms causes comparable mortality rates to E. coli in the vhp‐1 mutant, similar to the tol‐1, dbl‐1, or daf‐16 mutants. Thus, susceptibility to Mm primarily involves MAPK. While the skn‐1 mutant does not display the same bagging phenotype as the MAPK mutant, it displays increased depigmentation and reduced survival. Although these observations demonstrate the importance of skn‐1 in protection from mycobacteria, the more modest phenotype suggests that there are additional downstream mediators of MAPK protection yet to be identified. Further studies could identify additional downstream regulatory genes and the effectors involved in MAPK‐mediated protection from mycobacteria. Apparently, C. elegans responds differently to pathogenic and nonpathogenic mycobacteria through different levels of MAPK activation (Fig. 7). Nonpathogenic mycobacteria activate MAPK leading to bacterial killing and C. elegans survival, but pathogenic mycobacteria may utilize vhp‐1 to reduce activation of MAPK, leading to susceptibility and mortality. Similarly, mycobacterial virulence inversely correlates with levels of MAPK activation in mammalian macrophages (Roach and Schorey 2002; Schorey and Cooper 2003). The mechanisms involved in this differential activation of MAPK were previously unknown, demonstrating that analysis of interactions of pathogenic mycobacteria with C. elegans can provide insight into mechanistic differences that lead to pathogenesis. Further study of vhp‐1 in C. elegans is needed, since it is likely that its absence also affects other kinases that play a role in host defense.
Figure 7.
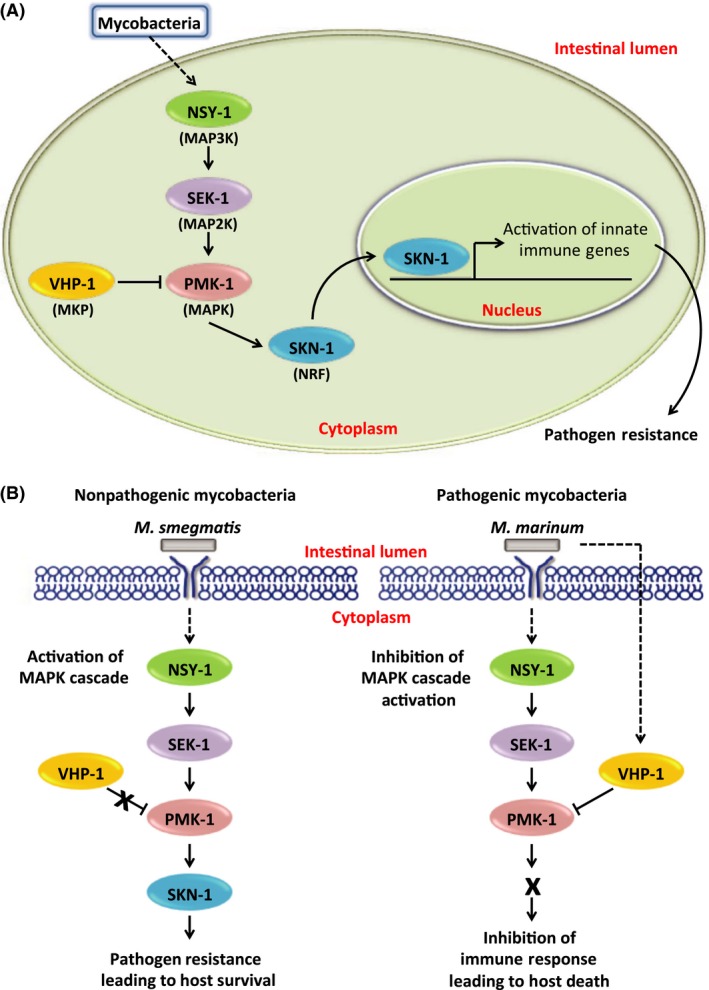
Mycobacterial mechanism of MAPK pathway control during C. elegans infection. (A) When C. elegans is infected with mycobacteria, the p38 MAPK pathway (PMK‐1) is activated, leading to an innate immune response through downstream regulators, including the NRF‐like regulator SKN‐1, that increase pathogen resistance. The MAPK phosphatase, VHP‐1, inhibits activation of MAPK, but is inactivated when foreign organisms are recognized, allowing MAPK‐mediated protection. (B) In contrast to nonpathogenic mycobacteria (M. smegmatis), pathogenic mycobacteria (M. marinum) may inhibit activation of MAPK through the MAPK phosphatase VHP‐1, allowing colonization and pathogenesis.
The ability of Mm to colonize C. elegans is due to interactions with the intestine, allowing elucidation of the bacterial ligands and receptors involved. We identified eight Mm mutants that are defective for infection of mammalian macrophages as well as causing less mortality in C. elegans, suggesting the mechanisms are relevant to mammalian pathogenesis. Colonization of the C. elegans intestine produces morphological changes in actin within villi, consistent with known interactions of pathogenic mycobacteria with actin during infection of macrophages (Guerin and de Chastellier 2000; Anes et al. 2003), suggesting that C. elegans can be used for study of mycobacterial molecular pathogenesis by allowing analysis of signaling pathways involved in innate immunity, mechanisms of resistance to ROS and lysosomes, and mechanisms of colonization and remodeling of actin. The tractability of C. elegans will facilitate mechanistic analysis of both the protective response to mycobacteria and virulence pathways, providing insight into novel strategies to prevent pathogenesis.
Conflicts of Interest
No authors have a conflict of interest.
Supporting information
Figure S1. Pathological changes in wild‐type (N2) C. elegans infected with bacteria.
Figure S2. Morphological characteristics of C. elegans (TP12) infected with E. coli (OP50).
Figure S3. Survival of TP12 after bacterial infection.
Figure S4. Pathological changes in TP12 C. elegans infected with bacteria.
Figure S5. Bacterial load in C. elegans (N2) determined by plating for CFU.
Figure S6 . C. elegans Infected with complemented MIM of M. marinum.
Figure S7. Impact of C. elegans pmk‐1 on infection with mycobacteria.
Figure S8. Pathological changes in pmk‐1 Mutant C. elegans infected with bacteria.
Figure S9. The C. elegans tol‐1, dbl‐1, and daf‐16 Pathways Do Not Impact Mycobacterial Infection.
Figure S10. Pathological changes in tol‐1 Mutant C. elegans infected with bacteria.
Figure S11. Pathological changes in dbl‐1 Mutant C. elegans infected with bacteria.
Figure S12. Pathological changes in daf‐16 Mutant C. elegans infected with bacteria.
Figure S13. Role of C. elegans skn‐1 in mycobacterial infection.
Figure S14. Pathological changes in skn‐1 Mutant C. elegans infected with bacteria
Figure S15. Role of C. elegans vhp‐1 in mycobacterial infection.
Figure S16. Pathological changes in vhp‐1 Mutant C. elegans infected with bacteria.
Table S1. Macrophage infection mutants (MIMs) of M. marinum.
Table S2. List of C. elegans RNAi constructs and mutant strains.
Table S3. Primers used for confirmation of C. elegans mutants and RNAi knock‐down.
Acknowledgments
We thank Dr. Stanislav Vitha and Richard Littleton at the Texas A&M Microscopy Imaging Center (MIC) and Robbie Schultz for assistance in transmission electron microscopy.
MicrobiologyOpen 2016; 5(3): 436–452
References
- Aballay, A. , Drenkard E., Hilbun L. R., and Ausubel F. M.. 2003. Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr. Biol. 13:47–52. [DOI] [PubMed] [Google Scholar]
- Anes, E. , Kuhnel M. P., Bos E., Moniz‐Pereira J., Habermann A., and Griffiths G.. 2003. Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat. Cell Biol. 5:793–802. [DOI] [PubMed] [Google Scholar]
- Ashrafi, K. , Chang F. Y., Watts J. L., Fraser A. G., Kamath R. S., Ahringer J., et al. 2003. Genome‐wide RNAi analysis of Caenorhabditis elegans fat regulatory genes. Nature 421:268–272. [DOI] [PubMed] [Google Scholar]
- Bishai, W. R. , Dannenberg A. M. Jr, Parrish N., Ruiz R., Chen P., Zook B. C., et al. 1999. Virulence of mycobacterium tuberculosis CDC1551 and H37Rv in rabbits evaluated by Lurie's pulmonary tubercle count method. Infect. Immun. 67:4931–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, S. 1974. The genetics of Caenorhabditis elegans . Genetics 77:71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez, V. , Mohri‐Shiomi A., Maadani A., Vega L. A., and Garsin D. A.. 2007. Oxidative stress enzymes are required for DAF‐16‐mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans . Genetics 176:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo, J. D. 1999. Exploring a novel perspective on pathogenic relationships. Trends Microbiol. 7:96–98. [DOI] [PubMed] [Google Scholar]
- Coleman, J. J. , and Mylonakis E.. 2009. The tangled web of signaling in innate immunity. Cell Host Microbe 5:313–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couillault, C. , and Ewbank J. J.. 2002. Diverse bacteria are pathogens of Caenorhabditis elegans . Infect. Immun. 70:4705–4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmons, S. W. , Klass M. R., and Hirsh D.. 1979. Analysis of the constancy of DNA sequences during development and evolution of the nematode Caenorhabditis elegans . Proc. Natl Acad. Sci. USA 76:1333–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann, I. , and Pujol N.. 2010. Innate immunity in C. elegans . Adv. Exp. Med. Biol. 708:105–121. [DOI] [PubMed] [Google Scholar]
- Evans, E. A. , Kawli T., and Tan M. W.. 2008. Pseudomonas aeruginosa suppresses host immunity by activating the DAF‐2 insulin‐like signaling pathway in Caenorhabditis elegans . PLoS Pathog. 4:e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares, H. , and Greenwald I.. 2001. Genetic analysis of endocytosis in Caenorhabditis elegans: coelomocyte uptake defective mutants. Genetics 159:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin, D. A. , Sifri C. D., Mylonakis E., Qin X., Singh K. V., Murray B. E., et al. 2001. A simple model host for identifying Gram‐positive virulence factors. Proc. Natl Acad. Sci. USA 98:10892–10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaziou, P. , Falzon D., Floyd K., and Raviglione M.. 2013. Global epidemiology of tuberculosis. Semin. Respir. Crit. Care Med. 34:3–16. [DOI] [PubMed] [Google Scholar]
- Guerin, I. , and de Chastellier C.. 2000. Pathogenic mycobacteria disrupt the macrophage actin filament network. Infect. Immun. 68:2655–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall, D. H. , Hartwieg E., and Nguyen K. C.. 2012. Modern electron microscopy methods for C. elegans . Methods Cell Biol. 107:93–149. [DOI] [PubMed] [Google Scholar]
- Hodgkin, J. , Kuwabara P. E., and Corneliussen B.. 2000. A novel bacterial pathogen, Microbacterium nematophilum, induces morphological change in the nematode C. elegans . Curr. Biol. 10:1615–1618. [DOI] [PubMed] [Google Scholar]
- Irazoqui, J. E. , Urbach J. M., and Ausubel F. M.. 2010. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat. Rev. Immunol. 10:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki, A. , and Medzhitov R.. 2010. Regulation of adaptive immunity by the innate immune system. Science 327:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, S. K. , Hernandez‐Abanto S. M., Cheng Q. J., Singh P., Ly L. H., Klinkenberg L. G., et al. 2007. Accelerated detection of mycobacterium tuberculosis genes essential for bacterial survival in guinea pigs, compared with mice. J. Infect. Dis. 195:1634–1642. [DOI] [PubMed] [Google Scholar]
- Janeway, C. A. Jr . 1992. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 13:11–16. [DOI] [PubMed] [Google Scholar]
- Jansen, W. T. , Bolm M., Balling R., Chhatwal G. S., and Schnabel R.. 2002. Hydrogen peroxide‐mediated killing of Caenorhabditis elegans by Streptococcus pyogenes . Infect. Immun. 70:5202–5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson, H. B. , von Hofsten A., and von Mecklenburg C.. 1984. Life cycle of the endoparasitic nematophagous fungus Meria coniospora: a light and electron microscopic study. Antonie Van Leeuwenhoek 50:321–327. [DOI] [PubMed] [Google Scholar]
- Jebamercy, G. , Vigneshwari L., and Balamurugan K.. 2013. A MAP Kinase pathway in Caenorhabditis elegans is required for defense against infection by opportunistic Proteus species. Microbes Infect. 15:550– 568. [DOI] [PubMed] [Google Scholar]
- Juffermans, N. P. , Verbon A., Belisle J. T., Hill P. J., Speelman P., van Deventer S. J., et al. 2000. Mycobacterial lipoarabinomannan induces an inflammatory response in the mouse lung. A role for interleukin‐1. Am. J. Respir. Crit. Care Med. 162:486–489. [DOI] [PubMed] [Google Scholar]
- Kawai, T. , and Akira S.. 2010. The role of pattern‐recognition receptors in innate immunity: update on Toll‐like receptors. Nat. Immunol. 11:373–384. [DOI] [PubMed] [Google Scholar]
- Kim, D. H. , Liberati N. T., Mizuno T., Inoue H., Hisamoto N., Matsumoto K., et al. 2004. Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK‐1 MAPK kinase and VHP‐1 MAPK phosphatase. Proc. Natl Acad. Sci. USA 101:10990–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingeter, L. M. , and Lin X.. 2012. C‐type lectin receptor‐induced NF‐kappaB activation in innate immune and inflammatory responses. Cell. Mol. Immunol. 9:105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinnijenhuis, J. , Oosting M., Joosten L. A., Netea M. G., and Van Crevel R.. 2011. Innate immune recognition of mycobacterium tuberculosis. Clin. Dev. Immunol. 2011:405310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson, K. L. , Hmama Z., Herrera‐Velit P., Rochford R., and Reiner N. E.. 1998. Lipoarabinomannan of mycobacterium tuberculosis promotes protein tyrosine dephosphorylation and inhibition of mitogen‐activated protein kinase in human mononuclear phagocytes. Role of the Src homology 2 containing tyrosine phosphatase 1. J. Biol. Chem. 273:645–652. [DOI] [PubMed] [Google Scholar]
- Komura, T. , Yasui C., Miyamoto H., and Nishikawa Y.. 2010. Caenorhabditis elegans as an alternative model host for legionella pneumophila, and protective effects of Bifidobacterium infantis. Appl. Environ. Microbiol. 76:4105–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul, A. , Herget T., Klebl B., and Ullrich A.. 2004. Interplay between mycobacteria and host signalling pathways. Nat. Rev. Microbiol. 2:189–202. [DOI] [PubMed] [Google Scholar]
- Kurz, C. L. , Chauvet S., Andres E., Aurouze M., Vallet I., Michel G. P., et al. 2003. Virulence factors of the human opportunistic pathogen Serratia marcescens identified by in vivo screening. EMBO J. 22:1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maadani, A. , Fox K. A., Mylonakis E., and Garsin D. A.. 2007. Enterococcus faecalis mutations affecting virulence in the Caenorhabditis elegans model host. Infect. Immun. 75:2634–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacQueen, A. J. , Baggett J. J., Perumov N., Bauer R. A., Januszewski T., Schriefer L., et al. 2005. ACT‐5 is an essential Caenorhabditis elegans actin required for intestinal microvilli formation. Mol. Biol. Cell 16:3247–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan‐Miklos, S. , Tan M. W., Rahme L. G., and Ausubel F. M.. 1999. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa‐Caenorhabditis elegans pathogenesis model. Cell 96:47–56. [DOI] [PubMed] [Google Scholar]
- Mallo, G. V. , Kurz C. L., Couillault C., Pujol N., Granjeaud S., Kohara Y., et al. 2002. Inducible antibacterial defense system in C. elegans . Curr. Biol. 12:1209–1214. [DOI] [PubMed] [Google Scholar]
- Mehta, P. K. , Pandey A. K., Subbian S., El‐Etr S. H., Cirillo S. L., Samrakandi M. M., et al. 2006. Identification of Mycobacterium marinum macrophage infection mutants. Microb. Pathog. 40:139–151. [DOI] [PubMed] [Google Scholar]
- Mosser, T. , Matic I., and Leroy M.. 2011. Bacterium‐induced internal egg hatching frequency is predictive of life span in Caenorhabditis elegans populations. Appl. Environ. Microbiol. 77:8189–8192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moy, T. I. , Mylonakis E., Calderwood S. B., and Ausubel F. M.. 2004. Cytotoxicity of hydrogen peroxide produced by Enterococcus faecium. Infect. Immun. 72:4512–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, H. , Yan B. S., Rojas M., Shebzukhov Y. V., Zhou H., Kobzik L., et al. 2005. Ipr1 gene mediates innate immunity to tuberculosis. Nature 434:767–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp, D. , Csermely P., and Soti C.. 2012. A role for SKN‐1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans . PLoS Pathog. 8:e1002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. O. , El‐Tarabily K. A., Ghisalberti E. L., and Sivasithamparam K.. 2002. Pathogenesis of Streptoverticillium albireticuli on Caenorhabditis elegans and its antagonism to soil‐borne fungal pathogens. Lett. Appl. Microbiol. 35:361–365. [DOI] [PubMed] [Google Scholar]
- Philips, J. A. , and Ernst J. D.. 2012. Tuberculosis pathogenesis and immunity. Annu. Rev. Pathol. 7:353–384. [DOI] [PubMed] [Google Scholar]
- Roach, S. K. , and Schorey J. S.. 2002. Differential regulation of the mitogen‐activated protein kinases by pathogenic and nonpathogenic mycobacteria. Infect. Immun. 70:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach, T. I. , Barton C. H., Chatterjee D., and Blackwell J. M.. 1993. Macrophage activation: lipoarabinomannan from avirulent and virulent strains of Mycobacterium tuberculosis differentially induces the early genes c‐fos, KC, JE, and tumor necrosis factor‐alpha. J. Immunol. 150:1886–1896. [PubMed] [Google Scholar]
- Schorey, J. S. , and Cooper A. M.. 2003. Macrophage signalling upon mycobacterial infection: the MAP kinases lead the way. Cell. Microbiol. 5:133–142. [DOI] [PubMed] [Google Scholar]
- Schultz, R. D. , Bennett E. E., Ellis E. A., and Gumienny T. L.. 2014. Regulation of extracellular matrix organization by BMP signaling in Caenorhabditis elegans . PLoS ONE 9:e101929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sem, X. , and Rhen M.. 2012. Pathogenicity of Salmonella enterica in Caenorhabditis elegans relies on disseminated oxidative stress in the infected host. PLoS ONE 7:e45417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh, M. U. , and Champion P. A.. 2010. To catch a killer. What can mycobacterial models teach us about Mycobacterium tuberculosis pathogenesis? Curr. Opin. Microbiol. 13:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivers, R. P. , Kooistra T., Chu S. W., Pagano D. J., and Kim D. H.. 2009. Tissue‐specific activities of an immune signaling module regulate physiological responses to pathogenic and nutritional bacteria in C. elegans . Cell Host Microbe 6:321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, V. , and Aballay A.. 2009. Regulation of DAF‐16‐mediated innate immunity in Caenorhabditis elegans . J. Biol. Chem. 284:35580–35587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenger, S. , and Modlin R. L.. 1999. T cell mediated immunity to Mycobacterium tuberculosis. Curr. Opin. Microbiol. 2:89–93. [DOI] [PubMed] [Google Scholar]
- Strohmeier, G. R. , and Fenton M. J.. 1999. Roles of lipoarabinomannan in the pathogenesis of tuberculosis. Microbes Infect. 1:709–717. [DOI] [PubMed] [Google Scholar]
- Tenor, J. L. , and Aballay A.. 2008. A conserved Toll‐like receptor is required for Caenorhabditis elegans innate immunity. EMBO Rep. 9:103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel, E. R. , Chu S. W., Reinke V., Lee S. S., Ausubel F. M., and Kim D. H.. 2006. p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans . PLoS Genet. 2:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, L. , Cerny R. L., and Cirillo J. D.. 2004. Evidence that hsp90 is involved in the altered interactions of Acanthamoeba castellanii variants with bacteria. Eukaryot. Cell 3:567–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. S. , Nam H. J., Seo M., Han S. K., Choi Y., Nam H. G., et al. 2011. OASIS: online application for the survival analysis of lifespan assays performed in aging research. PLoS ONE 6:e23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pathological changes in wild‐type (N2) C. elegans infected with bacteria.
Figure S2. Morphological characteristics of C. elegans (TP12) infected with E. coli (OP50).
Figure S3. Survival of TP12 after bacterial infection.
Figure S4. Pathological changes in TP12 C. elegans infected with bacteria.
Figure S5. Bacterial load in C. elegans (N2) determined by plating for CFU.
Figure S6 . C. elegans Infected with complemented MIM of M. marinum.
Figure S7. Impact of C. elegans pmk‐1 on infection with mycobacteria.
Figure S8. Pathological changes in pmk‐1 Mutant C. elegans infected with bacteria.
Figure S9. The C. elegans tol‐1, dbl‐1, and daf‐16 Pathways Do Not Impact Mycobacterial Infection.
Figure S10. Pathological changes in tol‐1 Mutant C. elegans infected with bacteria.
Figure S11. Pathological changes in dbl‐1 Mutant C. elegans infected with bacteria.
Figure S12. Pathological changes in daf‐16 Mutant C. elegans infected with bacteria.
Figure S13. Role of C. elegans skn‐1 in mycobacterial infection.
Figure S14. Pathological changes in skn‐1 Mutant C. elegans infected with bacteria
Figure S15. Role of C. elegans vhp‐1 in mycobacterial infection.
Figure S16. Pathological changes in vhp‐1 Mutant C. elegans infected with bacteria.
Table S1. Macrophage infection mutants (MIMs) of M. marinum.
Table S2. List of C. elegans RNAi constructs and mutant strains.
Table S3. Primers used for confirmation of C. elegans mutants and RNAi knock‐down.