

Here, Lemay et al. investigated whether the mechanism of transcription termination is evolutionarily conserved in S. pombe. They show that the S. pombe ortholog of Nrd1, Seb1, does not function in NNS-like termination but promotes polyadenylation site selection of coding and noncoding genes.
Keywords: 3′ end processing, Nrd1, polyadenylation, S. pombe, Seb1, transcription termination
Abstract
Termination of RNA polymerase II (RNAPII) transcription is associated with RNA 3′ end formation. For coding genes, termination is initiated by the cleavage/polyadenylation machinery. In contrast, a majority of noncoding transcription events in Saccharomyces cerevisiae does not rely on RNA cleavage for termination but instead terminates via a pathway that requires the Nrd1–Nab3–Sen1 (NNS) complex. Here we show that the Schizosaccharomyces pombe ortholog of Nrd1, Seb1, does not function in NNS-like termination but promotes polyadenylation site selection of coding and noncoding genes. We found that Seb1 associates with 3′ end processing factors, is enriched at the 3′ end of genes, and binds RNA motifs downstream from cleavage sites. Importantly, a deficiency in Seb1 resulted in widespread changes in 3′ untranslated region (UTR) length as a consequence of increased alternative polyadenylation. Given that Seb1 levels affected the recruitment of conserved 3′ end processing factors, our findings indicate that the conserved RNA-binding protein Seb1 cotranscriptionally controls alternative polyadenylation.
Termination of RNA polymerase II (RNAPII) transcription is a critical step of gene expression that is functionally associated with RNA 3′ end formation and release of the nascent transcript from the site of transcription. For most protein-coding genes, current data suggest a model in which transcription termination is initiated by the cotranscriptional recruitment of 3′ end processing factors to the C-terminal domain (CTD) of the largest subunit of RNAPII (Porrua and Libri 2015). The CTD consists of a succession of heptad repeats with the consensus amino acid sequence Y-S-P-T-S-P-S, which is subjected to a plethora of stage-dependent post-translational modifications that control the recruitment of various RNA processing factors (Corden 2013). One of these CTD modifications, phosphorylation of Ser2 (Ser2P), gradually increases as transcription elongation progresses and peaks at the 3′ end of mRNA-encoding genes. Ser2P is in fact important for the recruitment of 3′ end processing factors in both budding yeast (Saccharomyces cerevisiae) and human cells (Ahn et al. 2004; Kim et al. 2010; Nojima et al. 2015), consistent with the overall conservation of 3′ end processing factors (Xiang et al. 2014). The transfer of 3′ end processing factors from the transcription complex onto the nascent transcript involves the recognition of a functional polyadenylation [poly(A)] signal (PAS) composed of cis-acting RNA elements that define the site of pre-mRNA cleavage (Shi and Manley 2015). The nascent pre-mRNA is subsequently cleaved by an endonuclease (Ysh1 in S. cerevisiae; CPSF73 in humans), generating a free 3′ end for the poly(A) machinery. Endonucleolytic cleavage also generates an uncapped 5′ end to the RNA downstream from the cleavage site, providing an entry point for a 5′–3′ exonuclease (Rat1 in S. cerevisiae; Xrn2 in humans) that has been proposed to chase RNAPII and promote its dissociation from the DNA template, a mechanism of transcription termination referred to as the torpedo model (Kim et al. 2004b; Fong et al. 2015).
In addition to mRNA-coding genes, RNAPII is also responsible for the synthesis of many noncoding RNAs (ncRNAs), including small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and cryptic unstable transcripts (CUTs). In S. cerevisiae, transcription termination of these ncRNAs does not rely on the 3′ end processing machinery but on a mechanism that requires the activity of the Nrd1–Nab3–Sen1 (NNS) complex (Porrua and Libri 2015). Furthermore, whereas termination at protein-coding genes correlates with Ser2P, NNS recruitment to the CTD is influenced by Ser5P via the CTD interaction domain (CID) of Nrd1 (Gudipati et al. 2008; Vasiljeva et al. 2008a). The presence of specific RNA sequence motifs is crucial to subsequently engage the NNS complex onto nascent ncRNAs via the RNA-binding properties of Nrd1 and Nab3 (Creamer et al. 2011; Porrua et al. 2012; Schulz et al. 2013; Webb et al. 2014). Interestingly, transcription termination by the NNS complex does not appear to be associated with endonucleolytic cleavage of the nascent RNA but rather functions by a mechanism that dislodges RNAPII from the DNA template via the helicase activity of Sen1 (Porrua and Libri 2013). Another important feature of NNS-dependent termination is its functional association with the exosome complex of 3′–5′ exonucleases, which contributes to 3′ end trimming of snRNA/snoRNA precursors and the rapid degradation of products of pervasive transcription (Arigo et al. 2006; Thiebaut et al. 2006; Vasiljeva and Buratowski 2006).
Interestingly, genome-wide studies indicate that pervasive transcription is widespread in not only S. cerevisiae but also many other species, including humans (Jensen et al. 2013). However, despite the many studies that have underscored the critical role of the S. cerevisiae NNS complex in limiting the extent of pervasive transcription, the conservation of NNS-like transcription termination across eukaryotic species has remained elusive. Human SCAF8 and Schizosaccharomyces pombe Seb1 share a common protein domain architecture with S. cerevisiae Nrd1, including a conserved RNA recognition motif (RRM) and a CID (Meinhart and Cramer 2004). Fission yeast Seb1 was in fact shown to function in heterochromatin assembly via binding to ncRNAs originating from pericentromeric repeats (Marina et al. 2013), which is reminiscent of the silencing function of Nrd1 at ribosomal DNA and telomeric loci (Vasiljeva et al. 2008b). As yet, however, it remains unknown whether S. pombe Seb1 and human SCAF8 function in NNS-like transcription termination.
Here, we set out to characterize NNS-like transcription termination in S. pombe and disclose its functional relevance in transcriptome surveillance. Unexpectedly, proteomic analysis of Seb1-associated proteins found no evidence for an NNS-like complex but rather identified several mRNA 3′ end processing factors. Furthermore, transcriptome-wide analysis of Seb1–RNA associations revealed widespread binding downstream from poly(A) sites. The functional significance of this RNA-binding pattern was demonstrated by increased levels of alternative mRNA poly(A) in Seb1-depleted cells, resulting in global changes in 3′ untranslated region (UTR) lengths. Our data suggest that Seb1 controls poly(A) site selection by promoting the recruitment of specific cleavage/poly(A) factors at the 3′ ends of genes via a mechanism linked to transcription elongation kinetics. Our findings reveal that regulation of 3′ UTR length is a cotranscriptional process controlled by the recruitment of the Seb1 RNA-binding protein at the 3′ ends of genes.
Results
Global RNAPII transcription termination defects in Seb1-depleted cells
To examine whether an NNS-like complex exists in S. pombe, amino acid sequence comparisons against the fission yeast proteome identified gene products with substantial sequence homology with S. cerevisiae Nab3 and Nrd1: 22% and 29% identical (53% and 57% similar) to S. pombe SPAC3H8.09c (Nab3) and SPAC222.09 (Seb1), respectively. Interestingly, the fission yeast genome expresses two distinct Sen1 paralogs, SPAC6G9.10c (Sen1) and SPBC29A10.10c (Dbl8), which show 26% and 27% identity (58% similarity), respectively, to S. cerevisiae Sen1. Surprisingly, only the NRD1 homolog seb1 is essential for viability in S. pombe, which is in contrast to S. cerevisiae, where NRD1, NAB3, and SEN1 are all essential genes. We also found that the S. pombe dbl8Δ/sen1Δ double mutant was viable (data not shown).
The S. cerevisiae NNS complex is well known for its involvement in termination of noncoding transcripts such as snoRNA genes (Porrua and Libri 2015). To test whether this function is conserved in fission yeast, we measured RNAPII density along a snoRNA gene (Fig. 1A) by chromatin immunoprecipitation (ChIP) assay in strains in which genes encoding for putative orthologs of the budding yeast NNS complex were either deleted (nab3, sen1, or dbl8) or expressed under the control of the thiamine-sensitive nmt1 promoter in the case of the essential seb1 gene (Pnmt1-seb1). In wild-type cells, RNAPII ChIP signals showed the expected gradual decline as transcription progresses downstream from the snR3 gene (Fig. 1B). Deletion of nab3, sen1, and dbl8 as well as the dbl8Δ/sen1Δ double deletion did not markedly affect this RNAPII profile (Fig. 1B). In contrast, the depletion of Seb1 in thiamine-supplemented medium resulted in increased levels of RNAPII at the 3′ end of snR3 relative to the wild-type strain grown in the same conditions (Fig. 1B, see regions 4–5), consistent with transcription termination defects. A similar RNAPII profile was observed after Seb1 depletion using a CTD-independent ChIP approach that used a strain expressing an HA-tagged version of a core RNAPII component, Rpb3 (Fig. 1C). Readthrough transcription was also observed by analyzing the distribution of elongating RNAPII by a transcription run-on (TRO) assay, revealing increased production of nascent RNA at the 3′ end of snR3 in Seb1-depleted cells (Fig. 1D,E). Therefore, a substantial proportion of RNAPII that terminates downstream from snR3 in wild-type cells fails to terminate in Seb1-deficient cells. Importantly, global analysis of RNAPII levels in Seb1-depleted cells by ChIP-seq (ChIP combined with high-throughput sequencing) revealed widespread transcription termination defects that were not limited to noncoding genes (Fig. 1F) but were also apparent at protein-coding genes (Fig. 1G,H). Plotting the cumulative levels of RNAPII binding relative to annotated cleavage/poly(A) sites indicated a clear shift in polymerase density downstream from the noticeable decline observed in wild-type cells (Fig. 1I). This demonstrates that transcription termination defects are a general feature of Seb1-depleted cells. Importantly, the cumulative RNAPII profile of Seb1-depleted cells was not biased by a specific class of gene or restricted to a particular genomic arrangement, as mRNA-, snoRNA-, and snRNA-encoding genes all showed transcription termination defects as well as both tandem and convergent gene pairs (Supplemental Fig. S1). These results indicate that a deficiency in Seb1 causes widespread defects in RNAPII termination at coding and noncoding genes, which is in contrast to S. cerevisiae Nrd1, which mainly functions at ncRNAs. The functional divergence between S. pombe Seb1 and S. cerevisiae Nrd1 was further demonstrated by the inability of Seb1 to complement loss of Nrd1 functions in S. cerevisiae, while expression of Nrd1 did not complement loss of Seb1 functions in S. pombe (Supplemental Fig. S2A–E). We conclude that Seb1 and Nrd1 are not functional homologs.
Figure 1.

Transcription termination defects in Seb1-depleted cells. (A) Schematic of the snR3 snoRNA locus. Bars above the gene show the positions of PCR products used for ChIP analyses in B and C. p(A) refers to the poly(A) site of the 3′ extended precursor (Lemay et al. 2010). (B) ChIP analyses of RNAPII density along the snR3 gene using extracts prepared from wild-type and the indicated mutant strains. ChIP signals (percent input) were normalized to region 1. Error bars indicate SD. n = 3 biological replicates from independent cell cultures. (C) ChIP analyses of HA-tagged Rpb3 (Rpb3-3XHA) along the snR3 gene in Seb1-depleted cells (Pnmt1-seb1) or control (wild-type) cells. Error bars indicate SD. n = 3 biological replicates from independent cell cultures. (D) Schematic showing the position of probes (1–5) used for TRO assays along the snR3 snoRNA locus. (E) Representative TRO blot for snR3. (F–H) RNAPII profiles (ChIP-seq) across the snR3 (F), rps2 (G), and fba1 (H) genes for the indicated strains. (W) Watson strand; (C) Crick strand; (RPM) reads per million. (I) Cumulative RNAPII profile relative to poly(A) sites in the indicated strains. Curves show the sum of normalized ChIP-seq sequencing scores over a genomic region covering the major poly(A) site.
Seb1 associates with proteins involved in mRNA 3′ end processing and is enriched at the 3′ ends of genes
To elucidate the mechanism by which a deficiency in Seb1 results in transcription termination defects, we decided to identify Seb1-associated proteins using a functional His-TEV-protein A-tagged version of Seb1 (Seb1-HTP) expressed from its endogenous chromosomal locus. Purification of Seb1 (Fig. 2A) coupled to mass spectrometry resulted in the identification of 471 Seb1-associated proteins (Supplemental Table S1). Consistent with the conclusion that Seb1 does not function in NNS-like transcription termination, Nab3- and Sen1-specific peptides were not identified in the Seb1 purification, while only three Dbl8-specific peptides were detected (2.1% sequence coverage). We next used computer algorithms (Berriz et al. 2009) to distinguish functional protein classes within the top 10% of the Seb1-associated proteins. Notably, a significant number of proteins involved in mRNA 3′ end processing were enriched among the top 10% (P = 7.96−6, Fisher's exact test) (Fig. 2B; Supplemental Table S2): Rna14 from the cleavage factor 1A (CF1A) complex as well as Cft1, Cft2, Ysh1, and Pta1 from the cleavage and poly(A) factor (CPF) complex (Xiang et al. 2014). Reciprocal immunoprecipitation assays confirmed the nuclease-resistant association between Seb1 and Ysh1 (Fig. 2C) as well as between Seb1 and Cft2 (Fig. 2D). Collectively, 18 components predicted to be part of the S. pombe cleavage and poly(A) machinery were copurified with Seb1 (Supplemental Table S2).
Figure 2.

Seb1 interacts with the 3′ end processing machinery and is enriched at the 3′ ends of genes. (A) Coomassie blue staining of proteins copurified with Seb1-HTP (lane 2) and from a control untagged strain (lane 1). The arrow indicates the position of Seb1-HTP. (B) A subset of the top 10% of Seb1-associated proteins identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) is shown. The intensity represents the relative abundance (peptide intensity), while the percentage of coverage and the peptide number represent the unique peptide sequence coverage and the number of unique peptides, respectively. (C,D) Immunoblot analyses of whole-cell extracts (WCE) (lanes 1,2) and IgG-sepharose precipitates ([IP] IgG) (lanes 3–5) prepared from control Seb1-Myc cells or Seb1-Myc cells coexpressing a TAP tag version of Ysh1 (C) or Cft2 (D). (Lanes 4,5) Purification experiments were performed in the absence or presence of the benzonase nuclease. (E–G) ChIP-seq analysis of Seb1-HTP occupancy along the snR3 (E), fba1 (F), and rps2 (G) genes. (W) Watson strand; (C) Crick strand; (RPM) reads per million. (H) Heat map of Seb1 DNA-binding sites derived from ChIP-seq for all RNAPII transcribed genes. Genes were sorted by length and aligned at their transcription start sites (TSSs). The curved line represents the poly(A) sites. Strength of binding is coded from white (no binding) to dark blue (strong binding).
The purification of Seb1 also identified factors predicted to function in transcription termination, such as Dhp1, Rhn1, and Din1 (Fig. 2B; Supplemental Table S2), which share homology with Rat1, Rtt103, and Rai1, respectively—three proteins that are required for termination of RNAPII transcription in S. cerevisiae (Kim et al. 2004b). In addition, core components of RNAPI and RNAPII complexes (P = 2.552−5, Fisher's exact test) (Fig. 2B) as well as a significant number of proteins involved in ncRNA processing (P = 3.812−13, Fisher's exact test) (Fig. 2B) were identified in the top 10% of Seb1-associated proteins.
Binding profiles of mRNA 3′ end processing factors by ChIP tend to show recruitment at the 3′ ends of genes (Bentley 2014). Consistent with the copurification of Seb1 with mRNA 3′ end processing factors, ChIP-seq analysis of Seb1 indicated strong enrichment at the 3′ end of snoRNA-encoding (Fig. 2E) and mRNA-coding (Fig. 2F,G) genes, with genome-wide cross-linking in the vicinity of poly(A) sites (Fig. 2H). Notably, the 3′ end enrichment profile of Seb1 was irrespective of gene length or gene class (Fig. 2H; Supplemental Fig. S3A–D), consistent with the generalized transcription termination defects observed in Seb1-deficient cells (Fig. 1). In contrast, a 3′ end enrichment of Seb1 was not observed at RNAPI and RNAPIII transcribed genes (Supplemental Fig. S3E,F), suggesting a mode of 3′ end recruitment specific to RNAPII transcription. Together, the protein interaction network and the ChIP-seq analysis of Seb1 strongly support a general role in 3′ end processing and transcription termination.
Seb1 controls poly(A) site selection
According to current models of transcription termination (Porrua and Libri 2015), at least two mechanisms could account for the generalized increase in readthrough transcription in Seb1-deficient cells. First, Seb1 could promote 3′ end processing (PAS recognition and/or mRNA cleavage), which, according to the torpedo model, would result in termination defects in conditions of Seb1 deficiency. Alternatively, Seb1 could function subsequent to mRNA cleavage by promoting dissociation of RNAPII from the DNA template following passage through PAS elements. If Seb1 functions after pre-mRNA cleavage, 3′ end processing defects are not expected; in contrast, if Seb1 regulates pre-mRNA cleavage, defects in 3′ end processing are expected in Seb1-depleted cells. To distinguish between these models, we compared the landscape of poly(A) site selection between Seb1-deficient and control cells by 3′ region extraction and deep sequencing (3′ READS), an approach developed to map mRNA cleavage sites at the genome-wide level (Hoque et al. 2013). Out of 5393 genes with mappable poly(A) we found that 3013 genes (55%) showed a change in poly(A) site decision in Seb1-depleted cells relative to the control (Fig. 3A): Two-thousand-four-hundred-eighty-four genes (82%) showed lengthening of the 3′-most exon (Supplemental Table S3, red), whereas 529 genes (18%) had shorter 3′-most exons (Supplemental Table S3, blue). The preferential use of distal poly(A) sites in the seb1 mutant resulted in a significant increase in median 3′ UTR length (Fig. 3B): from 168 nucleotides (nt) in wild-type to 256 nt in Seb1-deficient cells. Importantly, the seb1 mutant showed increased heterogeneity in poly(A) site positions relative to wild-type cells: Analysis of relative abundance for the top three poly(A) sites (based on abundance in wild-type cells) for each gene indicated a significant decrease for the most abundant poly(A) site in the seb1 mutant relative to the wild type (Supplemental Fig. S4A); in contrast, secondary poly(A) sites were more frequently used in the seb1 mutant (Supplemental Fig. S4B,C). Examples of coding and noncoding genes whose cleavage site positions were strongly affected by a Seb1 deficiency are shown in Figure 3, C–H, and Supplemental Figure S4, D–K. In eukaryotes, alternative poly(A) (APA) has the potential to influence the stability, translation, and localization of a given mRNA through the inclusion or exclusion of cis-acting elements in the 3′ UTR (Tian and Manley 2013). We thus examined whether the changes in poly(A) site position observed in the seb1 mutant influenced mRNA abundance by analyzing standard RNA sequencing (RNA-seq) data. Interestingly, the 3′ UTR-lengthening phenotype detected in the seb1 mutant did not markedly change mRNA abundance (Fig. 3I). In sum, these results indicate that Seb1 functions in poly(A) site selection.
Figure 3.
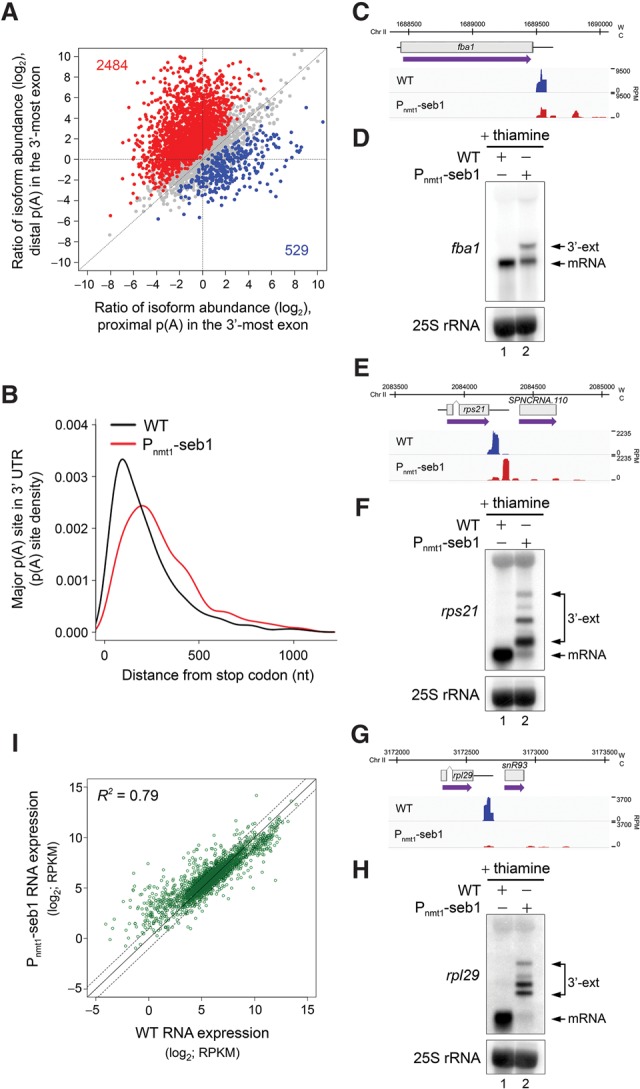
Seb1 levels affect poly(A) site selection. (A) Regulation of APA site utilization in the 3′-most exon as determined by 3'READS. The number of genes with a significantly lengthened 3′ UTR (red dots) and the number of genes with a significantly shortened 3′ UTR (blue dots) are indicated in the graph. Significantly regulated isoforms are those with a P-value of <0.05 (Fisher's exact test). Only the two most abundant isoforms for each gene were analyzed. (B) Distribution of 3'READS-derived poly(A) sites relative to the upstream stop codon. The major poly(A) site is the one with the highest number of reads per gene. Mapped poly(A) sites in Seb1-depleted cells, as compared with wild-type cells, are, on average, significantly more distant from the stop codon. P-value < 2.22−16 by Wilcoxon rank-sum test. (C–H) 3'READS profiles and Northern blot analyses of fba1 (C,D), rps21 (E,F), and rpl29 (G,H) genes in the indicated strains. (W) Watson strand; (C) Crick strand; (RPM) reads per million. 3′ extended transcripts that accumulate in the Seb1-depleted condition are shown (3′-ext). (I) RNA expression changes for transcripts with 3’ extended 3′ UTRs in wild-type and Seb1-depleted cells as measured by RNA-seq. (RPKM) Reads per kilobase per million. The coefficient of determination (R2) is indicated.
The CID and RRM domains of Seb1 are required for poly(A) site selection
Although our results suggest that Seb1 is not the functional homolog of S. cerevisiae Nrd1, Seb1 shares extensive homology with the CID (34% identity/70% similarity) and the RRM (46% identity/76% similarity) of Nrd1 (Supplemental Fig. S5A). In fact, Seb1 copurified with a GST-tagged version of the S. pombe CTD that was coexpressed in fission yeast (Materne et al. 2015) but not with a control GST fusion protein (Fig. 4A, lanes 11,12), consistent with the notion that Seb1 can interact with the CTD of Rpb1. In contrast, a pull-down assay of the GST-CTD did not recover a non-CID-containing protein (Fig. 4A, lane 7). We next tested whether the predicted CID domain of Seb1 was necessary for normal 3′ end processing/transcription termination by taking advantage of previously determined CTD–CID structures (Meinhart and Cramer 2004; Lunde et al. 2010; Kubicek et al. 2012). In these structures, the CTD adopts a β-turn conformation that docks into a CID hydrophobic pocket via a set of conserved residues that, based on sequence alignment, would involve Tyr64 (Y64), Asp67 (D67), and Arg71 (R71) of Seb1 (Supplemental Fig. S5A). We therefore generated a mutant allele that expressed a version of Seb1 (CIDmut) with substitutions at these particular residues. Wild-type and CIDmut alleles of seb1 were chromosomally integrated as a single copy into the Pnmt1-seb1 conditional strain, and the extent to which the CID mutant restored the transcription termination defects induced by depletion of endogenous Seb1 was examined by RNAPII ChIP assays along the rps2 model gene (Fig. 4B). It should be noted that the converging ret3 gene is barely transcribed by RNAPII (Fig. 1G) and is therefore unlikely to contribute to the ChIP signal downstream from rps2. As can be seen in Figure 4C, the CIDmut version of Seb1 showed increases in RNAPII levels at the 3′ end of rps2 that were similar to Seb1-depleted cells (Pnmt1-seb1+EV), suggesting that the ability of Seb1 to interact with the CTD of Rpb1 is important for its function in 3′ end processing. This conclusion was supported by RNA analyses, demonstrating altered poly(A) site selection in cells that expressed the CIDmut version of Seb1 (Fig. 4D, lane 4).
Figure 4.
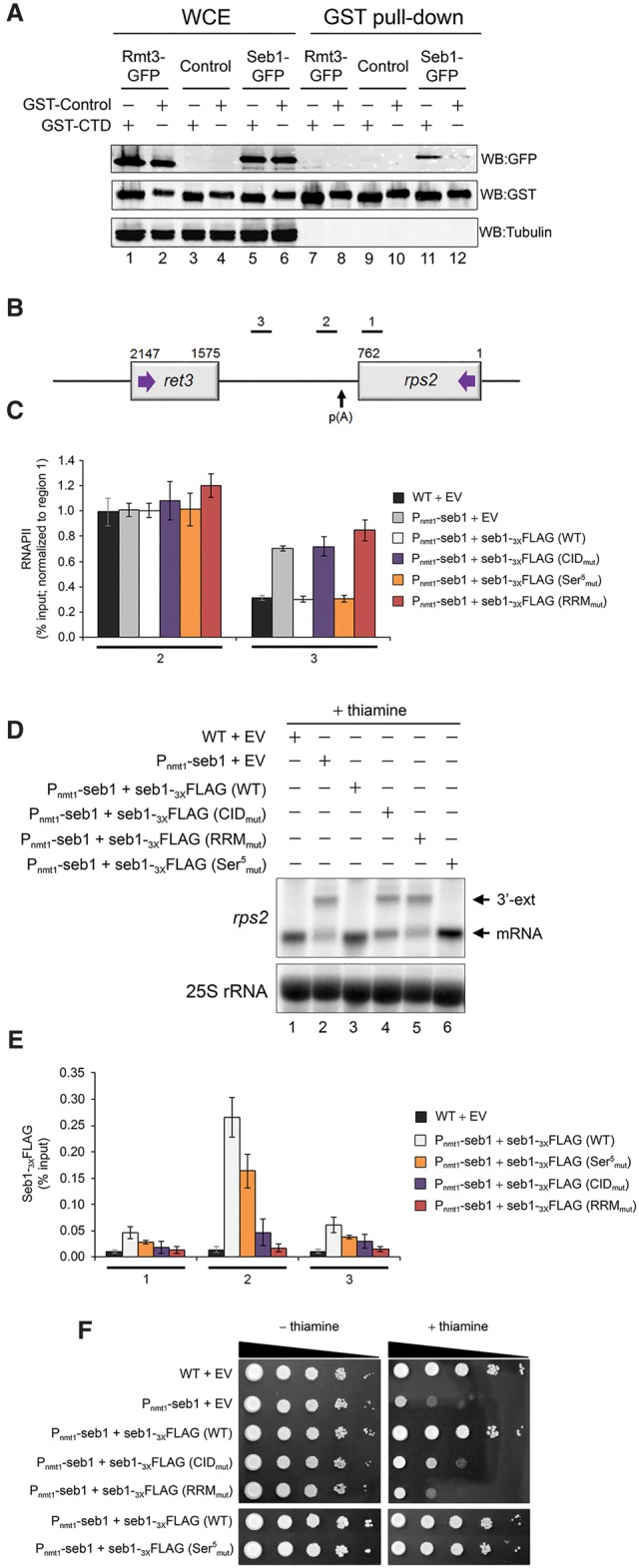
Seb1 requires functional CID and RRM domains for accurate 3′ end processing and transcription termination. (A) Immunoblot analysis of whole-cell extracts (WCE) (lanes 1–6) and glutathione-sepharose pull-downs (lanes 7–12) prepared from the indicated strains expressing either GST-CTD (odd-numbered lanes) or a control GST fusion protein (even-numbered lanes). (B) Bars above the rps2 gene show the positions of PCR products used for ChIP analyses. (C) RNAPII ChIP analysis using extracts prepared from the Pnmt1-seb1 conditional strain containing genomically integrated constructs that express the indicated versions of Flag-tagged Seb1 (wild type, CIDmut, Ser5mut, and RRMmut) (see the text; Supplemental Fig. S5a for description) as well as an empty control vector (EV). Cells were grown in the presence of thiamine to deplete endogenous Seb1. Error bars indicate SD. n = 3 biological replicates from independent cell cultures. (D) Northern blot analysis of rps2 mRNA from the indicated strains. The rps2 3′ extended transcripts are shown (3′-ext). (E) ChIP analyses of wild-type and mutant versions of Seb1-Flag along the rps2 gene. Control wild-type cells with an empty vector (black bars) were used as a negative control for the anti-Flag ChIP assays. Error bars indicate SD. n = 3 biological replicates from independent cell cultures. (F) Tenfold serial dilutions of the indicated strains were spotted on thiamine-free (left) or thiamine-containing (right) minimal medium.
In budding yeast, the preferential binding site of Nrd1 is formed after Ser5P of the CTD (Gudipati et al. 2008; Vasiljeva et al. 2008a). To test the possibility that Ser5P is important for Seb1 recruitment and function, we expressed a version of Seb1 with amino acid substitutions at conserved residues (Ser5mut) (Supplemental Fig. S5A) that were previously shown for S. cerevisiae Nrd1 to significantly decrease binding to the Ser5P CTD and were necessary for Nrd1-dependent RNA processing (Kubicek et al. 2012). Strikingly, the Ser5mut version of Seb1 fully restored the defects in transcription termination (Fig. 4C) and 3′ end processing (Fig. 4D, lane 6) induced by the depletion of endogenous Seb1, arguing against a role for Ser5P in Seb1 recruitment and function.
As Seb1 was shown to associate with ncRNA (Marina et al. 2013), we examined whether its function in 3′ end processing required RNA recognition. To test this, we substituted conserved residues in the Seb1 RRM that were shown for Nrd1 to be absolutely required for RNA binding (Supplemental Fig. S5A; Bacikova et al. 2014). Substitutions in the RRM domain of Seb1 (RRMmut) resulted in readthrough transcription (Fig. 4C) and affected poly(A) site selection (Fig. 4D, lane 5), similar to Seb1-deficient cells (Fig. 4D, lane 2). Moreover, we found that the CID and RRM mutants of Seb1 were affected in their recruitment at the 3′ end of rps2 (Fig. 4E, see region 2). In contrast, the Ser5mut version of Seb1 showed only a modest reduction in cross-linking (Fig. 4E), consistent with the absence of defects in 3′ end processing and transcription termination for this mutant. Importantly, the 3′ end processing/transcription termination defects observed for the CID and RRM mutants were not the consequence of a problem in protein stability (Supplemental Fig. S5B). The extent to which the different Seb1 mutants restored the growth defect induced by a Seb1 deficiency also correlated with the levels of 3′ end processing/transcription termination defects (Fig. 4F). Taken together, these data argue that the function of Seb1 in 3′ end RNA processing and transcription termination is enforced by a specific mode of recruitment at the 3′ ends of genes that involves its CID and RRM domains.
Seb1 binds GUA-containing motifs downstream from cleavage sites
The functional requirement of the Seb1 RRM domain for the selection of 3′ end cleavage sites prompted us to examine the position and sequence specificity implicated in Seb1 RNA recognition. We used our functional HTP-tagged version of Seb1 to map RNA interactions of Seb1 at the transcriptome-wide level by UV-induced RNA–protein cross-linking and analysis of cDNA by high-throughput sequencing (CRAC) (Granneman et al. 2009). Briefly, actively growing cells were UV-irradiated to forge covalent bonds between proteins and RNA and were subjected to affinity purification under stringent conditions to recover Seb1-associated RNAs (Supplemental Fig. S6A–C), which were analyzed by high-throughput sequencing. Only a very small number of mappable reads were recovered from the untagged negative control samples, roughly 400–1000 times less compared with the Seb1-tagged samples. A breakdown of hits from two independent CRAC experiments revealed that nearly 70% of Seb1-associated RNAs originate from protein-encoding genes and intergenic regions (Fig. 5A). Specifically, a genome-wide coverage plot showed that the majority of Seb1 RNA binding mapped downstream from genome annotations corresponding to ORFs or ncRNAs (Supplemental Fig. S6D), which is consistent with the genome-wide localization of Seb1 as determined by ChIP-seq. Examples of Seb1 binding downstream from mRNA cleavage sites as determined by CRAC are shown in Figure 5B. Globally, analysis of the hit distribution across annotated poly(A) sites revealed preferential binding of Seb1 50–100 nt downstream from cleavage sites (Fig. 5C, green plot and right axis), a profile that generally matched the distribution of mapped microdeletions (Fig. 5C; red plot and left axis), which can be used to precisely map protein–RNA interactions (Granneman et al. 2009)
Figure 5.

Seb1 binds to GUA-containing motifs downstream from poly(A) sites. (A) Distribution of Seb1-bound reads between transcript classes for two independent CRAC experiments. (B) Seb1 CRAC cDNA read distribution (red) and 3'READS profile (blue) of rps2 and pgk1 genes in a wild-type strain. (W) Watson strand; (C) Crick strand; (RPM) reads per million. (C) Cumulative Seb1 RNA-binding sites relative to annotated poly(A) sites. The green curve (right Y-axis) shows the number of reads per nucleotide position, which is a measure of the binding preference. The red curve (left Y-axis) shows the number of deletions per nucleotide position, which is an indication of direct cross-linking. (D) Sequence logo of Seb1 cross-linking sites derived from the WebLogo application (Crooks et al. 2004) using the top 10 pyMotif-derived k-mers from each CRAC experiment. (E) Average gene distribution of tetrameric motifs derived from the Seb1 CRAC data (GUAG and UGUA) and control tetramers with shuffled dinucleotides (AGGU and UAUG). (F) Schematic of the rps2-GFP-rps2 construct used to address the functional significance of the Seb1 consensus motif in poly(A) site selection. Shown is a 405-nt region that includes the last seven codons of the GFP mRNA (in green) as well as the major poly(A) site of the GFP-rps2 mRNA detected in wild-type (G shown in red; +89 from stop codon) and Seb1-depleted (T shown in blue; +376 from stop codon) cells, as determined by 3′ RACE. The AAUAAA poly(A) signals are italicized in orange. Sequences in bold show Seb1 consensus motifs with the GUA core underlined. In mutant #1, the GUA core of the three Seb1 binding motifs located upstream of the wild-type rps2 cleavage site was mutated to CAC, whereas mutant #2 introduced CAC mutations in the eight Seb1-binding motifs located downstream from the rps2 cleavage site. (G) Northern blot analysis using total RNA prepared from wild-type (lanes 1,2,4,5) and Seb1-deficient (lane 3) cells that express either wild-type (lanes 2,3) or mutant (mutant #1 [lane 4] and mutant #2 [lane 5]) versions of the GFP-rps2 construct. Cells were grown in the presence of thiamine. The blot was analyzed using probes specific for the GFP mRNA and 25S rRNA.
Next, we searched for overrepresented sequences in Seb1 read contigs using the pyMotif algorithm from the pyCRAC package (Webb et al. 2014). Among the top-scoring k-mers (4-mers to 8-mers) recovered from both CRAC experiments (Supplemental Table S4), a clear GUA trinucleotide core was identified, which was surrounded by A/U and A/G as 5′ and 3′ nucleotides, respectively (Fig. 5D). Remarkably, the identified Seb1 consensus sequence is nearly identical to the GUA(A/G)-binding motif previously reported for Nrd1 (Creamer et al. 2011; Wlotzka et al. 2011; Porrua et al. 2012; Schulz et al. 2013; Bacikova et al. 2014; Schaughency et al. 2014), suggesting that sequence-specific recognition by the RRM domains of Seb1 and Nrd1 has been conserved despite divergent roles in RNA metabolism. Mapping the top Seb1 tetramers identified by CRAC (GUAG and UGUA) (Supplemental Table S4) along all fission yeast genes showed a strong enrichment downstream from cleavage sites, whereas gene bodies were markedly depleted of Seb1-binding motifs (Fig. 5E). In contrast, control tetramers with similar base composition but lacking the GUA trinucleotide core showed relatively even distribution along genes (Fig. 5E). Consistent with a role in recruiting Seb1, we found that genes containing either GUAG or UGUA Seb1 motifs at the 3′ end produced significantly higher levels of readthrough transcripts in Seb1-depleted cells than genes without the motif (Supplemental Fig. S7C). Similarly, genes that demonstrated strong Seb1 RNA cross-linking at the 3′ end as determined by CRAC produced greater levels of readthrough transcripts in the absence of Seb1 than genes showing no Seb1 RNA interactions (Supplemental Fig. S7E).
To address the functional significance of the identified Seb1 consensus motif, we analyzed 3′ end processing of the GFP mRNA expressed under the control of the rps2 promoter (∼0.5 kb) and downstream (∼1.0 kb) elements (Fig. 5F). In wild-type cells, the GFP mRNA used the normal rps2 poly(A) site located 89 nt downstream from the stop codon (Fig. 5F,G, lane 2). In contrast, the GFP mRNA produced a 3′ extended transcript that used a distal poly(A) site located 376 nt downstream from the stop codon in Seb1-depleted cells (Fig. 5F,G, lane 3). Importantly, changing the consensus GUA trinucleotide motifs found downstream from the rps2 cleavage site to CAC (see Fig. 5F) resulted in the accumulation of a readthrough GFP mRNA identical to the one detected in Seb1-depleted cells (Fig. 5G, cf. lanes 3 and 5). However, changing GUA consensus motifs located upstream of the rps2 cleavage site did not markedly alter poly(A) site selection (Fig. 5G, lane 4). We also found that the presence of Seb1 consensus motifs was important for 3′ end processing of a snoRNA (Supplemental Fig. S8). Together, our results support a model in which Seb1 promotes poly(A) site selection by binding onto nascent transcripts via a mechanism that relies on the recognition of GUA-containing motifs present downstream from 3′ end cleavage sites.
Seb1 promotes the recruitment of cleavage and poly(A) factors
Given the copurification of Seb1 and cleavage/poly(A) factors (Fig. 2), the binding of Seb1 50–100 nt downstream from cleavage sites (Fig. 5), and its ability to favor proper poly(A) site selection (Fig. 3), we sought to investigate whether a deficiency in Seb1 influenced the recruitment of components of the 3′ end processing machinery. We thus monitored the recruitment of four independent factors that belong to the CF1A (Rna14 and Clp1) and CPF (Ysh1 and Cft2) complex by ChIP assays. TAP-tagged versions of Rna14, Clp1, Ysh1, and Cft2 were all functional, as no growth phenotype was detected compared with the control untagged strain (Supplemental Fig. S9A), and their expression was not affected by the depletion of Seb1 (Supplemental Fig. S9B–E). These 3′ end processing factors showed maximal recruitment near the poly(A) site of rps2 in normal cells (Fig. 6A–E, region 2), consistent with ChIP data from S. cerevisiae and human cells (Licatalosi et al. 2002; Kim et al. 2004a; Glover-Cutter et al. 2008). In contrast, this binding profile was lost for all tested proteins in Seb1-depleted cells, showing no accumulation around the poly(A) site and, accordingly, a flat distribution (Fig. 6A–E). Because rps2 (and protein-coding genes in general) showed slightly lower levels of total RNAPII in Seb1-deficient cells (Fig. 1G–I; Supplemental Fig. S1A), we normalized the recruitment data to total polymerase levels as measured by RNAPII ChIP signal performed with the same extracts. The normalized data revealed significant defects in Rna14 and Cft2 recruitment, whereas Clp1 and Ysh1 levels were similar between Seb1-deficient and control cells (Fig. 6F–I). Importantly, similar results were obtained for a snoRNA-encoding gene (Supplemental Fig. S9F–L), indicating that this defective recruitment pattern is not limited to mRNA-coding genes. We conclude that Seb1 functions in poly(A) site selection by promoting the cotranscriptional recruitment of components of the 3′ end processing machinery.
Figure 6.

Seb1 levels affect the cotranscriptional assembly of the cleavage/poly(A) machinery. (A) Bars above the rps2 gene show the positions of PCR products used for ChIP analyses. (B–E) ChIP assays of TAP-tagged versions of Rna14 (B), Clp1 (C), Ysh1 (D), and Cft2 (E) in wild-type and Seb1-depleted cells (Pnmt1-seb1). An untagged control strain was used to monitor the background signal of the ChIP assays. (F–I) Recruitment of 3′ end processing factors as a ratio of total RNAPII at the 3′ end of rps2 (region 2). ChIP signal of TAP-tagged 3′ end processing factors was divided by the total RNAPII signal at region 2. Region 2 was analyzed because it represents the location of maximal 3′ end processing factor recruitment. Error bars indicate SD. n = 3 biological replicates from independent cell cultures. (*) P < 0.05, Student's t-test.
Reduction of transcription elongation rates attenuates the poly(A) site selection defects of Seb1-deficient cells
Despite the absence of a global reduction in RNA levels (Fig. 3I), a general reduction in total RNAPII levels was observed in gene bodies in Seb1-deficient cells (Fig. 1F–I; Supplemental Fig. S1). Such an observation may be indicative of a change in the transcription elongation rate. To determine whether transcription kinetics contributes to poly(A) site selection by Seb1, we grew wild-type and Seb1-depleted cells in the presence of 6-azauracil (6-AU), which slows down transcription elongation and increases RNAPII pausing (Mason and Struhl 2005). Treatment of wild-type cells with 6-AU resulted in a slight decrease in mRNA levels (Fig. 7A,B, cf. lanes 1 and 2), consistent with reduced transcription kinetics. Notably, the addition of 6-AU to Seb1-deficient cells resulted in a marked changed in the ratio between proximal and distal mRNA isoforms compared with untreated cells. Specifically, 6-AU partially restored the altered poly(A) site selection resulting from a Seb1 deficiency by increasing the levels of the normal short isoform, concurrent with reducing the levels of the distal isoform (Fig. 7A,B, cf. lanes 3 and 4). These results show that slowing down transcription rates lessens the need for Seb1 in selecting proper 3′ end cleavage sites, suggesting an important role for RNAPII elongation kinetics in Seb1-dependent poly(A) site selection.
Figure 7.
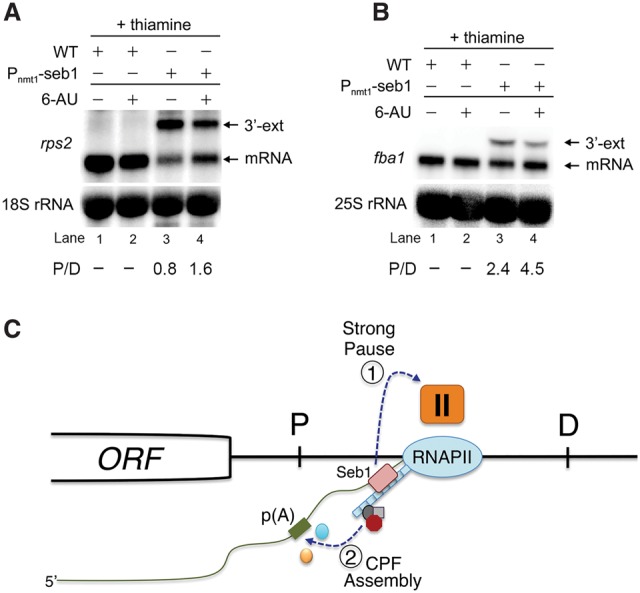
Transcription kinetics contributes to Seb1-dependent poly(A) site selection. (A,B) Northern blot analysis of total RNA prepared from wild-type (lanes 1,2) and Seb1-depleted (lanes 3,4) cells that were treated (lanes 2,4) or not treated (lanes 1,3) with 6-AU. Blots were probed for rps2 (A) and fba1 (B) mRNAs. Ratios of proximal (P) relative to distal (D) mRNA isoforms are indicated (average from two independent experiments). (C) Model for Seb1-dependent poly(A) site selection. The passage of RNAPII through a poly(A) site is thought to induce a change in the kinetics of transcription elongation, including pausing of the RNAPII complex (Nag et al. 2006; Grosso et al. 2012; Davidson et al. 2014; Fusby et al. 2015; Nojima et al. 2015). We propose that the cooperative binding of Seb1 to the RNAPII CTD and to RNA motifs clustered downstream from poly(A) sites positively contributes to RNAPII pausing (1), thereby promoting poly(A) site recognition and assembly of a cleavage-competent cleavage/poly(A) (CPF) complex (2). In the absence of Seb1, RNAPII pausing is leaky, increasing the frequency of RNAPII complexes that reach distal (D) poly(A) sites.
Discussion
In this study, we identified an unexpected role for Seb1 in poly(A) site selection and termination of RNAPII transcription. We also provide a framework of how Seb1 cotranscriptionally promotes accurate 3′ end processing, thereby controlling the length of 3′ UTRs. These findings are significant, as they provide novel insights into the poorly understood mechanism that coordinates transcription and cleavage site selection, which underlie gene regulation by APA.
NNS-like transcription termination is not evolutionarily conserved
In S. cerevisiae, transcription termination of noncoding genes does not depend on the mRNA 3′ end processing machinery but generally relies on a pathway that requires the NNS complex, which targets released transcripts to the RNA exosome for degradation or processing (Porrua and Libri 2015). To date, however, the conservation of NNS-like transcription termination has remained elusive, as a distinctive NNS complex has not been described in metazoans. Instead, a complex consisting of the nuclear cap-binding proteins and ARS2 (the CBCA complex) appears to share functional similarities to the S. cerevisiae NNS complex by connecting transcription of noncoding genes to exosome-dependent degradation in humans (Andersen et al. 2013; Hallais et al. 2013). Accordingly, the CBCA complex suppresses the production of readthrough transcripts from several classes of noncoding genes by promoting transcription termination in a manner dependent on the distance from the transcription start site (Andersen et al. 2013; Hallais et al. 2013), which is similar to NNS-dependent termination in S. cerevisiae (Gudipati et al. 2008; Vasiljeva et al. 2008a). In contrast to metazoans, S. pombe possesses putative orthologs of all NNS components: Seb1, Nab3, and Sen1/Dbl8. However, our proteomic analysis of Seb1 did not reveal the presence of a typical NNS-like complex. Instead, we found a significant enrichment of proteins involved in mRNA 3′ end cleavage and poly(A). Such a physical connection with the 3′ end processing machinery was not detected in proteomic analyses of Nrd1 (Vasiljeva and Buratowski 2006), arguing that Seb1 and Nrd1 have functionally diverged. Indeed, our results indicate that Seb1 and Nrd1 cannot functionally complement each other (Supplemental Fig. S2). Together with the absence of strong termination defects in cells deleted for nab3, sen1, and dlb8 (Fig. 1), our findings argue that a machinery distinct from the budding yeast NNS complex connects ncRNA transcription to exosome-dependent RNA decay in fission yeast. Accordingly, a complex that is reminiscent of the human CBCA–NEXT complex has recently been described in S. pombe. This complex, known as MTREC or NURS (Lee et al. 2013; Egan et al. 2014; Zhou et al. 2015), binds to noncoding, unwanted, and misprocessed transcripts and targets them for degradation by the nuclear exosome. Our study thus suggests that the NNS–exosome connection that functions in the recognition and degradation of aberrant transcripts in S. cerevisiae has functionally diverged in the S. pombe lineage and has been lost over the course of metazoan evolution.
Seb1 controls poly(A) site selection
Recent transcriptome-wide studies indicate that multiple poly(A) sites are used in most eukaryotic genes, as demonstrated in humans (Hoque et al. 2013) as well as budding (Ozsolak et al. 2010) and fission (Mata 2013; Schlackow et al. 2013) yeasts. This process, known as APA, is emerging as a major layer of gene regulation, allowing the inclusion or exclusion of sequences that control the localization, stability, and translation of mRNAs (Tian and Manley 2013). As yet, however, the mechanism of poly(A) site recognition and how poly(A) site selection is modulated remain poorly understood. Notably, our study disclosed a key role for the Seb1 RNA-binding protein in cleavage site selection in vivo, showing that a deficiency in Seb1 results in widespread changes in 3′ UTR lengths as a consequence of increased APA. The direct role of Seb1 in poly(A) site selection is supported by several observations: (1) Seb1 is specifically recruited at the 3′ end of RNAPII transcribed genes (Fig. 2; Supplemental Fig. S3), (2) subunits of the 3′ end processing machinery copurify with Seb1 (Fig. 2), (3) Seb1 binds nascent transcripts 50–100 nt downstream from cleavage sites (Fig. 5), and (4) RNA-seq data and Western blotting analysis indicate that a Seb1 deficiency does not affect the expression of genes encoding for components of the 3′ end processing machinery (Supplemental Fig. S9; data not shown). Our results therefore suggest that the heterogeneity of PAS usage in fission yeast (Mata 2013; Schlackow et al. 2013) is not a purely random process but can be modulated by controlling Seb1 levels.
Although Seb1 globally affected poly(A) site selection, our standard RNA-seq data together with numerous Northern blot validations did not reveal significant changes in mRNA abundance in Seb1-depleted cells. These observations argue that Seb1 is not required for the cleavage reaction itself but rather to correctly position the cleavage site. To promote accurate 3′ end processing, we found that Seb1 required a functional CID. A version of Seb1 with CID substitutions at conserved residues shown to be involved in CTD interactions in related CTD-binding proteins (Meinhart and Cramer 2004; Lunde et al. 2010) abolished Seb1 enrichment at the 3′ end processing site and impaired poly(A) site selection (Fig. 4). Our structure–function analysis further indicated that the Ser5 phosphorylated form of the CTD (Ser5-P) is not the prevalent means by which recruitment of Seb1 is achieved, in contrast to S. cerevisiae Nrd1 (Gudipati et al. 2008; Vasiljeva et al. 2008a). This result is not unexpected, however, given that Ser5-P marks occur predominantly in the early elongation phase of the transcription cycle, whereas the recruitment of Seb1 is specific to the 3′ ends of genes. Accordingly, it is tempting to speculate that the CID-dependent enrichment of Seb1 near 3′ end processing sites depends on Ser2P of the CTD, which is known to be predominant at the 3′ ends of genes in S. cerevisiae and humans (Ahn et al. 2004; Kim et al. 2010; Grosso et al. 2012). However, CTD phosphorylation dynamics remain poorly understood in S. pombe, and specific phosphorylation marks were in fact shown to behave differently than in other organisms (Cassart et al. 2012).
Our study also indicated that binding of Seb1 to (A/U)GUA(A/G)-containing motifs 50–100 nt downstream from the poly(A) site was important for Seb1 recruitment and 3′ end cleavage site selection. Notably, this Seb1-binding signature downstream from the poly(A) site explains the reported lack of interaction between Seb1 and the act1 mRNA (Marina et al. 2013), as Seb1 is bound to the 3′ fragment following cleavage instead of the mature polyadenylated mRNA. Collectively, our findings support a model in which Seb1 is recruited to 3′ end processing sites via interactions with the CTD of the RNAPII elongation complex and specific RNA motifs clustered downstream from the poly(A) site as they emerge from the transcribing polymerase. Such a cooperative contribution of CID and RRM domains in the spatio–temporal recruitment of RNA processing factors is not uncommon during mRNA 3′ end processing (Dichtl et al. 2002; Kyburz et al. 2003) and also underlies Nrd1-dependent recruitment (Gudipati et al. 2008).
How does Seb1 promote the selection of poly(A) sites? Our data indicated that Seb1 is important for the cotranscriptional organization of properly assembled cleavage and poly(A) (CPF) complexes at the 3′ ends of genes (Fig. 6; Supplemental Fig. S9). We also showed that slowing down transcription rates and increasing RNAPII pausing frequency using 6-AU attenuated the cleavage site selection defects of Seb1-depleted cells (Fig. 7). Together, these findings suggest a model in which binding of Seb1 to clustered RNA motifs downstream from poly(A) sites modulates transcription elongation kinetics (RNAPII pausing), enabling the recruitment and assembly of cleavage-competent CPF complexes (Fig. 7C). Defects in the cotranscriptional assembly of CPF complexes as a result of changes in transcription kinetics at the 3′ ends of genes may allow competition between poly(A) sites as they emerge from the elongating polymerase, therefore providing a greater opportunity for the use of distal cleavage sites. A model in which Seb1-dependent poly(A) site selection is mechanistically linked to transcription elongation kinetics is supported by recent studies showing that RNAPII pausing influences the choice between APA sites (Fusby et al. 2015; Oktaba et al. 2015). Furthermore, definition of a minimal downstream element (DSE) important for RNAPII pausing in S. pombe (Aranda and Proudfoot 1999) previously identified an 18-base-pair (bp) region that contains two copies of the pentanucleotide ATGTA, which is similar to the Seb1 RNA-binding motif determined by CRAC (Fig. 5D). However, our data are also consistent with an alternative model in which Seb1 helps to recruit and/or assemble the 3′ end processing machinery, consequently influencing RNAPII pausing at the 3′ ends of genes. However, since proteomic analyses indicate that Seb1 is not a core subunit of the fission yeast 3′ end processing complex (Vanoosthuyse et al. 2014), we favor a model in which Seb1 promotes RNAPII pausing, thereby transiently associating with the 3′ end processing machinery via a paused transcription elongation complex (Fig. 7C). Although Seb1 presumably affects transcription kinetics at the 3′ end of genes, distinctive RNAPII peaks are still detected downstream from poly(A) sites in the Seb1-depleted strain (Fig. 1I), suggesting that RNAPII can still pause, albeit less efficiently, in the absence of Seb1. Accordingly, the possibility that other factors act redundantly or cooperatively with Seb1 to maximize RNAPII pausing and termination is plausible, as several fail-safe transcription termination pathways have been described (Lemay and Bachand 2015).
Our ChIP analysis of Seb1-deficient cells detected a reduction in the occupancy of Rna14 and Cft2, which are subunits of the evolutionarily conserved CstF and CPSF complexes (Xiang et al. 2014), respectively, at the 3′ ends of genes. However, Seb1 levels did not affect the recruitment of every component of CstF and CPSF complexes (Clp1 and Ysh1). Although the molecular basis underlying the specificity of 3′ end factor recruitment by Seb1 remains to be determined, these data suggest that CstF and CPSF components may not be recruited to genes as complete preformed complexes but may require stepwise assembly processes that occur cotranscriptionally, which is consistent with previous work (Chao et al. 1999; Johnson et al. 2009; Mayer et al. 2012). Consistent with this idea, a complex containing Cft2 and other CPF components can be isolated independently of Ysh1 in S. cerevisiae (Ghazy et al. 2012). In addition, the observation that Seb1 levels did not impair the overall recruitment of Ysh1, which is the fission yeast homolog of the human endonuclease CPSF73, is also consistent with the conclusion that Seb1 is not essential for the cleavage reaction itself but is necessary to correctly position the site of cleavage.
Remarkably, our findings argue for the involvement of the mRNA cleavage and poly(A) machinery in S. pombe snoRNA 3′ end processing, which is in contrast to snoRNA 3′ end formation in S. cerevisiae that relies mostly on the NNS complex (Porrua and Libri 2015). Accordingly, we found that Seb1 and mRNA 3′ end processing factors were generally recruited at the 3′ ends of both mRNA and snoRNA genes. We also observed termination defects and altered recruitment of mRNA 3′ end processing factors at both mRNA and snoRNA genes in Seb1-depleted cells. Consistent with this idea, snoRNAs display mRNA-like features in fission yeast. For instance, mature snoRNAs are produced from polyadenylated precursors as part of their maturation cycle, which requires the activity of the canonical poly(A) polymerase (Pla1) and the nuclear poly(A)-binding protein Pab2 (Lemay et al. 2010). Moreover, recent genome-wide mapping of poly(A) sites in fission yeast revealed that the most prevalent cis elements associated with cleavage site identification are common between mRNA and snoRNA genes (Mata 2013; Schlackow et al. 2013). Interestingly, the apparent role of Seb1 in fission yeast 3′ end processing mirrors functions of Pcf11 in S. cerevisiae, a protein that also interacts with the CTD of RNAPII (Barilla et al. 2001). Similarly to Seb1, Pcf11 is involved in transcription termination of both coding RNAs and ncRNAs as well as in poly(A) site selection (Grzechnik et al. 2015). Despite these similarities, we found that the expression of S. pombe Seb1 did not rescue a Pcf11 deficiency in S. cerevisiae (Supplemental Fig. S2F–H), suggesting that S. pombe Seb1 and S. cerevisiae Pcf11 contribute to different aspects of 3′ end processing.
The importance of precisely selecting the correct poly(A) site is key during embryonic development and is of primordial importance for human health (Curinha et al. 2014). The identification of Seb1 as an essential factor that can influence 3′ end processing decisions in a cotranscriptional manner is an important advance in understanding the interplay between transcription and APA regulation. Given the similarities between S. pombe and human poly(A) sites (Mata 2013; Schlackow et al. 2013), including use of the canonical AAUAAA hexamer, together with the fact that metazoans include several proteins that possess both CIDs and RRM domains (Corden 2013), we predict that the links between transcription and poly(A) site selection described in fission yeast are likely to apply in higher eukaryotes.
Materials and methods
Yeast strains and media
A list of all S. pombe and S. cerevisiae strains used in this study is provided in Supplemental Table S5. Fission yeast cells were grown at 30°C in yeast extract medium with adenine, uracil, and amino acid supplements (YES) or in Edinburgh minimal medium (EMM) supplemented with adenine, uracil, and the appropriate amino acids.
ChIP assays
ChIP-qPCR and ChIP-seq experiments were performed as described previously (Lemay et al. 2014). The antibodies used are described in the Supplemental Material.
Protein analyses
Analysis of protein expression and affinity purification methods are described in details in the Supplemental Material.
3′READS analysis
The 3′READS method used in this study was performed and analyzed as previously described using total S. pombe RNA (Hoque et al. 2013).
CRAC assays
CRAC was performed as previously described (Granneman et al. 2009) using S. pombe cells grown in YES medium to an OD600 of 0.45–0.5 and UV-irradiated in the Megatron UV cross-linker for 220 sec. Additional details can be found in the Supplemental Material.
Computational methods
Reads obtained from Illumina HiSeq runs were quality-filtered according to the Illumina pipeline. Detailed computational methods for RNA-seq, ChIP-seq, and CRAC analyses are described in the Supplemental Material.
Accession codes
ChIP-seq and RNA-seq data are accessible using the ArrayExpress archive under accession codes E-MTAB-2237 and E-MTAB-4827. The data from the CRAC and 3′READS analyses can be accessed through Gene Expression Omnibus accession codes GSE82326 and GSE75753, respectively.
Supplementary Material
Acknowledgments
We thank D. Hermand, D. Libri, L. Minvielle-Sebastia, and R. Wellinger for strains, plasmids, and reagents; S.R. Atkinson (University College London) for RNA-seq libraries; and the sequencing platforms of McGill University, the Génome Québec Innovation Centre, and Edinburgh Genomics. This work used the computing resources of the UK Medical Bioinformatics partnership (UK MED-BIO; aggregation, integration, visualisation and analysis of large, complex data), which is supported by the Medical Research Council (grant no. MR/L01632X/1) and Imperial College High Performance Computing Service (http://www.imperial.ac.uk/admin-services/ict/self-service/research-support/hpc). This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSREC) to F.B., a Wellcome Trust Senior Investigator Award to J.B., the UK Medical Research Council to S.M., the National Institute of General Medical Sciences (GM084089) to B.T., and a Wellcome Trust Research and Career Development Grant (091549) to S.G. F.B. is supported as a Canada Research Chair in Quality of Gene Expression.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.280222.116.
References
- Ahn SH, Kim M, Buratowski S. 2004. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell 13: 67–76. [DOI] [PubMed] [Google Scholar]
- Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, Verheggen C, Schein A, Bunkenborg J, Poser I, Hallais M, et al. 2013. The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol 20: 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda A, Proudfoot NJ. 1999. Definition of transcriptional pause elements in fission yeast. Mol Cell Biol 19: 1251–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arigo JT, Eyler DE, Carroll KL, Corden JL. 2006. Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell 23: 841–851. [DOI] [PubMed] [Google Scholar]
- Bacikova V, Pasulka J, Kubicek K, Stefl R. 2014. Structure and semi-sequence-specific RNA binding of Nrd1. Nucleic Acids Res 42: 8024–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barilla D, Lee BA, Proudfoot NJ. 2001. Cleavage/polyadenylation factor IA associates with the carboxyl-terminal domain of RNA polymerase II in Saccharomyces cerevisiae. Proc Natl Acad Sci 98: 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley DL. 2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berriz GF, Beaver JE, Cenik C, Tasan M, Roth FP. 2009. Next generation software for functional trend analysis. Bioinformatics 25: 3043–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassart C, Drogat J, Migeot V, Hermand D. 2012. Distinct requirement of RNA polymerase II CTD phosphorylations in budding and fission yeast. Transcription 3: 231–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao LC, Jamil A, Kim SJ, Huang L, Martinson HG. 1999. Assembly of the cleavage and polyadenylation apparatus requires about 10 seconds in vivo and is faster for strong than for weak poly(A) sites. Mol Cell Biol 19: 5588–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corden JL. 2013. RNA polymerase II C-terminal domain: tethering transcription to transcript and template. Chem Rev 113: 8423–8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creamer TJ, Darby MM, Jamonnak N, Schaughency P, Hao H, Wheelan SJ, Corden JL. 2011. Transcriptome-wide binding sites for components of the Saccharomyces cerevisiae non-poly(A) termination pathway: Nrd1, Nab3, and Sen1. PLoS Genet 7: e1002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curinha A, Oliveira Braz S, Pereira-Castro I, Cruz A, Moreira A. 2014. Implications of polyadenylation in health and disease. Nucleus 5: 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, Muniz L, West S. 2014. 3′ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev 28: 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichtl B, Blank D, Sadowski M, Hubner W, Weiser S, Keller W. 2002. Yhh1p/Cft1p directly links poly(A) site recognition and RNA polymerase II transcription termination. EMBO J 21: 4125–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan ED, Braun CR, Gygi SP, Moazed D. 2014. Post-transcriptional regulation of meiotic genes by a nuclear RNA silencing complex. RNA 20: 867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong N, Brannan K, Erickson B, Kim H, Cortazar MA, Sheridan RM, Nguyen T, Karp S, Bentley DL. 2015. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol Cell 60: 256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusby B, Kim S, Erickson B, Kim H, Peterson ML, Bentley DL. 2015. Coordination of RNA polymerase II pausing and 3′ end processing factor recruitment with alternative polyadenylation. Mol Cell Biol 36: 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghazy MA, Gordon JM, Lee SD, Singh BN, Bohm A, Hampsey M, Moore C. 2012. The interaction of Pcf11 and Clp1 is needed for mRNA 3′-end formation and is modulated by amino acids in the ATP-binding site. Nucleic Acids Res 40: 1214–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover-Cutter K, Kim S, Espinosa J, Bentley DL. 2008. RNA polymerase II pauses and associates with pre-mRNA processing factors at both ends of genes. Nat Struct Mol Biol 15: 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granneman S, Kudla G, Petfalski E, Tollervey D. 2009. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci 106: 9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosso AR, de Almeida SF, Braga J, Carmo-Fonseca M. 2012. Dynamic transitions in RNA polymerase II density profiles during transcription termination. Genome Res 22: 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzechnik P, Gdula MR, Proudfoot NJ. 2015. Pcf11 orchestrates transcription termination pathways in yeast. Genes Dev 29: 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipati RK, Villa T, Boulay J, Libri D. 2008. Phosphorylation of the RNA polymerase II C-terminal domain dictates transcription termination choice. Nat Struct Mol Biol 15: 786–794. [DOI] [PubMed] [Google Scholar]
- Hallais M, Pontvianne F, Andersen PR, Clerici M, Lener D, Benbahouche Nel H, Gostan T, Vandermoere F, Robert MC, Cusack S, et al. 2013. CBC-ARS2 stimulates 3′-end maturation of multiple RNA families and favors cap-proximal processing. Nat Struct Mol Biol 20: 1358–1366. [DOI] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, Tian B. 2013. Analysis of alternative cleavage and polyadenylation by 3′ region extraction and deep sequencing. Nat Methods 10: 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen TH, Jacquier A, Libri D. 2013. Dealing with pervasive transcription. Mol Cell 52: 473–484. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Cubberley G, Bentley DL. 2009. Cotranscriptional recruitment of the mRNA export factor Yra1 by direct interaction with the 3′ end processing factor Pcf11. Mol Cell 33: 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Ahn SH, Krogan NJ, Greenblatt JF, Buratowski S. 2004a. Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J 23: 354–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Krogan NJ, Vasiljeva L, Rando OJ, Nedea E, Greenblatt JF, Buratowski S. 2004b. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432: 517–522. [DOI] [PubMed] [Google Scholar]
- Kim H, Erickson B, Luo W, Seward D, Graber JH, Pollock DD, Megee PC, Bentley DL. 2010. Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat Struct Mol Biol 17: 1279–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek K, Cerna H, Holub P, Pasulka J, Hrossova D, Loehr F, Hofr C, Vanacova S, Stefl R. 2012. Serine phosphorylation and proline isomerization in RNAP II CTD control recruitment of Nrd1. Genes Dev 26: 1891–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyburz A, Sadowski M, Dichtl B, Keller W. 2003. The role of the yeast cleavage and polyadenylation factor subunit Ydh1p/Cft2p in pre-mRNA 3′-end formation. Nucleic Acids Res 31: 3936–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NN, Chalamcharla VR, Reyes-Turcu F, Mehta S, Zofall M, Balachandran V, Dhakshnamoorthy J, Taneja N, Yamanaka S, Zhou M, et al. 2013. Mtr4-like protein coordinates nuclear RNA processing for heterochromatin assembly and for telomere maintenance. Cell 155: 1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, Bachand F. 2015. Fail-safe transcription termination: because one is never enough. RNA Biol 12: 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay JF, D′Amours A, Lemieux C, Lackner DH, St-Sauveur VG, Bahler J, Bachand F. 2010. The nuclear poly(A)-binding protein interacts with the exosome to promote synthesis of noncoding small nucleolar RNAs. Mol Cell 37: 34–45. [DOI] [PubMed] [Google Scholar]
- Lemay JF, Larochelle M, Marguerat S, Atkinson S, Bahler J, Bachand F. 2014. The RNA exosome promotes transcription termination of backtracked RNA polymerase II. Nat Struct Mol Biol 21: 919–926. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Geiger G, Minet M, Schroeder S, Cilli K, McNeil JB, Bentley DL. 2002. Functional interaction of yeast pre-mRNA 3′ end processing factors with RNA polymerase II. Mol Cell 9: 1101–1111. [DOI] [PubMed] [Google Scholar]
- Lunde BM, Reichow SL, Kim M, Suh H, Leeper TC, Yang F, Mutschler H, Buratowski S, Meinhart A, Varani G. 2010. Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat Struct Mol Biol 17: 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina DB, Shankar S, Natarajan P, Finn KJ, Madhani HD. 2013. A conserved ncRNA-binding protein recruits silencing factors to heterochromatin through an RNAi-independent mechanism. Genes Dev 27: 1851–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason PB, Struhl K. 2005. Distinction and relationship between elongation rate and processivity of RNA polymerase II in vivo. Mol Cell 17: 831–840. [DOI] [PubMed] [Google Scholar]
- Mata J. 2013. Genome-wide mapping of polyadenylation sites in fission yeast reveals widespread alternative polyadenylation. RNA Biol 10: 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Materne P, Anandhakumar J, Migeot V, Soriano I, Yague-Sanz C, Hidalgo E, Mignion C, Quintales L, Antequera F, Hermand D. 2015. Promoter nucleosome dynamics regulated by signalling through the CTD code. Elife 4: e09008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Heidemann M, Lidschreiber M, Schreieck A, Sun M, Hintermair C, Kremmer E, Eick D, Cramer P. 2012. CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 336: 1723–1725. [DOI] [PubMed] [Google Scholar]
- Meinhart A, Cramer P. 2004. Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 430: 223–226. [DOI] [PubMed] [Google Scholar]
- Nag A, Narsinh K, Kazerouninia A, Martinson HG. 2006. The conserved AAUAAA hexamer of the poly(A) signal can act alone to trigger a stable decrease in RNA polymerase II transcription velocity. RNA 12: 1534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso AR, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. 2015. Mammalian NET-seq reveals genome-wide nascent transcription coupled to RNA processing. Cell 161: 526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oktaba K, Zhang W, Lotz TS, Jun DJ, Lemke SB, Ng SP, Esposito E, Levine M, Hilgers V. 2015. ELAV links paused Pol II to alternative polyadenylation in the Drosophila nervous system. Mol Cell 57: 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM. 2010. Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143: 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Libri D. 2013. A bacterial-like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat Struct Mol Biol 20: 884–891. [DOI] [PubMed] [Google Scholar]
- Porrua O, Libri D. 2015. Transcription termination and the control of the transcriptome: why, where and how to stop. Nat Rev Mol Cell Biol 16: 190–202. [DOI] [PubMed] [Google Scholar]
- Porrua O, Hobor F, Boulay J, Kubicek K, D'Aubenton-Carafa Y, Gudipati RK, Stefl R, Libri D. 2012. In vivo SELEX reveals novel sequence and structural determinants of Nrd1–Nab3–Sen1-dependent transcription termination. EMBO J 31: 3935–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaughency P, Merran J, Corden JL. 2014. Genome-wide mapping of yeast RNA polymerase II termination. PLoS Genet 10: e1004632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlackow M, Marguerat S, Proudfoot NJ, Bahler J, Erban R, Gullerova M. 2013. Genome-wide analysis of poly(A) site selection in Schizosaccharomyces pombe. RNA 19: 1617–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz D, Schwalb B, Kiesel A, Baejen C, Torkler P, Gagneur J, Soeding J, Cramer P. 2013. Transcriptome surveillance by selective termination of noncoding RNA synthesis. Cell 155: 1075–1087. [DOI] [PubMed] [Google Scholar]
- Shi Y, Manley JL. 2015. The end of the message: multiple protein–RNA interactions define the mRNA polyadenylation site. Genes Dev 29: 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiebaut M, Kisseleva-Romanova E, Rougemaille M, Boulay J, Libri D. 2006. Transcription termination and nuclear degradation of cryptic unstable transcripts: a role for the nrd1–nab3 pathway in genome surveillance. Mol Cell 23: 853–864. [DOI] [PubMed] [Google Scholar]
- Tian B, Manley JL. 2013. Alternative cleavage and polyadenylation: the long and short of it. Trends Biochem Sci 38: 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoosthuyse V, Legros P, van der Sar SJ, Yvert G, Toda K, Le Bihan T, Watanabe Y, Hardwick K, Bernard P. 2014. CPF-associated phosphatase activity opposes condensin-mediated chromosome condensation. PLoS Genet 10: e1004415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Buratowski S. 2006. Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol Cell 21: 239–248. [DOI] [PubMed] [Google Scholar]
- Vasiljeva L, Kim M, Mutschler H, Buratowski S, Meinhart A. 2008a. The Nrd1–Nab3–Sen1 termination complex interacts with the Ser5-phosphorylated RNA polymerase II C-terminal domain. Nat Struct Mol Biol 15: 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiljeva L, Kim M, Terzi N, Soares LM, Buratowski S. 2008b. Transcription termination and RNA degradation contribute to silencing of RNA polymerase II transcription within heterochromatin. Mol Cell 29: 313–323. [DOI] [PubMed] [Google Scholar]
- Webb S, Hector RD, Kudla G, Granneman S. 2014. PAR-CLIP data indicate that Nrd1-Nab3-dependent transcription termination regulates expression of hundreds of protein coding genes in yeast. Genome Biol 15: R8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlotzka W, Kudla G, Granneman S, Tollervey D. 2011. The nuclear RNA polymerase II surveillance system targets polymerase III transcripts. EMBO J 30: 1790–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Tong L, Manley JL. 2014. Delineating the structural blueprint of the pre-mRNA 3′-end processing machinery. Mol Cell Biol 34: 1894–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhu J, Schermann G, Ohle C, Bendrin K, Sugioka-Sugiyama R, Sugiyama T, Fischer T. 2015. The fission yeast MTREC complex targets CUTs and unspliced pre-mRNAs to the nuclear exosome. Nat Commun 6: 7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.