

Abstract
The 23S rRNA methyltransferase RrmJ (FtsJ) is responsible for the 2′-O methylation of the universally conserved U2552 in the A loop of 23S rRNA. This 23S rRNA modification appears to be critical for ribosome stability, because the absence of functional RrmJ causes the cellular accumulation of the individual ribosomal subunits at the expense of the functional 70S ribosomes. To gain insight into the mechanism of substrate recognition for RrmJ, we performed extensive site-directed mutagenesis of the residues conserved in RrmJ and characterized the mutant proteins both in vivo and in vitro. We identified a positively charged, highly conserved ridge in RrmJ that appears to play a significant role in 23S rRNA binding and methylation. We provide a structural model of how the A loop of the 23S rRNA binds to RrmJ. Based on these modeling studies and the structure of the 50S ribosome, we propose a two-step model where the A loop undocks from the tightly packed 50S ribosomal subunit, allowing RrmJ to gain access to the substrate nucleotide U2552, and where U2552 undergoes base flipping, allowing the enzyme to methylate the 2′-O position of the ribose.
Modified nucleotides have been identified in nearly every cell and organism analyzed so far (11). Of the almost 100 posttranscriptional modifications that have been characterized in ribonucleotides, about one-third are present in rRNAs (30). Of those, the majority are methylation reactions on either the base or the 2′-O-ribose of the respective nucleotide. Most of these modifications cluster in conserved regions of functionally important rRNA domains and have been suggested to be important for the structure and function of the ribosome (4, 12, 13).
Although the chemical nature of the individual rRNA modifications has been known for some years, most of the enzymes that are responsible for these modifications have not yet been identified. In the case of Escherichia coli 23S rRNA, which has at least 14 different methylated nucleotides, only 5 modifying enzymes have been identified so far. These are the three base-modifying methyltransferases, RrmA (m1G745) (15), RumA (m5U1939) (1), and RumB (m5U747) (23), as well as the two unrelated 2′-O-ribose methyltransferases, RlmB (Gm2251) (22) and RrmJ (Um2552) (6, 7).
Our study focuses on the highly conserved RrmJ (FtsJ) protein, which is the first identified heat-inducible 2′-O-methyltransferase of E. coli (6, 7). While the deletion of most of the known 23S rRNA methyltransferases has been shown not to affect E. coli growth or ribosome assembly, the deletion of RrmJ has been found to affect both activities (6, 7). The rrmJ deletion strain, which no longer harbors the highly conserved Um2552 modification in the A loop of the ribosome, accumulates large amounts of free 30S and 50S ribosomal subunits at the expense of functional 70S ribosomes. This ribosome defect appears to be due specifically to a lack of methyltransferase activity, because the expression of active-site RrmJ mutants in the rrmJ deletion strain does not rescue this phenotype (16). The ribosome defect is, furthermore, thought to be the reason for the decreased translational efficiency of S30 lysates prepared from the rrmJ deletion strain (8) as well as for the significant growth disadvantage that is exerted by this strain (6). This finding was consistent with those of earlier studies, which demonstrated that the incorporation of 23S rRNA carrying a U2552 mutation into ribosomes affected cell growth and peptidyl transferase activity (20, 29).
RrmJ is one of the few RNA-modifying enzymes that has been shown to act very late in the maturation process of the ribosome (6, 7). Only fully assembled 50S ribosomal subunits prepared from the rrmJ deletion strain appear to serve as substrates for RrmJ in vitro, while naked 23S rRNA or ∼40S ribosomal particles that are prepared from the rrmJ deletion strain are not methylated by purified RrmJ (6). This finding suggested that either the correct folding of the 23S rRNA or additional protein-protein interactions are necessary for the substrate recognition.
While E. coli cells have only one RrmJ homologue, eukaryotic cells usually have several. Yeast cells, for instance, have been found to harbor three RrmJ homologues: Trm7p in the cytosol (28), Mrm2p in mitochondria (26), and Spb1p in the nucleus (27). Whereas the mitochondrial and nuclear RrmJ homologues function as rRNA methyltransferases, the cytosolic Trm7p has been shown to be responsible for two 2′-O-ribose methylations at position 32 (Cm32) and 34 (Gm34) in the anticodon loop of certain yeast tRNAs. The ability of yeast RrmJ homologues to recognize either rRNA or tRNA corroborates reports about a possible dual substrate specificity for E. coli RrmJ, which has been found to methylate tRNAs in vitro in addition to 23S rRNA (6).
How methyltransferases recognize and bind their target sequences has not been well established for the majority of known enzymes. Only very few cocrystallization studies of RNA methyltransferases with their substrates have revealed how RNA is associated with the proteins. One of these proteins is the mRNA 2′-O-methyltransferase VP39 from vaccinia virus, whose structure closely resembles that of RrmJ, which has been solved in complex with S-adenosylmethionine (AdoMet) (6). The structure of VP39 has been solved in complex with the reaction product S-adenosylhomocysteine and a 5′ m7G-capped, single-stranded RNA hexamer (18). The modeling of the mRNA substrate analogue of VP39 into the structure of RrmJ assisted us in identifying the active site residues in RrmJ and in proposing a reaction mechanism (16).
We used extensive mutagenesis studies to analyze the putative 23S rRNA substrate binding site in RrmJ. Sequence comparisons and an analysis of the structure of RrmJ allowed us to identify a number of residues that are potentially involved in rRNA binding. We introduced these mutations into wild-type RrmJ and functionally characterized the mutant proteins in vivo and in vitro. We identified a highly conserved, positively charged ridge that appears to serve as the RNA substrate binding site of RrmJ. Furthermore, we discovered that the isolated unmodified A loop serves as the minimal methylation substrate of wild-type RrmJ in vitro. In situ modeling studies of the A-loop structure into the proposed substrate binding site of RrmJ suggested a base flipping mechanism for RrmJ, which is postulated to be important for the methylation process.
MATERIALS AND METHODS
Site-directed mutagenesis.
Site-directed mutagenesis was performed according to the QuickChange protocol (Stratagene, La Jolla, Calif.). Wild-type rrmJ was cloned into pET11a to generate pHB1 (6), which was used as template DNA for all mutations. All introduced mutations were confirmed by DNA sequencing. Table 1 lists all plasmids and strains that were generated. For protein expression and purification, the pET11a derivatives were introduced into a BL21 strain that contained a deletion of the rrmJ gene (JUH47) (16). To perform phenotypical studies, the plasmids were introduced into the rrmJ deletion strain HB23. The expression analysis of all of the mutants was performed as previously described (16).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Genotype or relevant features | Source or reference |
|---|---|---|
| MG1655 | rph-1 | Lab collection |
| HB24 | MG1655, zgi-203::Tn10, Tcr | 6 |
| HB23 | MG1655, zgi-203::Tn10 Tcr, rrmJΔ567 | 6 |
| HB25 | HB23, pHB1 | 6 |
| HB1 | BL21(DE3), pHB1 | 6 |
| BL21(DE3) | F−ompT hsdSB (rB− mB−) gal dcm(DE3) | Novagene |
| JUH47 | BL21(DE3), rrmJΔ567 Tcr | 16 |
| pET11a | pBR322 derived | Novagene |
| pHB1 | pET11a rrmJ | 6 |
| pJUH12 | pET11a rrmJ K38A | 16 |
| pJUH3 | pET11a rrmJ D20A | This study |
| pHB30 | pET11a rrmJ Y22A | This study |
| pJUH5 | pET11a rrmJ R32A/R34A | This study |
| pJUH6 | pET11a rrmJ F37A/L39A | This study |
| pJUH7 | pET11a rrmJ Q67A/Y68A | This study |
| pJUH20 | pET11a rrmJ D136N | This study |
| pJUH22 | pET11a rrmJ F166A | This study |
| pJUH23 | pET11a rrmJ K189A | This study |
| pJUH10 | pET11a rrmJ R194A | This study |
| pHB31 | pET11a rrmJ S197A | This study |
Purification of the RrmJ mutants.
The purification of the RrmJ mutants was performed as previously described (16). The protein concentration was determined by UV absorbance by using an extinction coefficient of 1.0 for a 1 mg/ml solution at A280 for all mutant proteins (6).
Analytical polysome profiles.
Analytical polysome profiles of the wild-type strain HB24, the rrmJ deletion strain HB23, and HB23 expressing the individual rrmJ mutant proteins under stringent and nonstringent salt conditions were performed and analyzed as previously described (6, 16).
Preparation of ribosomal subunits and methylation assay.
The preparation of the 50S ribosomal subunits of the rrmJ deletion strain HB23 and analysis of the methyl transfer activity of wild-type RrmJ and the mutant proteins was performed as previously described (16). RrmJ (either 100 or 200 nM) in 50 mM HEPES-KOH (pH 7.5), 85 mM NH4Cl, 3 mM MgCl2, and 2 mM β-mercaptoethanol was used. For Km measurements, the initial rate for methylation was measured in the presence of 50 μM [3H]AdoMet (85.0 Ci/mmol; Amersham Biosciences) and various concentrations of 50S ribosomal subunits, ranging from 0.25 to 10 μM. At defined time points (2.5, 5, 7.5, and 10 min) after initiation of the methylation reaction at 37°C, 8-μl aliquots were taken and the [3H]methyl incorporation was determined as described previously (6). The slopes of the methyl incorporation versus time were calculated. Methylation reactions with the A loop as the substrate were performed in 50 mM HEPES-KOH (pH 7.5), 50 mM NaCl, 10 mM EDTA, 1 mM dithiothreitol, 40 U of RNase inhibitor RNasin (Promega), and 0.1 mg of bovine serum albumin/ml at 37°C. RrmJ (5 μM) was incubated with 50 μM [3H]AdoMet (85 Ci/mmol), and the reaction was started by the addition of 1 μM purified A loop (nucleotides 2545 to 2563 of E. coli 23S rRNA; kindly provided by Joseph Puglisi, Stanford University) (3). At defined time points after the methylation reaction was initiated, 8-μl aliquots were taken and supplemented with 40 μg of tRNA (Sigma) as the carrier for precipitation, and the [3H]methyl incorporation was determined as described previously (6).
AdoMet titration.
To determine the amount of AdoMet present in the various RrmJ mutant preparations, we used an AdoMet titration curve (16). In short, a 23 μM solution of the AdoMet-free D83A mutant RrmJ protein was prepared, and 1 μM AdoMet (ɛ257 = 15,400 M−1 cm−1) was added per titration step (16). After each AdoMet addition, the optical density at 280 nm (OD280)/OD260 ratio of the protein-AdoMet mixture was determined. After volume corrections, these ratios were plotted against the amounts of titrated AdoMet. The OD280/OD260 ratios of the purified RrmJ mutants were then determined, and the amounts of bound AdoMet were calculated.
RrmJ binding to the ribosome.
The preparation of 50S ribosomal subunits from the wild type (HB24), the rrmJ deletion strain expressing no RrmJ (HB23), or the RrmJ-D136N mutant protein was performed under stringent, high-salt conditions (1 mM MgCl2 and 200 mM NH4Cl) as described previously (6). Aliquots of 2 μM 50S ribosomal subunits alone or supplemented with various concentrations of purified wild-type RrmJ were prepared. Ten microliters of each of those aliquots was loaded onto a Tris-glycine sodium dodecyl sulfate-14% polyacrylamide gel (Invitrogen), and Western blot analysis was performed with polyclonal antibodies against RrmJ.
RESULTS
Rational RrmJ mutant design.
The conservation of an amino acid residue in a protein is a strong indicator of its importance in the structure and/or function of the protein. Residues that are exposed to the surface of a protein are usually highly variable unless they are involved in the catalytic mechanism of the protein, in its structural integrity, or in substrate and/or cofactor binding. To investigate the potential substrate binding site of the 23S rRNA methyltransferase RrmJ, we therefore performed extensive site-directed mutagenesis, replacing the majority of highly conserved residues in RrmJ. We spared only those residues that have been shown to be involved in AdoMet binding and/or the catalytic site of RrmJ (16) as well as those that we predicted to play a structural role in the protein fold (Fig. 1).
FIG. 1.

Rational mutant design. The 27 most highly conserved residues in RrmJ are shown in bold and underlined letters. These amino acids have been identified as highly conserved by aligning a set of 1,112 nonredundant paralog sequences to a set of 29 likely RrmJ orthologs (6). The black dots above the RrmJ sequence indicate the residues that have been identified as involved in AdoMet binding (6). Stars mark the three amino acids that have been shown to form the catalytic triad (16). The filled cylinders above the sequence depict the amino acids which are either predicted to play a structural role in RrmJ or reside on the opposite site of the RrmJ molecule. The open arrows mark all highly conserved residues that were mutated in previous studies (16). The closed arrows depict the residues that were mutated in this study.
Of the 27 most highly conserved amino acids in RrmJ (6), 11 amino acids either directly contact the cofactor AdoMet or are active site residues (6, 16). An analysis of the crystal structure of RrmJ allowed us to predict, furthermore, that the highly conserved residues P127, F161, F182, P190, and S193 are likely to perform structural functions in the methyltransferase, either by forming the AdoMet binding pocket (e.g., P127) or by building the hydrophobic core of the α/β protein (e.g., F161 and F182). Q119 and G159 also were excluded from our mutagenesis studies, because both amino acids are located on the opposite site of the RrmJ molecule and appeared, therefore, rather unlikely to be involved in substrate binding. These considerations left us with nine highly conserved residues (D20, Y22, R32, R34, Q67, D136, F166, R194, and S197), eight of which we replaced individually or pair-wise with alanine residues. We decided to replace D136 with an asparagine residue to maintain the size of this residue.
To expand our mutagenesis scheme, we also used a structure-based sequence alignment with the structurally most closely related 2′-O-methyltransferase, VP39. The structure of VP39 has been solved in complex with the mRNA substrate analogue 5′ m7G-capped RNA hexamer (18). Studies modeling the mRNA substrate analogue onto the RrmJ structure revealed four additional, somewhat less-conserved residues in RrmJ that suggested involvement in the substrate association as well. These considerations led us to make additional alanine substitutions for the residues F37, L39, Y68, and K189 in RrmJ.
In vivo phenotype of the RrmJ mutants.
To first analyze how the individual mutations affect the in vivo function of RrmJ, we transformed the plasmids encoding the respective RrmJ mutants into the rrmJ deletion strain HB23. We used quantitative Western blot analysis to determine the expression levels of the various mutant proteins and found that the mutant proteins were all soluble and expression levels were similar to that of a single copy of wild-type RrmJ expressed from its normal chromosomal location (data not shown). This finding excluded the possibility that any observed in vivo phenotype was due to inappropriately low or high RrmJ mutant protein levels. The wild-type-like expression levels of the RrmJ mutant proteins has been observed before and is due to their leaky expression from the uninduced pET11a vector (16).
To evaluate the in vivo function of our RrmJ mutant proteins, we first analyzed their growth phenotype in liquid Luria-Bertani medium at 37°C. As summarized in Table 2, wild-type E. coli strains show a doubling time of 25 min at 37°C, while the rrmJ deletion strain grows significantly slower, with a doubling time of more than 50 min. This severe growth defect is clearly due to a defect in the methyltransferase activity of RrmJ, because expression of the active site mutant RrmJ-K38A in the rrmJ deletion strain did not rescue the growth defect (Table 2). We then analyzed the growth of the individual RrmJ mutants. Three of the mutants revealed a significantly compromised in vivo activity. The doubling time for the RrmJ-F166A mutant was 40 min, the doubling time for the RrmJ-R32A/R34A mutant was 43 min, and the doubling time for the RrmJ-D136N mutant was 45 min; all of these were much longer than the doubling time determined for an rrmJ deletion strain expressing wild-type RrmJ, which was 25 min. Noteworthy, all other mutant strains also showed slightly longer doubling times than the wild-type strain, ranging from 32 to 38 min each. This finding suggested that the replacement of any of the conserved amino acids might impair the in vivo function of RrmJ to some extent.
TABLE 2.
Summary of the in vivo and in vitro activities of RrmJ and the mutantsa
| Mutation | Doubling time at 37°C (min) | Ribosome profile for condition indicated
|
AdoMet/protein (%) | Apparent kcat (min−1) | Apparent Km (μM) | Kcat/Km (μM−1 min−1) | |
|---|---|---|---|---|---|---|---|
| Associating | Dissociating | ||||||
| Wild type | 25 | +++ | +++ | 76 | 0.064 | 0.7 ± 0.05 | 0.080 |
| rrmJ | 53 | (+) | (+) | ||||
| K38Ab | 58 | (+) | (+) | 68 | <0.001 | ||
| D20A | 36 | +++ | +++ | 81 | 0.036 ± 0.007 | 1.5 ± 0.4 | 0.024 |
| Y22A | 32 | +++ | ++ | 78 | 0.059 ± 0.004 | 0.7 | 0.084 |
| R32A/34A | 43 | + | + | 72 | 0.017 ± 0.013 | 2.7 ± 0.6 | 0.006 |
| F37A/L39A | 38 | +++ | +++ | ND | ND | ND | ND |
| Q67A/Y68A | 38 | +++ | +++ | ND | ND | ND | ND |
| D136N | 45 | + | + | 71 | 0.034 ± 0.007 | 0.5 ± 0.2 | 0.068 |
| F166A | 40 | + | +++ | 40 ± 4 | ND | ND | ND |
| K189A | 32 | ++ | +++ | 65 | 0.007 ± 0.002 | 0.5 | 0.014 |
| R194A | 33 | +++ | ++ | 59 | 0.051 ± 0.010 | 1.5 ± 0.4 | 0.034 |
| S197A | 34 | +++ | ++ | 74 | 0.058 ± 0.004 | 1.4 ± 0.01 | 0.041 |
The apparent kcat and Km values are the means and standard deviations of results from at least three independent experiments. The in vivo data presented for wild-type RrmJ were obtained with the rrmJ deletion strain HB23 expressing wild-type rrmJ from a pET11a plasmid (HB25). Growth of this strain is very similar to that of the corresponding wild-type E. coli strain (MG1655), but it accumulates slightly higher levels of 30S and 50S ribosomal subunits in lysates prepared under associating conditions and of 40S ribosomal subunits in lysates prepared under dissociating salt conditions. Western blot analysis revealed that all mutant variants were soluble and were expressed in similar amounts as the single copy of wild-type RrmJ from its normal chromosomal location in MG1655. ND, not determined; (+), +, ++, or +++, ribosome profiles are severely, moderately, slightly, or not impaired in lysates prepared under the indicated salt condition.
See reference 16.
Ribosome profiles of the RrmJ mutant strains.
The growth defect of strains lacking functional RrmJ was proposed to be due to the absence of the highly conserved Um2552 modification in the A loop of the 23S rRNA, which causes either assembly or stability problems of the 70S ribosome and leads to an impaired translational efficiency. To investigate to what extent the growth defect of the mutant strains correlates with the ribosome profiles for these strains, we prepared lysates of the rrmJ mutant strains and compared the ribosome profiles under nonstringent and stringent salt conditions to the lysate of the rrmJ deletion strain expressing wild-type RrmJ from a plasmid (HB25). This strain accumulates slightly higher levels of 30S, 40S, and 50S ribosomal subunits than a wild-type E. coli strain that expresses RrmJ from its chromosomal copy (6). Because all of the RrmJ variants we used are expressed in this background strain, we decided to use this rescue strain, HB25, as the appropriate “wild-type” control in all of our in vivo experiments. The ribosome defect of an rrmJ deletion strain is revealed by the accumulation of large amounts of 30S and 50S ribosomal subunits in lysates prepared under nonstringent salt conditions (Fig. 2, left diagrams), and by the population of ∼40S ribosomal particles in lysates prepared under stringent salt conditions (Fig. 2, right diagrams) (6).
FIG. 2.

In vivo activity of the RrmJ mutants. Polysome profiles of rrmJ deletion strain HB23 expressing no RrmJ or expressing wild-type RrmJ or the mutant proteins from a plasmid to wild-type protein levels. The ribosome profiles were analyzed under either associating, nonstringent salt conditions (10 mM MgCl2, 100 mM NH4Cl) (left panel) or dissociating, stringent salt conditions (1 mM MgCl2, 200 mM NH4Cl) (right panel). The ribosome profiles of lysates prepared from the individual strains (dark broken lines) are compared to the polysome profile of an rrmJ deletion strain expressing wild-type (wt) RrmJ (light broken lines).
We found that the degree of ribosome stabilities observed in the individual mutant strains corresponded only to a certain extent to the degree of growth of the respective mutant strains in liquid culture. The three mutant strains that showed the slowest growth in liquid Luria-Bertani medium (R32A/R34A, D136N, and F166A) indeed had the strongest ribosome defect. R32A/R34A and D136N mutant strains accumulated larger amounts of 30S and 50S ribosomal subunits than wild-type strains under nonstringent salt conditions, and had a significant amount of ∼40S ribosomal particles under stringent salt conditions. The F166A mutant protein accumulated more ribosomal subunits without populating the ∼40S particles. All other mutant strains, however, showed ribosome profiles that were either only slightly different from or identical to the ribosome profiles prepared from wild-type strains (Fig. 2; Table 2). This result was in contrast to the growth disadvantages that we observed in all of these strains and suggested that the growth defects observed in the rrmJ deletion strain might not be directly connected to defects in ribosome assembly and/or stability.
Kinetic characterization of the RrmJ mutant proteins.
Biochemical characterization of the mutated RrmJ proteins was necessary to further elucidate which residues might be involved in substrate binding. We therefore decided to purify all mutant proteins, excluding only the two RrmJ variants that had substitutions for less-conserved amino acids (F37A/L39A and Q67A/Y68A) and showed ribosome profiles that were indistinguishable from wild-type ribosome profiles (Table 2). All other mutant proteins were expressed as soluble proteins to near wild-type protein levels in the transformed rrmJ deletion strain, indicating that the mutations did not significantly alter the stability of the proteins. To obtain additional evidence that the respective mutations did not cause a major change in the conformation of the mutant proteins, we determined the amount of AdoMet that remained associated with each of the eight mutant RrmJ proteins after their purification (16). Wild-type RrmJ appears to have a very high affinity for its cofactor, because AdoMet is still bound to more than 70% of the protein after its purification. As shown in Table 2, at least 59% and up to 81% of AdoMet was associated with all but one of the RrmJ mutant proteins after their purification, confirming that the overall structural integrity of the mutant proteins very likely has been maintained. Only the RrmJ-F166A mutant protein, which showed one of the most severe growth and ribosome defects when expressed in the rrmJ deletion strain in vivo, was associated with significantly less AdoMet after purification. This result suggested a decrease in AdoMet binding affinity and/or the presence of a certain amount of an inactive yet stably folded RrmJ mutant species in our preparation that is no longer able to bind AdoMet. We therefore decided to not use this mutant protein for our further in vitro studies.
To test the enzymatic activities of the various RrmJ mutant proteins in vitro, we investigated their ability to methylate 50S ribosomal subunits prepared from the rrmJ deletion strain (HB23). By using a 100 nM concentration of enzyme and keeping both substrates, AdoMet, and 50S ribosomal subunits in large excess, we determined the apparent kcat values for each of the purified mutant proteins at 37°C. As shown in Fig. 3 and summarized in Table 2, four of the mutant proteins revealed at least a 50% decrease in their kcat values, with the RrmJ variants harboring a mutation in R32/R34 and K189 showing the most significant decrease in enzymatic activity. Mutations in D20 and D136 also led to slight decrease in kcat values, while mutations in Y22, R194 and S197 did not dramatically alter the turnover numbers of RrmJ under the conditions tested.
FIG. 3.
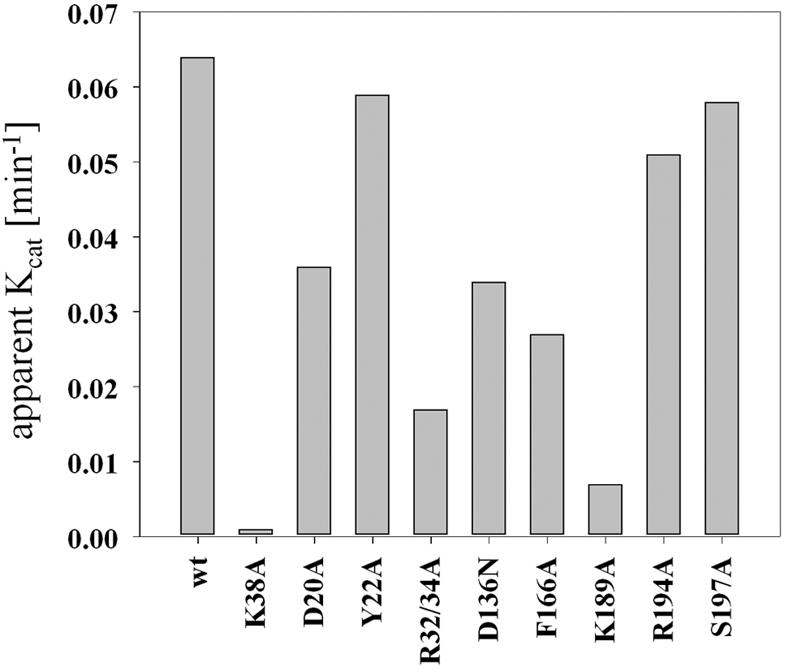
In vitro activity of the various RrmJ mutants. A 100 nM concentration of purified enzyme was incubated with 50 μM [3H]AdoMet (200 μC/ml) in methylation buffer (50 mM HEPES-KOH [pH 7.5], 85 mM NH4Cl, 3 mM MgCl2, 2 mM β-mercaptoethanol, 40 U of RNasin) at 37°C. The methylation reaction was started by the addition of either 5 μM or 8 μM 50S subunits prepared from the rrmJ deletion strain HB23. At defined time points, aliquots were taken and the [3H]methyl incorporation was determined as described previously. wt, wild type.
Next, we determined the apparent Km values of 50S ribosomal subunits for each of the purified RrmJ mutants. For these experiments, we kept the AdoMet concentration at 50 μM and varied the concentration of 50S ribosomal subunits from 0.2 to 10 μM. As shown in Table 2, an increased apparent Km value for 50S ribosomal subunits was found for four of our RrmJ variants. These RrmJ mutants harbored substitutions for the surface-exposed amino acids D20, R32/R34, R194, or S197, implicating these amino acids as potentially involved in the binding of the 23S rRNA. The observed changes in apparent Km values for 50S subunit binding in RrmJ mutant proteins were very similar to the changes in Kd and Km values that have been determined for substrate binding mutants of the 23S rRNA dimethyltransferase ErmC′ (24). For ErmC′ mutant proteins, Kd and Km values increased about 1.6- to 5-fold. This finding indicated that RNA binding is a highly cooperative, multivalent process, in which the mutation of single amino acids in the substrate binding site might not alter the substrate binding affinity as dramatically as do amino acid substitutions in the substrate binding site of other non-nucleic acid binding proteins.
In vitro enzymatic activity and in vivo ribosome profile: a good but not absolute correlation.
A comparison of the in vitro and in vivo activities of RrmJ and the mutants indicated a good but not absolute correlation between changes in kcat and Km values of the purified RrmJ variants and alterations in the ribosome profiles of the corresponding mutant strains. The two most obvious outliers in this correlation were the RrmJ-K189A and the RrmJ-D136N mutant proteins. The RrmJ-K189A mutant strain revealed no apparent ribosome defect in vivo and no significant change in Km for 23S rRNA but had an almost 10-fold decrease in apparent kcat value (Table 2). This result could be explained by the possibility that analysis of the ribosome profiles under steady-state conditions might not be sufficiently sensitive to detect these changes in the catalytic activity of RrmJ. The RrmJ-D136N mutant protein, on the other hand, showed a very severe growth disadvantage and ribosome defect in vivo but had wild-type kcat and Km values in vitro. To investigate whether the D136N mutant protein was indeed inactive in vivo, we analyzed the methylation status of 23S rRNA prepared from RrmJ-D136N expressing mutant strains. Our rationale was that if the D136N mutant protein was inactive in vivo, the 50S ribosomal subunits would be unmethylated and would serve as in vitro substrates for wild-type RrmJ much like 50S ribosomal subunits prepared from the rrmJ deletion strain. If, on the other hand, the D136N mutant protein was active in vivo, the ribosomal subunits would be methylated and, like 50S ribosomal subunits prepared from wild-type strains, would no longer serve as in vitro substrates (Fig. 4). We prepared 50S ribosomal subunits from the rrmJ deletion strain expressing the RrmJ-D136N mutant protein and tested the ability of wild-type RrmJ to methylate these ribosomal subunits in vitro. As shown in Fig. 4, 50S ribosomal subunits prepared from the rrmJ deletion strain expressing the D136N mutant showed the same incorporation of methyl groups upon incubation at 37°C as subunits prepared from the strain that lacked RrmJ altogether (Fig. 4). This result clearly showed that the D136N mutant is indeed unable to methylate 23S rRNA of 50S ribosomal subunits in vivo, which agreed well with the observed ribosome defect of the D136N-expressing rrmJ deletion strain.
FIG. 4.

RrmJ is associated with 50S ribosomal subunits in vivo. Methyl incorporation into 2 μM 50S ribosomal subunits prepared from wild-type strain HB24 (circles), rrmJ deletion strain HB23 (triangles), or rrmJ deletion strain HB23 expressing the D136N mutant protein (squares), either in the absence (open symbols) or in the presence (closed symbols) of 300 nM purified wild-type RrmJ. At defined time points, aliquots were taken and the [3H]methyl incorporation was determined as described previously. Inset: RrmJ is bound to 50S ribosomal subunits. Analysis of the RrmJ content in 2 μM 50S ribosomal subunits prepared from the wild-type strain (HB24) (lane 1), the rrmJ deletion strain (HB23) (lane 2), or the rrmJ deletion strain expressing the D136N mutant from a plasmid (lane 3). To evaluate how much RrmJ is bound to the ribosomal subunits, 50S ribosomal subunits prepared from the rrmJ deletion strain (HB23) were supplemented with 50 nM (lane 4), 100 nM (lane 5), or 200 nM (lane 6) concentrations of purified RrmJ. Aliquots of the ribosomal subunits were loaded onto a sodium dodecyl sulfate-14% polyacrylamide gel, and Western blot analysis was performed with antibodies against RrmJ.
RrmJ binds to 50S ribosomal subunits in vivo.
Interestingly, the analysis of the methyl incorporation into ribosomal subunits of D136N mutant cells was dependent only in part on the presence of exogenous wild-type RrmJ. Incubation of the D136N-50S ribosomal subunits at 37°C in the presence of [3H]AdoMet but in the absence of purified RrmJ led to a substantial methylation of the 23S rRNA (Fig. 4). This finding was in contrast to that for the 50S ribosomal subunits prepared from the rrmJ deletion strain, whose methylation was absolutely dependent on the presence of exogenous RrmJ (Fig. 4), and to the results obtained with 50S ribosomal subunits prepared from the wild-type strain, which did not show any methyl incorporation independent of the presence of purified RrmJ (Fig. 4). These results suggested that a methyltransferase activity was present in the 50S ribosomal subunits prepared from the rrmJ deletion strain expressing the RrmJ-D136N protein, which only became active under the chosen in vitro methylation conditions.
The purified D136N mutant protein showed near wild-type activity in vitro when tested on unmethylated 50S ribosomal subunits prepared from the rrmJ deletion strain. This finding, together with the observation that the yeast RrmJ homologue Mrm2p cofractionates with mitochondrial 21S rRNA, led us to investigate whether the observed methyltransferase activity was indeed the RrmJ-D136N mutant protein bound to its 50S ribosomal subunits, which might become active under our chosen in vitro conditions. As shown in the inset to Fig. 4, Western blot analyses of 50S ribosomal subunits from the rrmJ wild-type strain and the rrmJ deletion strain expressing the D136N mutant protein showed that E. coli RrmJ stays associated with the 50S ribosomal subunit during their preparation under stringent, high-salt conditions (1 mM MgCl2, 200 mM NH4Cl). No RrmJ was detected in the 30S subunits or the intact 70S ribosomes (data not shown). The amount of bound D136N mutant protein, in combination with the observed slightly lower in vitro activity of the purified D136N mutant (Fig. 3), explained very well the methylation activity that we observed with isolated 50S ribosomal subunits prepared from the D136N-expressing rrmJ deletion strain. This finding suggested that there might be a lack of efficient substrate release of D136N from 50S subunits in vivo, which could cause the apparent inactivity of the D136N mutant protein in vivo but might be less apparent under our chosen in vitro conditions.
Modeling of the A-loop structure onto the surface of RrmJ.
U2552, the methylation target of RrmJ, is one of the five residues that constitute the A loop in the peptidyltransferase center of the ribosome. Both the crystal structure (2) and the solution structure (nucleotides 2548 to 2560) (3) of the A loop have been solved. To model the solution structure of the A loop into RrmJ, the A loop was initially positioned manually by using the crystal structure of the mRNA 2′-O-methyltransferase VP39 from vaccinia virus bound with the reaction product S-adenosylhomocysteine and 5′ m7G-capped single-stranded RNA hexamer as a guide (18). The RNA was positioned by first overlaying the ribose and phosphate atoms of the reactive nucleotides into the active site of RrmJ (Fig. 5A). This positioning gave close agreements between the individual atom positions in the active site between U2552 of the A-loop RNA model and the 5′ m7G-capped RNA of VP39. However, the rest of the A loop clashed with protein residues. Most noteworthy, the double-helical stalk of the A loop clashed severely with the protruded helix 4 of RrmJ (Fig. 5A).
FIG. 5.

Modeling of the A loop onto the surface of RrmJ. (A) In situ modeling of the A-loop structure (nucleotides 2548 to 2560) as solved by NMR (3) onto the surface of RrmJ. Modeling is based on overlaying the U2552 reactive nucleotide into the active site of RrmJ with the 5′ m7G-capped reactive nucleotide in the VP39 structure. Without turning the A loop, the double helical stalk of the A loop clashes severely with the extended α4 helix of RrmJ (red model). To avoid this clashing, the A loop is turned by 85 degrees (blue model). Now, phosphodiesters occupy the same location in the model of RrmJ as do phosphodiesters of the 5′ m7G-capped RNA hexamer in the VP39 structure. (B) Base flipping of U2552. The model shows U2552 in the closed position (yellow), where the 2′ hydroxyl group of the ribose is not accessible to the methyl donor AdoMet, and in the flipped configuration (red), where the 2′ hydroxyl group is in the optimal position to be methylated. (C) Solvent-accessible surface of RrmJ colored by distance to ligand atoms from U2552-flipped A-loop RNA (blue ribbon) and AdoMet (shown as stick model). The surface is colored by distance from nearby atoms. Dark areas are too far away from any atoms outside the surface to be bonded. White or light areas are close enough for hydrophobic van der Waals interactions. Pink areas are close enough for hydrogen bonds. The figure was made with Protein Explorer (25).
To accommodate the A loop, we then kept the phosphodiesters of the reactive U2552 in position and rotated the A loop by approximately 85 degrees. This rotation prevented all clashes between the A loop and RrmJ protein (Fig. 5A). The phosphodiesters of the double helix now fit snugly into a deep cleft of RrmJ. Importantly, RNA-protein interactions involved exactly those residues (R34, R194, and S197) in RrmJ that when mutated to alanine residues exerted an increased Km for 23S rRNA binding (Fig. 5C). This finding suggested that these amino acids are indeed involved in the binding and positioning of 23S rRNA in RrmJ. In this model, the phosphodiesters now occupy the same location as the phosphodiesters of the 5′ m7G-capped RNA hexamer in the VP39 structure. Additionally, the minor groove of the A-loop RNA is now positioned to sit nicely atop helix 4 of RrmJ, rather than clashing with the protein.
Interestingly, however, once the A loop was turned to fit into the protein, the 2′-hydroxyl group of U2552 was no longer accessible to the active site of RrmJ (Fig. 5B). These observations suggested a base-flipping mechanism for U2552 methylation in which the A-loop RNA first binds into the RrmJ binding site and then undergoes a base-flipping rotation of U2552 by 85 degrees, thereby placing the 2′-hydroxyl in position for the methylation.
The base-flipped model of the A-loop RNA was constructed and positioned into the putative binding site of RrmJ (Fig. 5B). Both base-flipped U2552 and unflipped models were then energy-minimized by positional and simulated annealing by using the CNX program (5). During these refinements, the protein atoms were allowed to adjust to the RNA model. Only two residues were found to move slightly. The side chains of R194 and R196, both located on the surface of RrmJ, are required to change conformation in order for the A-loop RNA model to position optimally into the substrate binding cleft. These residues are then closely positioned next to the phosphate backbone of the model A loop.
Very similar results were obtained when we modeled the crystal structure of the A loop into RrmJ, because the major differences between the A-loop conformations in the crystal structure and in the solution structure involve parts of the A loop that do not contact RrmJ. The phosphate backbone of the A loop, however, which makes most of the contacts with RrmJ, is not significantly different in the two structures.
The unmodified A loop: the minimal substrate for RrmJ.
Modeling studies revealed that the A loop can be modeled into the substrate binding site we identified. This finding suggested that the A loop might serve as an in vitro substrate for RrmJ. We therefore performed in vitro methylation assays using the same unmodified A loop that was used in the nuclear magnetic resonance (NMR) (3) and our modeling studies (Fig. 5). The Um2552-methylated A loop was used as the control (3). As shown in Fig. 6, we observed a significant RrmJ-mediated methyl incorporation into the nonmodified A loop (Fig. 6). The methylated A loop, on the other hand, did not serve as an in vitro substrate for purified RrmJ (Fig. 6), strongly suggesting that the methylation of the A loop by RrmJ was indeed a specific process. Because the methyl incorporation into the A loop was slow compared to the methyl incorporation into 50S ribosomal subunits, we wanted to exclude the possibility that the observed A-loop methylation is due to small amounts of a different methyltransferase that might contaminate our wild-type RrmJ preparation. We therefore tested the active site mutant RrmJ-K38A, and found that this mutant protein was completely unable to methylate the unmodified A loop in vitro (data not shown). Together, these results show that RrmJ can specifically recognize, bind, and methylate the A loop when it adopts its solution structure. The slow methyl incorporation, however, might be due to missing contact sites that are present in the 50S ribosomal subunit.
FIG. 6.
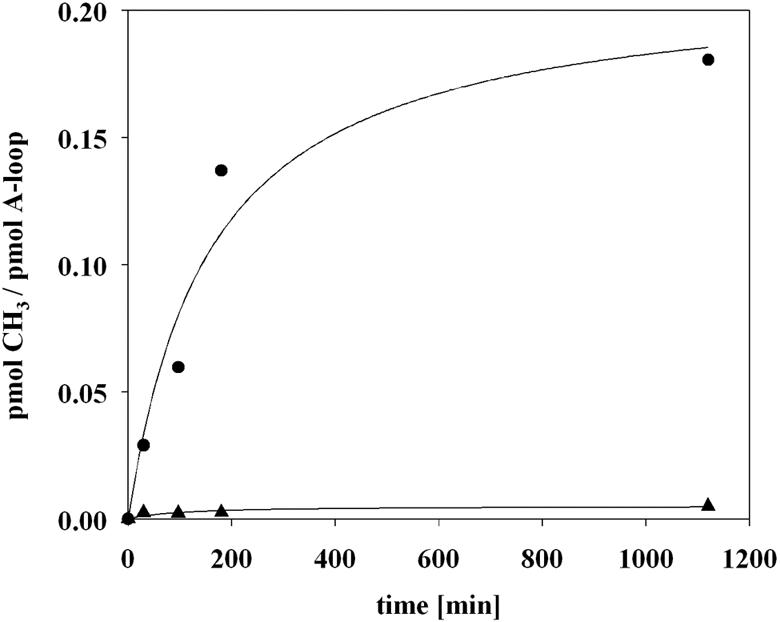
Nonmethylated, isolated A loop: a minimal substrate for RrmJ in vitro. A 5 μM concentration of wild-type RrmJ was incubated with a 1 μM concentration of nonmodified (circles) or methylated (triangles) A loop (nucleotides 2545 to 2563) and 50 μM [3H]AdoMet (200 μCi/ml) in methylation buffer (50 mM HEPES-KOH [pH 7.5], 50 mM NaCl, 10 mM EDTA, 1 mM dithiothreitol, 40 U of RNasin, and 0.1 mg of bovine serum albumin/ml) at 37°C. Aliquots were taken at defined time points, and the [3H]methyl incorporation was measured as described previously.
An analysis of the ribosome structure suggested that RrmJ is unable to access the A loop unless it is looped out of the ribosome. Such reversible undocking of the A loop has been proposed before and was suggested to occur as a regular step in tRNA selection and accommodation (3). Our findings confirm these considerations and suggest that the A loop adopts its solution structure when looped out of the ribosome, where it can be methylated by RrmJ. This provides an excellent explanation of how RrmJ is able to methylate a buried 23S rRNA residue so late in the maturation process of the ribosome.
DISCUSSION
In this study, we have used structure predictions, structural modeling approaches, and structure-based sequence alignments to identify highly conserved, surface-exposed residues that could be involved in the binding of the 2′-O-methyltransferase RrmJ to its substrate 23S rRNA. We mutated the residues identified, and after careful in vivo and in vitro analysis we are now proposing that a positively charged ridge built by the highly conserved arginines 32, 34, and 194 plays an important role in the coordination of the rRNA sugar phosphate backbone. In addition to this ridge, the highly conserved serine 197 appears to contribute to the binding of the RNA substrate. Moreover, based on the overall high similarity between the structures of VP39 and RrmJ, we also cannot exclude the possibility that the N terminus of RrmJ might be involved in the substrate coordination. An analysis of the crystal structure of VP39 revealed that the N terminus of VP39 lies like a lid on top of the RNA binding region, thereby retaining the RNA substrate in its correct position (19). This finding implies that a certain amount of flexibility of the N terminus is necessary in order to bind and release the substrate, which is in accord with the findings that the N terminus of RrmJ is not rigid enough to crystallize in the empty state of the enzyme (6).
Both the crystal and solution structures of the 19-residue A loop were then modeled into RrmJ. In neither case was it possible to position the 2′-hydroxyl of U2552 into the active site of RrmJ without significant clashes with the surrounding protein. However, for both the crystal and NMR structures, which differ mainly in those parts of the RNA that do not contact the protein, it was possible to model the A loop into RrmJ by overlaying the U2552 nucleotide in the active site with the 5′ m7G-capped reactive nucleotide in the VP39 structure and then simply turning the whole A loop into a cleft in the surface of RrmJ. This turning positioned the phosphodiesters into the same position as the phosphodiesters of the mRNA hexamer substrate in VP39. The residues that are in closest contact with the A-loop structure in our model were found to be the active site residue K38, and the surface residues Arg34, G131, F166, R194, and S197. Except for the less-conserved G131, all amino acids are highly conserved and have been found to alter the catalytic activity of the enzyme (K38), the apparent AdoMet affinity (F166), or the apparent Km for substrate binding (R34, R194, and S197) when mutated. The finding that the A-loop modeling positions exactly those surface exposed amino acids closest to the A loop that have been found to cause increased Km values for 23S rRNA when mutated in RrmJ strongly suggested that we have indeed identified the 23S rRNA binding site in RrmJ. This model now positions the ribose of the target U2552 next to the active site Lys164 and to the reactive methyl group of AdoMet. The suggested substrate binding site in RrmJ, which is very similar to the known substrate binding site of VP39, also resembles other predicted RNA binding sites in methyltransferases. Mutagenesis studies and structural predictions of ErmC′, the 23S rRNA dimethyltransferase from Bacillus subtilis, for instance, suggested a positively charged patch consisting of three arginine residues and one threonine residue, which positions the 23S rRNA right next to the catalytic site of the protein (24). Moreover, the recently solved crystal structure of a putative RNA methyltransferase from Mycobacterium tuberculosis, Rv2118c, displays a groove lined with positively charged residues which is wide enough to fit the RNA (14).
The methylation target of RrmJ, U2552, is one of five highly conserved A-loop residues that form part of the peptidyltransferase center in the ribosome. The residues of the A loop are thought to play a role in tRNA selection and accommodation. In the context of the ribosome, U2552 is positioned on the bottom of a deep cleft and, although it is solvent accessible, it appears not to be accessible to the 23-kDa protein RrmJ. This finding raised the intriguing question about the mechanism of substrate recognition and binding by RrmJ. It was suggested that docking of the A loop might be a reversible process and that undocking is required for tRNA binding (14). Our findings that the A loop, when it adopts its solution structure, can be modeled into the substrate binding site of RrmJ and, even more importantly, can be recognized by RrmJ as a substrate in vitro support this hypothesis. This reversible undocking of the A loop from the 50S ribosome could explain how RrmJ can gain access to the A-loop structure and can methylate U2552 so late in the maturation process of the ribosome assembly. It could, furthermore, represent the rate-limiting step, which was suggested to be the reason for the low turnover rates that have been observed for RrmJ in vitro (14).
Modeling studies of the A loop into the putative substrate binding site of RrmJ showed us that a base flipping of U2552 is likely to be required for the methylation reaction. A base flip would conserve both the active site orientation of the methylated U2552 and the phosphodiester contacts, as well as prevent a clash with the extended α4 helix, which is uniquely longer in RrmJ and VP39 than in structurally related catechol O-methyltransferases (14). Base flipping was first discovered when a DNA cytosine 5-methyltransferase was cocrystallized with its DNA substrate (21). Since then, base flipping has been shown or suggested for many other DNA-modifying enzymes. Now there are several indications that base flipping is a mechanism that can occur in RNA, as well (10). Base flipping of RNA has recently been shown to be induced by the binding of initiation factor IF1 to the 30S ribosomal subunit. Here, the binding of IF1 causes the bases A1492 and A1493 of the 16S rRNA to flip out of helix 44. This flipping buries the bases in specific pockets of initiation factor 1 (9). Furthermore, tRNA- and rRNA-modifying enzymes, which make modifications within base-paired regions, have been suggested to induce base flipping as well. The recently solved structure of the tRNA pseudouridine synthase TruB, in complex with its tRNA substrate, revealed that the target base U55 flips out completely, thereby disrupting the U55-G18 base pair and, with that, the tertiary structure of the tRNA (17). This action could give misfolded tRNAs the chance to correct their tertiary structure. The target nucleotide U2552 of RrmJ forms an unusual pyrimidine-pyrimidine base pair with C2556, which gives the A loop its compact loop structure (3). A base flip would also disrupt this base pair and, possibly, the A-loop structure. Our discovery that the A loop can be used as a minimal substrate in vitro now allows us to perform crystallization experiments with RrmJ substrate complexes, which should give us more insight into the methylation events and more evidence that RrmJ is indeed the first known rRNA methyltransferase that induces base flipping.
Acknowledgments
We thank Hans Bügl for initiating this project. We thank Scott Blanchard and Joseph Puglisi for providing us with purified A loops. We thank James Bardwell for critically reading the manuscript.
National Institutes of Health grant GM065318 and a Burroughs Wellcome Fund Career Award to U.J. supported this work.
REFERENCES
- 1.Agarwalla, S., J. T. Kealey, D. V. Santi, and R. M. Stroud. 2002. Characterization of the 23S rRNA m5U1939 methyltransferase from Escherichia coli. J. Biol. Chem. 277:8835-8840. [DOI] [PubMed] [Google Scholar]
- 2.Ban, N., P. Nissen, J. Hansen, P. B. Moore, and T. A. Steitz. 2000. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289:905-920. [DOI] [PubMed] [Google Scholar]
- 3.Blanchard, S. C., and J. D. Puglisi. 2001. Solution structure of the A loop of 23S ribosomal RNA. Proc. Natl. Acad. Sci. USA 98:3720-3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brimacombe, R., P. Mitchell, M. Osswald, K. Stade, and D. Bochkariov. 1993. Clustering of modified nucleotides at the functional center of bacterial ribosomal RNA. FASEB J. 7:161-167. [DOI] [PubMed] [Google Scholar]
- 5.Brunger, A. T., P. D. Adams, G. M. Clore, W. L. DeLano, P. Gros, R. W. Grosse-Kunstleve, J. S. Jiang, J. Kuszewski, M. Nilges, N. S. Pannu, R. J. Read, L. M. Rice, T. Simonson, and G. L. Warren. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. Sect. D 54:905-921. [DOI] [PubMed] [Google Scholar]
- 6.Bügl, H., E. B. Fauman, B. L. Staker, F. Zheng, S. R. Kushner, M. A. Saper, J. C. Bardwell, and U. Jakob. 2000. RNA methylation under heat shock control. Mol. Cell 6:349-360. [DOI] [PubMed] [Google Scholar]
- 7.Caldas, T., E. Binet, P. Bouloc, A. Costa, J. Desgres, and G. Richarme. 2000. The FtsJ/RrmJ heat shock protein of Escherichia coli is a 23 S ribosomal RNA methyltransferase. J. Biol. Chem. 275:16414-16419. [DOI] [PubMed] [Google Scholar]
- 8.Caldas, T., E. Binet, P. Bouloc, and G. Richarme. 2000. Translational defects of Escherichia coli mutants deficient in the Um2552 23S ribosomal RNA methyltransferase RrmJ/FTSJ. Biochem. Biophys. Res. Commun. 271:714-718. [DOI] [PubMed] [Google Scholar]
- 9.Carter, A. P., W. M. Clemons, Jr., D. E. Brodersen, R. J. Morgan-Warren, T. Hartsch, B. T. Wimberly, and V. Ramakrishnan. 2001. Crystal structure of an initiation factor bound to the 30S ribosomal subunit. Science 291:498-501. [DOI] [PubMed] [Google Scholar]
- 10.Cheng, X., and R. M. Blumenthal. 2002. Cytosines do it, thymines do it, even pseudouridines do it—base flipping by an enzyme that acts on RNA. Structure 10:127-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curran, J. F. 1998. Modified nucleotides in translation, p. 493-516. In H. Grosjean and R. Benne (ed.), Modification and editing of RNA. ASM Press, Washington, D.C.
- 12.Green, R., and H. F. Noller. 1996. In vitro complementation analysis localizes 23S rRNA posttranscriptional modifications that are required for Escherichia coli 50S ribosomal subunit assembly and function. RNA 2:1011-1021. [PMC free article] [PubMed] [Google Scholar]
- 13.Grosjean, H., G. Bjork, and B. E. H. Maben. 1995. Nucleotide modification and base conversion of RNA: summary and outlook. Biochimie 77:3-6.7541253 [Google Scholar]
- 14.Gupta, A., P. H. Kumar, T. K. Dineshkumar, U. Varshney, and H. S. Subramanya. 2001. Crystal structure of Rv2118c: an AdoMet-dependent methyltransferase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol. 312:381-391. [DOI] [PubMed] [Google Scholar]
- 15.Gustafsson, C., and B. C. Persson. 1998. Identification of the rrmA gene encoding the 23S rRNA m1G745 methyltransferase in Escherichia coli and characterization of an m1G745-deficient mutant. J. Bacteriol. 180:359-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hager, J., Staker, B. L., Bügl, H., and Jakob, U. 2002. Active site in RrmJ, a heat shock induced methyltransferase. J. Biol. Chem. 277:41978-41986. [DOI] [PubMed] [Google Scholar]
- 17.Hoang, C., and A. R. Ferre-D'Amare. 2001. Cocrystal structure of a tRNA Psi55 pseudouridine synthase: nucleotide flipping by an RNA-modifying enzyme. Cell 107:929-939. [DOI] [PubMed] [Google Scholar]
- 18.Hodel, A. E., P. D. Gershon, and F. A. Quiocho. 1998. Structural basis for sequence-nonspecific recognition of 5′-capped mRNA by a cap-modifying enzyme. Mol. Cell 1:443-447. [DOI] [PubMed] [Google Scholar]
- 19.Hodel, A. E., P. D. Gershon, X. Shi, and F. A. Quiocho. 1996. The 1.85 A structure of vaccinia protein VP39: a bifunctional enzyme that participates in the modification of both mRNA ends. Cell 85:247-256. [DOI] [PubMed] [Google Scholar]
- 20.Kim, D. F., and R. Green. 1999. Base-pairing between 23S rRNA and tRNA in the ribosomal A site. Mol. Cell 4:859-864. [DOI] [PubMed] [Google Scholar]
- 21.Klimasauskas, S., S. Kumar, R. J. Roberts, and X. Cheng. 1994. HhaI methyltransferase flips its target base out of the DNA helix. Cell 76:357-369. [DOI] [PubMed] [Google Scholar]
- 22.Lövgren, J. M., and P. M. Wikström. 2001. The rlmB gene is essential for formation of Gm2251 in 23S rRNA but not for ribosome maturation in Escherichia coli. J. Bacteriol. 183:6957-6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Madsen, C. T., J. Mengel-Jorgensen, F. Kirpekar, and S. Douthwaite. 2003. Identifying the methyltransferases for m5U747 and m5U1939 in 23S rRNA using MALDI mass spectrometry. Nucleic Acids Res. 31:4738-4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maravic, G., J. M. Bujnicki, M. Feder, S. Pongor, and M. Flogel. 2003. Alanine-scanning mutagenesis of the predicted rRNA-binding domain of ErmC′ redefines the substrate-binding site and suggests a model for protein-RNA interactions. Nucleic Acids Res. 31:4941-4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martz, E. 2002. Protein Explorer: easy yet powerful macromolecular visualization. Trends Biochem. Sci. 27:107-109. [DOI] [PubMed] [Google Scholar]
- 26.Pintard, L., J. M. Bujnicki, B. Lapeyre, and C. Bonnerot. 2002. MRM2 encodes a novel yeast mitochondrial 21S rRNA methyltransferase. EMBO J. 21:1139-1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pintard, L., D. Kressler, and B. Lapeyre. 2000. Spb1p is a yeast nucleolar protein associated with Nop1p and Nop58p that is able to bind S-adenosyl-l-methionine in vitro. Mol. Cell. Biol. 20:1370-1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pintard, L., F. Lecointe, J. M. Bujnicki, C. Bonnerot, H. Grosjean, and B. Lapeyre. 2002. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J. 21:1811-1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porse, B. T., and R. A. Garrett. 1995. Mapping important nucleotides in the peptidyl transferase centre of 23 S rRNA using a random mutagenesis approach. J. Mol. Biol. 249:1-10. [DOI] [PubMed] [Google Scholar]
- 30.Rozenski, J., P. F. Crain, and J. A. McCloskey. 1999. The RNA Modification Database: 1999 update. Nucleic Acids Res. 27:196-197. [DOI] [PMC free article] [PubMed] [Google Scholar]