

Abstract
Zinc is an essential nutrient for all forms of life. Within cells, most zinc is bound to protein. Because zinc serves as a catalytic or structural cofactor for many proteins, cells must maintain zinc homeostasis under severely zinc-deficient conditions. In yeast, the transcription factor Zap1 controls the expression of genes required for uptake and mobilization of zinc, but to date the fate of existing zinc-binding proteins under zinc starvation remains poorly understood. Autophagy is an evolutionarily conserved cellular degradation/recycling process in which cytoplasmic proteins and organelles are sequestered for degradation in the vacuole/lysosome. In this study, we investigated how autophagy functions under zinc starvation. Zinc depletion induced non-selective autophagy, which is important for zinc-limited growth. Induction of autophagy by zinc starvation was not directly related to transcriptional activation of Zap1. Instead, TORC1 inactivation directed zinc starvation-induced autophagy. Abundant zinc proteins, such as Adh1, Fba1, and ribosomal protein Rpl37, were degraded in an autophagy-dependent manner. But the targets of autophagy were not restricted to zinc-binding proteins. When cellular zinc is severely depleted, this non-selective autophagy plays a role in releasing zinc from the degraded proteins and recycling zinc for other essential purposes.
Keywords: autophagy, metal, TOR complex (TORC), yeast, zinc, starvation
Introduction
Autophagy is a conserved process in which cytoplasmic proteins and organelles are sequestered for degradation within the vacuole/lysosome (1, 2). During autophagy, cytoplasmic components are enwrapped by autophagosomes and delivered to the vacuole, where the contents are degraded by various hydrolytic enzymes, including proteases, lipases, nucleases, phosphatases, and glucosidases. The resultant compounds are transported out of the vacuole and recycled for use in biosynthesis and energy production. Autophagy plays important roles in multiple cellular processes (3). Autophagy occurs constitutively at low levels, serving as an important means for quality control by removing excessive or damaged proteins and organelles. Nutrient starvation significantly stimulates autophagy, contributing to cell survival by recycling constituents and maintaining energy levels (4). Several kinds of nutrient starvation stimulate autophagy, which is thought to provide nutrients from internal reservoirs when external nutrients are unavailable. Although bulk degradation via autophagy is principally a non-selective process, selective targeting of proteins and organelles by autophagy also plays important roles in cellular physiology (5).
To respond appropriately to environmental conditions, cells must respond to nutrient signals and make appropriate changes in their metabolism. In the face of severe depletion of several nutrients, cells arrest at the G1 stage of the cell cycle and remain in a quiescent state (6). In yeast, various forms of nutrient depletion also trigger autophagy (7). Nitrogen starvation is one of the strongest of such stimuli, and autophagy is indispensable for recycling intracellular amino acids. Target of rapamycin complex 1 (TORC1) negatively regulates this process by phosphorylating Atg13, which plays a key role in inducing autophagy (8). In response to nitrogen deprivation, TORC1 is inactivated, resulting in dephosphorylation of Atg13. Dephosphorylated Atg13 forms a complex with Atg1 and its regulatory complex, Atg17-Atg29-Atg31 (9), which is thought to trigger autophagosome formation (10, 11). Previous reports showed that depletion of glucose, sulfate, and several amino acids also induces autophagy (7, 12). Other forms of nutrient starvation or stress might also invoke autophagy, but the mechanism and physiological roles of these diverse nutritional perturbations remain elusive.
Transition metal ions such as iron, copper, zinc, and manganese are essential for life in all organisms. Most cellular zinc is tightly bound to proteins; consequently, the level of free zinc is predicted to be quite low. In most organisms, more than 5% of proteins bind zinc. Zinc serves as a catalytic and/or structural cofactor for many proteins. These include DNA or RNA polymerases, ribosomal proteins, transcription factors harboring zinc finger domains, central metabolic enzymes such as alcohol dehydrogenases, superoxide dismutase, and metalloproteases (13). Given that zinc is widely incorporated into essential proteins, abnormal zinc homeostasis causes serious problems for the cell.
Cells must maintain zinc homeostasis in the face of fluctuations in zinc availability. Pioneering work by Eide and co-workers (14) in budding yeast, Saccharomyces cerevisiae, revealed the mechanisms of transcriptional control under zinc-depleted conditions. To overcome zinc deficiency, the transcription factor Zap1 activates its own transcription and controls the expression of genes needed for uptake and mobilization of zinc. However, the fate of existing zinc-binding proteins upon zinc starvation remains poorly understood. Recently, the same group performed a systematic analysis that revealed genes that are essential or important during zinc starvation (15). Among them, many ATG genes were identified, but the exact mechanism by which autophagy helps cells tolerate zinc starvation remains to be elucidated. Here, we investigated the function of autophagy under zinc starvation.
Results
Autophagy is induced during zinc starvation
To determine the relationship between zinc homeostasis and autophagy, we analyzed the growth of cells in synthetic defined medium (SD),3 lacking zinc [SD(−zinc)]. To minimize metabolic requirements, in this work we used strains derived from the prototrophic wild-type strain X2180. Cells were grown in SD to mid-log phase (optical density [OD600] about 1) and then transferred to SD or SD(−zinc) at an OD600 of 0.05. When cultured in SD(−zinc), cells grew slowly for about six generations, and then stopped growing. The observation that zinc was a limiting factor for cell growth is consistent with the fact that zinc provides structural and catalytic cofactors for many essential proteins.
Next, we compared the growth of the wild-type strain and autophagy-defective atg2Δ cells. In standard zinc-replete conditions (SD), atg2Δ cells grew at nearly the same rate as the wild-type strain. However, under zinc-free conditions (SD(−zinc)) impairment of growth was more severe in atg2Δ than in the wild type. As zinc starvation continued (>15 h), cell growth of atg2Δ cells began to slow further, and these cells plateaued at a lower density (Fig. 1A), suggesting that autophagy plays an important role in supporting growth under zinc-depleted conditions.
Figure 1.
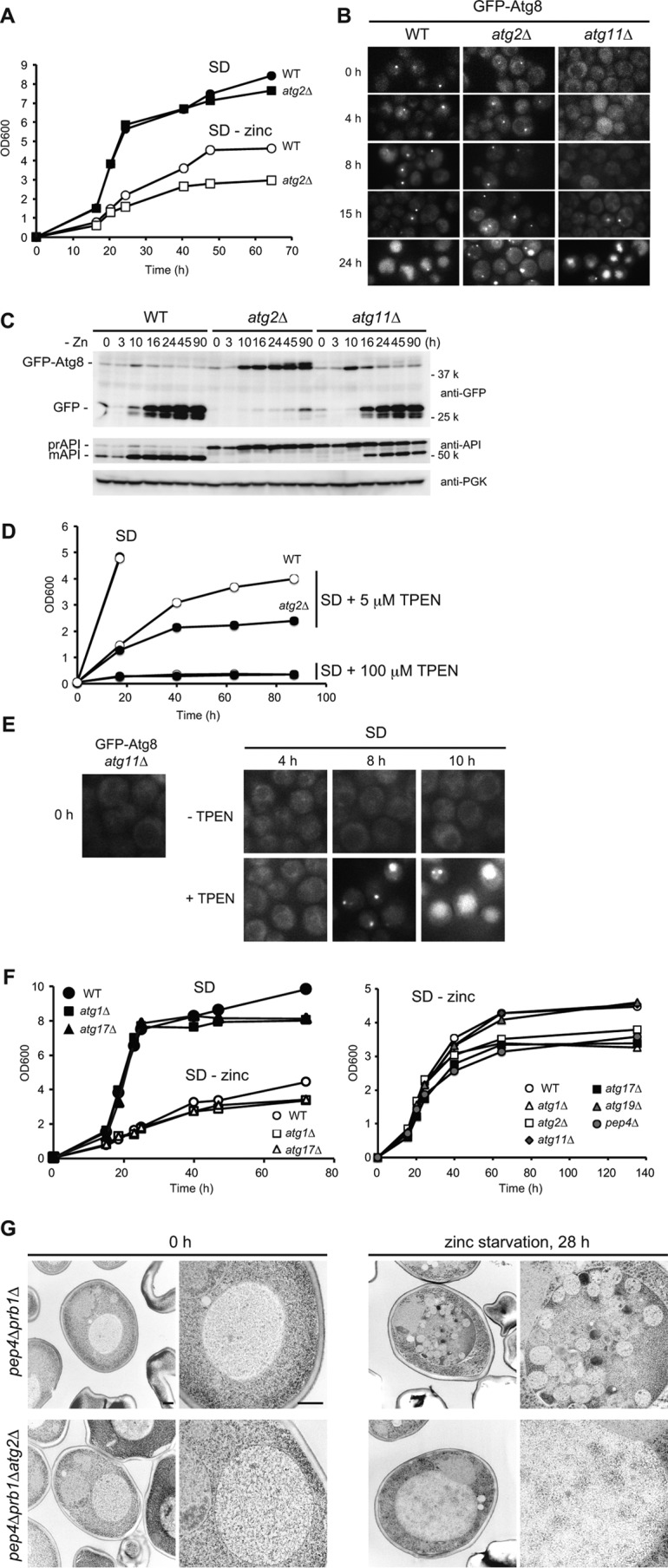
Autophagy is induced upon zinc starvation. A, growth in SD or SD(−zinc). Wild-type or atg2Δ cells were pre-grown in SD to mid-log phase (A600 =1) and then inoculated into fresh SD or SD(−zinc) at A600 = 0.05. Growth was monitored by measuring absorbance at 600 nm. B, GFP-Atg8 localization to the PAS and transport to the vacuole. Wild-type, atg2Δ, or atg11Δ cells expressing GFP-Atg8 were cultured as in A, and GFP-Atg8 was analyzed by fluorescence microscopy. C, processing of GFP-Atg8. Cells were cultured as in B. At the indicated time points, lysates were prepared and analyzed by Western blotting. D, zinc depletion by TPEN. Wild-type or atg2Δ cells were pre-grown in SD to log phase (A600 = 1), and then inoculated into fresh SD with or without TPEN (5 or 100 μm) at an initial A600 of 0.05. At the indicated time points, cell density was measured. E, microscopic observation of GFP-Atg8 expressed in atg11Δ cells in the absence (−TPEN) or presence of TPEN (+100 μm) in SD. F, growth of several types of atg mutants in SD or SD(−zinc). Wild-type, atg1Δ, atg2Δ, atg11Δ, atg17Δ, atg19Δ, or pep4Δ cells were cultured as in A. G, electron microscopic analyses. To observe autophagic bodies in the vacuoles, pep4Δprb1Δ or pep4Δprb1Δatg2Δ cells were used. The cells were cultured in SD(−zinc) for 0 or 28 h and then examined by transmission electron microscopy. Scale bar, 500 nm.
It was not clear how autophagy contributes to growth when zinc is unavailable from the environment. To determine whether autophagy was induced during zinc starvation, we performed several experiments to monitor autophagy. Standard and quantitative assays using alkaline phosphatase (pho8Δ60) as an autophagic reporter (16) were not applicable for this purpose, because zinc is required for the enzymatic activity and stability of Pho8 (see below and Ref. 17). As an alternative, we examined autophagy by fluorescence microscopy using GFP-Atg8 as a marker. Most Atg proteins, including Atg8, localize to the pre-autophagosomal structure (PAS) thought to be the site of autophagosomal formation (18). In addition to starvation-induced autophagy, the yeast Saccharomyces cerevisiae has a constitutive autophagy-like system, the Cvt pathway, for which Atg11 functions as the adaptor (19). In wild-type cells, GFP-Atg8-positive dots represent the PAS for both the Cvt pathway and autophagy, whereas in atg11Δ cells the dots represent only autophagosome formation (9). As autophagy proceeds, GFP-Atg8 is transported to the vacuole.
Using this system, we asked whether autophagy was induced by zinc starvation. In atg11Δ cells, GFP-Atg8 was not observed at the PAS after 8 h of zinc starvation but clearly localized there at 15 h. At 24 h, GFP-Atg8 clearly stained vacuoles in both wild-type and atg11Δ cells, but not in atg2Δ cells (Fig. 1B). Next, we prepared lysates from these cells and assessed the time of initiation of autophagy by immunoblot analyses. Induction of GFP-Atg8 was apparent at 10 h, and processed GFP was detected at 16 h in both wild-type and atg11Δ cells but not in atg2Δ cells (Fig. 1C), which correlated well with microscopic observations (Fig. 1B). To further validate the results, we examined another autophagy marker, API, a selective cargo for Cvt pathway during vegetative growth, for which ATG11 is essential. When autophagy is induced, pro-API is transported to the vacuole via autophagosomes, where it is processed into its mature form in an Atg17-dependent manner. In atg11Δ cells, maturation of API was not detectable before 10 h, but after 16 h mature API was clearly present. Therefore, we concluded that autophagy is induced after roughly 15–16 h in our zinc-free medium. It should be noted that, relative to nitrogen starvation, in which autophagy is promptly induced, the induction of autophagy during zinc starvation was characterized by a delay (see below).
Next, we tested the effects of a cell-permeant zinc chelator, N,N,N,N-tetrakis-[2-pyridyl-methyl]ethylenediamine (TPEN). Addition of TPEN to SD inhibited cell growth in a dose-dependent manner (Fig. 1D). High doses of TPEN (100 μm) completely inhibited growth of both wild-type and atg2Δ cells. By contrast, low doses of TPEN (5 μm) partially sustained cell growth. Under this condition, atg2Δ grew more slowly than the wild type, corresponding well to the strains' relative growth in SD(−zinc) (Fig. 1A). We next investigated whether TPEN treatment would induce autophagy by monitoring GFP-Atg8. In atg11Δ cells, GFP-Atg8 was not observed at the PAS at 4 h of TPEN treatment (Fig. 1E). However, at 8 h, GFP-Atg8 started to localize to the PAS, and after 10 h it was clearly observed within vacuoles. Compared with SD(−zinc), induction of autophagy was induced earlier, probably because the high dose of TPEN increased the severity of zinc starvation. Taken together, these findings suggest that the timing of autophagy induction in response to zinc depletion is variable, depending on the degree of cellular zinc deficiency.
We next investigated whether selective autophagy would function under zinc-free conditions. Like atg2Δ, cells deleted for ATG1 or ATG17 had growth defects (Fig. 1F). By contrast, deletion of ATG11 or ATG19, both of which are responsible for selective autophagy, had little effect on growth. Autophagic degradation requires Pep4 and Prb1, both of which are vacuolar proteases, and their absence causes accumulation of autophagic bodies in the vacuole. Similar growth defects were observed in pep4Δ cells, suggesting that degradation of autophagosome contents, rather than sequestration per se, is required to support growth. Therefore, we concluded that non-selective bulk autophagy is essential for zinc-deficient growth.
Furthermore, we performed electron microscopy to further characterize the mode of autophagy induced by zinc starvation. Many autophagic bodies accumulated in pep4Δprb1Δ, but not pep4Δprb1Δatg2Δ, after zinc starvation for 28 h (Fig. 1G). In the vacuoles of pep4Δprb1Δ cells, we observed autophagic bodies containing cytosolic ribosomes, as well as some mitochondrion-related structures. Based on the ultrastructural analyses, we concluded that non-selective autophagy is induced under zinc starvation.
Zinc depletion triggers initiation of autophagy
As mentioned above, the initiation of autophagy under zinc starvation is delayed. To investigate whether zinc starvation would result in limitation of some other nutrient that is the true trigger for autophagy, we took several approaches to determine whether induction of autophagy is induced by limitation of zinc per se.
First, we used ICP-MS analysis to measure cellular zinc and other divalent cations (iron and copper) before and after transfer to zinc-free medium. Wild-type and atg2Δ cells had similar ion content before zinc starvation (Fig. 2A, 0 h). After zinc starvation for 24 h, zinc levels dropped in both cells (Fig. 2A, 24 h), whereas the level of copper increased, and the iron level remained almost constant. Therefore, our zinc-free medium specifically decreased the cellular content of zinc but did not decrease the levels of copper and iron. Second, we measured the concentration of glucose in the media. A significant level of glucose (more than 1.5%) persisted in zinc-free media for up to 24 h, indicating that induction of autophagy was not caused by glucose limitation (Fig. 2B). Third, we added excess glucose, ammonium sulfate, amino acids, or a mixture of the latter two nitrogenous compounds to zinc-free medium and examined the influence of these nutrients on induction of autophagy. Indeed, neither GFP-Atg8 localization to the PAS nor its transport to the vacuole were affected by the presence of these nutrients (Fig. 2C, left). Furthermore, even if each nutrient was introduced after 15 h of zinc starvation, autophagy was still induced without any delay (Fig. 2C, right). Finally, we asked whether zinc starvation-induced autophagy would be abolished by supplementation with zinc. When zinc was introduced at 15 h, by which autophagy had already been induced, GFP-Atg8 localization to the PAS disappeared within 30 min (Fig. 2D). On the contrary, nitrogen starvation-induced autophagy was unresponsive to zinc supplementation (Fig. 2E). Therefore, we concluded that autophagy induced in zinc-free medium is primarily triggered by depletion of zinc per se, not due to limitation of some other nutrient, such as glucose or amino acids.
Figure 2.

Zinc starvation-induced autophagy is primarily caused by limitation of zinc itself but not of other nutrients. A, elemental analysis by ICP-MS. Wild-type and atg2Δ cells were pre-grown in SD to log phase (A600 = 1) and then inoculated into fresh SD(−zinc) at an initial A600 of 0.05. Before (left) or after 24 h of zinc starvation (right), cells were harvested, and zinc, iron, copper, and phosphorus contents were measured by ICP-MS. Wild type; white bar, atg2Δ; black bar. Note that phosphorus is presented in a different scale. Error bars represent the standard deviation. B, glucose content remaining in culture media. Wild-type or atg2Δ cells were cultured in SD(−zinc). At each time point, glucose content in the media was measured. Error bars represent the standard deviation. C–E, microscopic observation of GFP-Atg8 expressed in atg11Δ cells under zinc starvation with extra nutrients (nut). Cells were cultured in SD(−zinc) with extra nutrients (glucose, ammonium sulfate, or amino acid (aa) mixture, as indicated) for 15 or 24 h. At the indicated time points, GFP-Atg8 was monitored by fluorescence microscopy, e.g. 15 h + nut. 9 h indicates that addition of nutrient was performed after 15 h of zinc starvation and that GFP-Atg8 was observed 9 h later. D and E, microscopic observation of GFP-Atg8 localization in atg11Δ cells in response to zinc. D, zinc-starved cells (−Zn, 15 h) were supplied with 2 μm zinc for 30 min and then observed under a fluorescence microscope. E, nitrogen-starved cells (−N, 1 h) were supplied with 2 μm or 1 mm zinc for 30 min and examined as above.
Zinc starvation-induced autophagy is not directly controlled by Zap1
The transcriptional response to zinc-limited conditions has been extensively studied (14). In response to zinc deficiency, Zap1 senses cellular zinc status and regulates expression of many target genes, including ZAP1 itself. Among the targets of Zap1 are genes encoding zinc transporters, as well as factors involved in mobilization of zinc from the vacuole. Therefore, we investigated the relationship between the Zap1-mediated transcriptional response and autophagy.
In our zinc-free media, Zap1 was clearly up-regulated after 3–6 h of zinc starvation (Fig. 3A). In addition, we monitored the expression of Adh1 and Adh4, both of which are Zap1-targeted alcohol dehydrogenases. Under standard nutrient conditions, Adh1 is highly expressed and functions as a major alcohol dehydrogenase, whereas Adh4 expression is almost undetectable. Upon zinc starvation, they exhibit the opposite expression patterns: Zap1 induces expression of ADH4 but shuts off that of ADH1 (20). Given that Adh1 binds two zinc atoms, and Adh4 is predicted to use iron or alternatively one zinc atom, it is possible that cells can free zinc for other essential functions by switching isozymes (14). To explore this possibility, we examined the expression of Adh1 and Adh4. Consistent with Zap1 expression, Adh1 was down-regulated, and Adh4 was up-regulated after 6 h of zinc starvation (Figs. 3, A and B, and 5A). Deletion of ATG2 did not affect the Zap1-mediated response, indicating that autophagy does not perturb regulation of Zap1. Conversely, we investigated whether deletion of ZAP1 would affect autophagy. In zap1Δ cells, autophagy was induced earlier (4–8 h) than in wild-type cells (15 h) (Figs. 1B and 3C), as with TPEN treatment (Fig. 1E). Taken together, these observations elucidate the regulation of zinc homeostasis under zinc starvation (Fig. 3B). During the early steps of zinc starvation, cells transcriptionally up-regulate Zap1 and its target genes. Subsequently, if severely zinc-depleted conditions persist, non-selective autophagy is induced to support growth.
Figure 3.
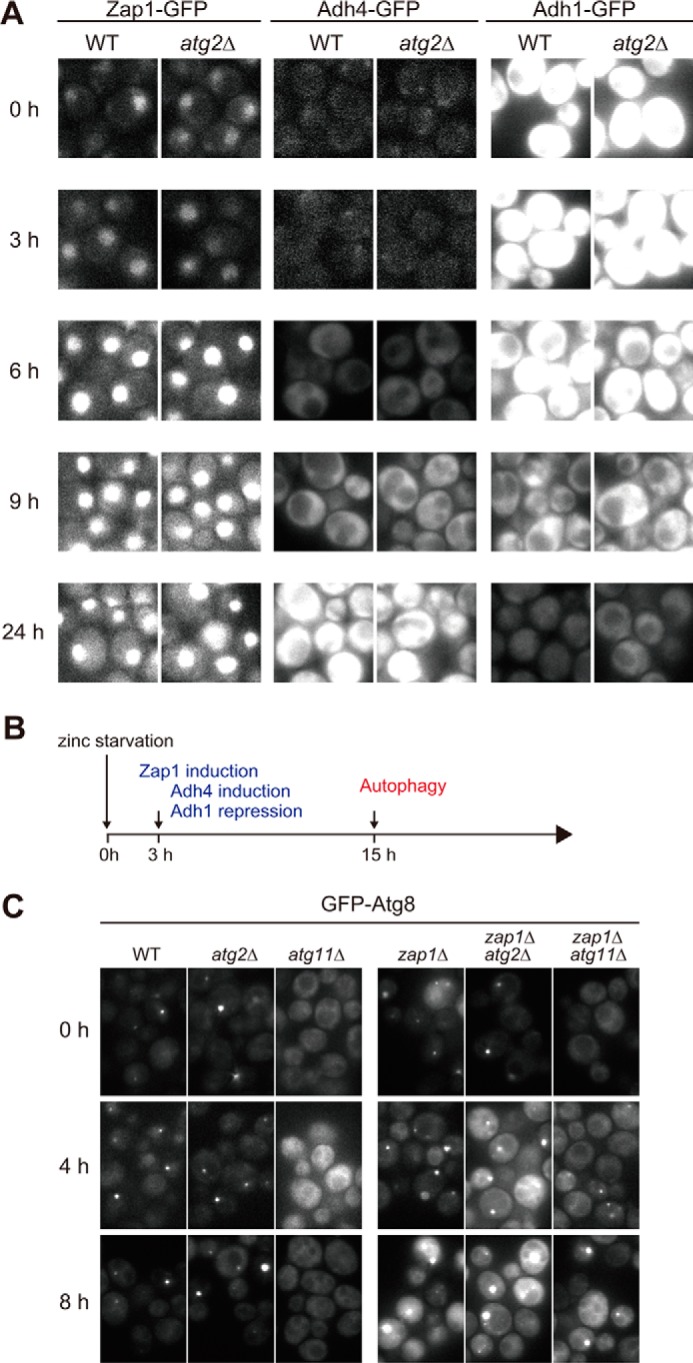
Autophagy is not directly related to Zap1 induction. A, microscopic observation of Zap1-GFP, Adh4-GFP, or Adh1-GFP in zinc-free media. Cells expressing Zap1-GFP, Adh4-GFP, and Adh1-GFP in wild-type or atg2Δ cells were cultured in SD(−zinc) for the indicated times and then observed by fluorescence microscopy. B, time course of Zap1 induction and autophagy following zinc starvation. C, microscopic observation of GFP-Atg8 in the absence of Zap1. Wild-type, atg2Δ, atg11Δ, zap1Δ, zap1Δatg2Δ, and zap1Δatg11Δ cells expressing GFP-Atg8 were transferred to SD(−zinc) at time 0. At the indicated time points, GFP-Atg8 was analyzed by fluorescence microscopy.
Figure 5.

Autophagy enables cells to scavenge zinc by degrading abundant proteins. A–C, cells in 0.5-ml aliquots of SD (−zinc) were collected at the indicated time points. Note that as equivalent volumes of cell culture were sampled for this experiment, protein loading increases with each time point as cell density increases with growth. Therefore, equivalent band intensities of Adh1 actually represent the degradation of this protein over time. A, amount of Adh1-GFP or Adh4-GFP in wild-type or atg2Δ cells under zinc starvation. 0.5-ml aliquots were collected at the indicated time points from cell cultures. Lysates were prepared and analyzed by Western blotting. B, amount of Fba1-GFP or Pgk1-GFP in wild-type or atg2Δ cells under zinc starvation. Western blotting was performed as in A. C, amount of Rpl37A-GFP, Rpl37B-GFP, or Pho8 in wild-type or atg2Δ cells under zinc starvation. Western blotting was performed as in A.
TORC1 is inactivated under zinc starvation
Given that autophagy was not directly linked to Zap, we sought to identify the signal that induces autophagy upon zinc starvation. First, we examined the phosphorylation of Atg13, with or without zinc, as a function of TORC1 activity. The phosphorylation/dephosphorylation of Atg13 is generally estimated by assessing the mobility shift of the protein by immunoblotting (8). Up to 7 h of zinc starvation, Atg13 migration was slow, but after 15 h the migration of the protein was strikingly faster (Fig. 4A). Phosphatase treatment diminished the slower migrating (0 h) and faster migrating (15 h) forms of Atg13 (Fig. 4B). These results indicate that the observed mobility shift is caused by phosphorylation and that Atg13 is hypo-phosphorylated after 15 h of zinc starvation. When zinc was supplied to zinc-starved cells, Atg13 returned to the hyperphosphorylated state within 5 min (Fig. 4C). Furthermore, cycloheximide did not affect the recovery of phosphorylation, indicating that the hyperphosphorylation was not due to de novo synthesis of Atg13. To confirm this observation, we investigated Sch9, a physiological substrate of TORC1. Zinc starvation for 15 h caused dephosphorylation of Sch9 (Fig. 4D). Moreover, as with Atg13, this modification was quickly reversed following the addition of zinc (Fig. 4D). To validate that TORC1 inactivation is involved in the induction of autophagy, we performed an experiment using a hyperactive Tor1 mutant, TORL2134M. It was previously shown that TOR1L2134M, which has a point mutation within the kinase domain, significantly delayed inactivation of TORC1 under nitrogen limitation, as judged by phosphorylation of Sch9 (21). We therefore asked whether autophagy is blocked or delayed in TOR1L2134M cells under zinc starvation. We examined autophagy using TOR1WT and TOR1L2134M cells at 0, 14, and 23 h of zinc starvation and found that TOR1L2134M cells showed delayed autophagic activity compared with TOR1WT cells (Fig. 4E). Furthermore, TOR1L2134M cells showed reduced extent of autophagy. Therefore, it is likely that TORC1 inactivation triggers zinc starvation-induced autophagy via dephosphorylation of Atg13.
Figure 4.

Regulation of zinc starvation-induced autophagy by TORC1. A–D, phosphorylation status of Atg13 and Sch9 under zinc starvation conditions. A, cells expressing Atg13 tagged with 6×HA were cultured in SD(−zinc). At the indicated time points, lysates were prepared, and the migration of Atg13 was monitored by Western blotting using an anti-HA antibody. B, lysates of cells cultured in SD(−zinc) for 0 or 15 h were incubated with or without λ-protein phosphatases (PPase). C, zinc-starved cells (−zinc 15 h) were supplied with 2 μm zinc for 5 or 10 min in the presence or absence of cycloheximide (CHX) (10 μg/ml). D, cells expressing Sch9 tagged with 6×HA were analyzed as in C. E, processing of GFP-Atg8 in atg11Δ cells expressing a Tor1 hyperactive mutant (TOR1L2134M) or Tor1WT under zinc starvation for 0, 14, and 24 h.
Zinc proteins are degraded by autophagy
To date, the effect of transcriptional shut off of ADH1 by Zap1 on Adh1 protein level has not been investigated in detail. Because Adh1 is one of the most abundant zinc proteins, it serves as an effective reservoir of zinc, which could be exploited by the cell when zinc is required. To determine whether Adh1 is degraded by autophagy, we constructed wild-type and atg2Δ cells chromosomally expressing Adh1-GFP under the control of its own promoter. To chase Adh1-GFP, we monitored the amounts of Adh1-GFP before and after zinc starvation in 0.5-ml aliquots of cell culture. In wild-type cells, Adh1-GFP was subjected to degradation after 15 h of starvation (Fig. 5A), and after 48 h about half of Adh1-GFP was degraded. In atg2Δ cells, however, the amount of Adh1-GFP was unchanged. To validate this result, we monitored the level of endogenous Adh1 using anti-Adh1 antibody (Fig. 5, A–C). As with Adh1-GFP, a significant amount of Adh1 was degraded in an autophagy-dependent manner (Fig. 5, A–C). Next, we investigated Adh4-GFP. Although expression of ADH4 was positively regulated by Zap1 (Fig. 3A), a significant portion of Adh4-GFP was inevitably targeted to degradation via autophagy.
We hypothesized that zinc-binding proteins may be preferential targets for destruction via autophagy. To explore this idea, we monitored two abundant glycolytic proteins, Fba1 and Pgk1, the former of which is a zinc protein. We constructed wild-type and atg2Δ cells chromosomally expressing Fba1-GFP and Pgk1-GFP under the control of the native promoters. Not only Fba1-GFP but also Pgk1-GFP were degraded by autophagy (Fig. 5B). Importantly, the kinetics of degradation of these proteins were almost identical, suggesting that zinc starvation-induced autophagy does not specifically target zinc-binding proteins.
Our EM analyses showed that large quantities of ribosomes were sequestered in autophagic bodies (Fig. 1G). Ribosomes are highly abundant structures, and cellular ribosome contents correlate closely with growth rate. Structural analyses of ribosomes revealed that each ribosome binds at least eight zinc ions (22), indicating that ribosomes are a potentially rich source of zinc. Accordingly, we investigated whether ribosomal zinc proteins were degraded by autophagy. To this end, we focused on Rpl37, which has been structurally validated as a zinc finger protein (13, 23). Because yeast has two genes encoding L37, RPL37A and RPL37B, we tagged both proteins with GFP. Both proteins were also degraded by autophagy (Fig. 5C). Based on these findings, we speculate that by degrading major zinc-binding proteins and ribosomes via autophagy, cells can liberate zinc for other essential uses.
To further test the idea that autophagy increases the bioavailability of zinc under severe starvation, we focused on Pho8, a vacuolar phosphatase/nucleotidase (24, 25). Previous studies revealed that zinc status controls Pho8 accumulation and activity (17); thus, we asked whether autophagy-derived zinc would protect Pho8 from degradation under zinc-free conditions. To answer this question, we compared the level of Pho8 in wild-type and atg2Δ cells. Levels of Pho8 were similar between wild-type and atg2Δ cells after 15 h of zinc starvation. After 48 h of zinc starvation, however, atg2Δ cells contained lower levels of Pho8 than wild-type cells (Fig. 5C). This observation suggests that autophagy maintains the stability of Pho8 by degrading existing zinc-binding proteins, liberating their zinc ions. Although we could not identify other zinc proteins whose stability and activity are highly dependent on autophagy under zinc starvation, we conclude that cells use non-selective autophagy to scavenge zinc by digesting abundant proteins and ribosomes under severe zinc starvation (see “Discussion”).
Discussion
Two-stage response enables cells to cope with zinc starvation
Because zinc is essential for the growth of all organisms, cells need to maintain zinc homeostasis when zinc availability fluctuates. Most cellular zinc is bound to protein, and excess zinc is stored in the vacuole/lysosome. In this study, we defined a two-stage response that allows cells to cope with zinc shortages (Fig. 3B). The first is the sensing of zinc deficiency and a subsequent Zap1-mediated transcriptional response, as reported previously (26). As cells are exposed to zinc starvation, Zap1 activates expression of genes needed for zinc uptake and mobilization of zinc from the vacuole. In addition, Zap1 represses ADH1 and instead induces ADH4 expression. Following activation of Zap1, cells can grow for several generations, although cellular zinc levels inevitably decrease as cell division proceeds. When cellular zinc levels drop below critical levels, cells enter the second stage of the response, which involves the induction of autophagy (Fig. 1). By degrading existing proteins, autophagy enables the cell to scavenge zinc during zinc starvation. Indeed, cell growth was compromised in cells lacking any of the ATG genes required for bulk autophagy (Fig. 1, A and F). We assume that by degrading existing proteins, autophagy liberates zinc for other essential functions. We showed that whereas induction of autophagy is not directly related to Zap1, deletion of ZAP1 causes earlier limitation of zinc, leading to accelerated induction of autophagy (Fig. 3C). A similar effect was observed following TPEN treatment (Fig. 1E). These observations demonstrate that the timing of autophagy induction in response to zinc starvation is strictly regulated by cellular zinc content.
Signals linking zinc starvation and TORC1
We provided evidence that TORC1 inactivation is linked to zinc starvation-induced autophagy (Fig. 4). How then does zinc starvation transmit a signal to TORC1? Although TORC1 components (Tor1, Tor2, Kog1, Tco89, and Lst8) in yeast are not known to be zinc-binding proteins, regulation of mTORC1 by zinc has been reported in mammals (27, 28). Therefore, it is possible that zinc starvation directly inhibits TORC1 activity. Another possibility is that zinc starvation indirectly inactivates TORC1 via factor(s) upstream of TORC1. If so, some alternative protein or metabolic change may regulate TORC1 activity. Further studies are needed to clarify the molecular mechanism by which zinc starvation inactivates TORC1.
Timing, mode, and magnitude of zinc starvation-induced autophagy
As discussed above, we revealed that non-selective autophagy during zinc starvation is characterized by a delay in induction (Fig. 1C). No such delay is observed for nitrogen starvation-induced autophagy. This likely reflects the fact that cellular zinc pools can suppress autophagy for some period of time. However, once autophagy is triggered, the magnitude of autophagy induced by zinc starvation seems to be as high as that induced by nitrogen starvation, as assessed by GFP-Atg8 cleavage assay and EM analyses (Fig. 1, C and G). We also showed that autophagy induced by zinc starvation is not restricted to zinc-binding proteins. As bulk autophagy proceeds, a significant amount of zinc ions would be released, primarily from the most abundant zinc proteins, such as the ribosomal proteins Adh1 and Fba1 (Fig. 5). Because cellular ribosome content correlates closely with growth rate, excess ribosomes are unnecessary under zinc starvation. Thus, autophagy helps the cell re-arrange its components in a manner optimized for zinc-limited growth.
Importantly, we found that Pho8 levels were clearly reduced in autophagy-deficient cells under zinc starvation (Fig. 5C). Given that the stability of Pho8 is regulated by zinc, this result suggests that autophagy protects Pho8 from degradation by increasing the bioavailability of zinc. Similarly, we anticipate that autophagy would ensure appropriate functions of other zinc proteins essential for zinc starvation.
Comparison of zinc and other transition metals
Autophagy is often described as a recycling system for proteins. Here, we propose that autophagy also functions as a recycling system for zinc. In the current version of the Saccharomyces Genome Database, ∼300 proteins are listed as zinc ion binding, and these zinc proteins are widely distributed and localized throughout cells. In this regard, induction of non-selective autophagy and recycling of zinc is reasonable from a nutritional point of view. In contrast to zinc proteins, the number of iron- and copper-binding proteins is relatively small, and most of these proteins are mitochondrial; nonetheless, we wondered whether depletion of other transition metals, such as iron and copper, would also induce autophagy. In contrast to the situation with zinc, however, we are unable to observe autophagy in SD(−iron) or SD(−copper) conditions (data not shown). At present, we do not know why zinc starvation in particular induces autophagy. However, in the accompanying paper (36), we report an intimate relationship between autophagy and iron.
Previous work in mammalian cells has indicated that the zinc chelator TPEN inhibits basal, H2O2-induced (29), tamoxifen-induced (30), and ethanol-induced autophagy (31), and that zinc supplementation stimulates autophagic activity (32). The reason for these apparent discrepancies between yeast and mammalian cells in response to zinc starvation is not clear. One possible explanation is that machinery essential for autophagy in mammals, such as the Atg proteins, is more sensitive to zinc depletion than that of yeast. In this study, in yeast we followed autophagy under severe zinc depletion. Further studies are required to understand how zinc controls autophagy in other organisms under various physiological conditions and their molecular details.
Experimental procedures
Yeast strains and media
Yeast strains used in this study are listed in Table 1. Strains were generated using one-step gene disruption or replacement methods as described previously (33, 34). All deletion and epitope-tagged strains constructed in this study were validated by PCR. Cells were grown in minimal synthetic defined medium (SD; 0.17% yeast nitrogen base without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose). Yeast nitrogen base was prepared according to Wickerham and Burton's formulation (37). SD(−zinc) was identical to SD except that it lacked zinc. SD(−N) was the same as SD but lacked ammonium sulfate. For Fig. 2C, supplementation with 3× Glu and 3× N represents 6% glucose and 1.5% ammonium sulfate, respectively. Supplementation with 3× amino acids represents 3× Yeast Synthetic Drop-out Medium supplements without uracil (Sigma, Y1501). All media used were buffered with 50 mm MES/KOH. TPEN (Sigma) was used as a zinc chelator.
Table 1.
Yeast strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| X2180-1B | MATα SUC2 mal mel gal2 CUP1 | Yeast Genetic Stock Center |
| MMY2 | MATα SUC2 mal mel gal2 CUP1 atg1Δ::kanMX6 | This study |
| MMY3 | MATα SUC2 mal mel gal2 CUP1 atg2Δ::kanMX6 | 25 |
| MMY7 | MATα SUC2 mal mel gal2 CUP1 atg11Δ::kanMX6 | 25 |
| MMY9 | MATα SUC2 mal mel gal2 CUP1 atg17Δ::kanMX6 | 25 |
| MMY10 | MATα SUC2 mal mel gal2 CUP1 atg19Δ::kanMX6 | 25 |
| MMY46 | MATα SUC2 mal mel gal2 CUP1 pep4Δ::zeoNT3 | This study |
| MMY98 | MATα SUC2 mal mel gal2 CUP1 atg8Δ::GFP-ATG8::hphNT1 | 25 |
| MMY104 | MATα SUC2 mal mel gal2 CUP1 atg8Δ::GFP-ATG8::hphNT1 atg2Δ::kanMX6 | 25 |
| MMY108 | MATα SUC2 mal mel gal2 CUP1 atg8Δ::GFP-ATG8::hphNT1 atg11Δ::kanMX6 | 25 |
| TMK971 | MATα SUC2 mal mel gal2 CUP1 pep4Δ::kanMX6 prb1Δ::hphNT1 | 25 |
| MMY156 | MATα SUC2 mal mel gal2 CUP1 pep4Δ::zeoNT3 prb1Δ::hphNT1 atg2Δ::kanMX6 | 25 |
| TMK993 | MATα SUC2 mal mel gal2 CUP1 ZAP1-GFP::kanMX6 | This study |
| MMY245 | MATα SUC2 mal mel gal2 CUP1 ZAP1-GFP::kanMX6 atg2Δ::natNT2 | This study |
| TMK979 | MATα SUC2 mal mel gal2 CUP1 ADH1-GFP::kanMX6 | This study |
| TMK981 | MATα SUC2 mal mel gal2 CUP1 ADH1-GFP::kanMX6 atg2Δ::hghNT1 | This study |
| TMK987 | MATα SUC2 mal mel gal2 CUP1 ADH4-GFP::kanMX6 | This study |
| TMK989 | MATα SUC2 mal mel gal2 CUP1 ADH4-GFP::kanMX6 atg2Δ::hghNT1 | This study |
| TMK811 | MATα SUC2 mal mel gal2 CUP1 ATG13–6HA::kanMX4 | This study |
| TMK830 | MATα SUC2 mal mel gal2 CUP1 FBA1-GFP::kanMX6 | This study |
| TMK854 | MATα SUC2 mal mel gal2 CUP1 FBA1-GFP::kanMX6 atg2Δ::hghNT1 | This study |
| TMK834 | MATα SUC2 mal mel gal2 CUP1 PGK1-GFP::kanMX6 | This study |
| TMK858 | MATα SUC2 mal mel gal2 CUP1 PGK1-GFP::kanMX6 atg2Δ::hghNT1 | This study |
| MMY476 | MATα SUC2 mal mel gal2 CUP1 SCH9–6HA::kanMX4 | This study |
| MMY469 | MATα SUC2 mal mel gal2 CUP1 atg8Δ::GFP-ATG8::hphNT1 zap1Δ::natNT2 | This study |
| MMY472 | MATα SUC2 mal mel gal2 CUP1 atg8Δ::GFP-ATG8::hphNT1 atg2Δ::kanMX6 zap1Δ::natNT2 | This study |
| MMY473 | MATα SUC2 mal mel gal2 CUP1 atg8Δ::GFP-ATG8::hphNT1 atg11Δ::kanMX6 zap1Δ::natNT2 | This study |
| SMY36 | MATα SUC2 mal mel gal2 CUP1 RPL37A-GFP::kanMX4 | This study |
| SMY41 | MATα SUC2 mal mel gal2 CUP1 RPL37A-GFP::kanMX4 atg2Δ::natNT2 | This study |
| SMY37 | MATα SUC2 mal mel gal2 CUP1 RPL37B-GFP::kanMX4 | This study |
| SMY38 | MATα SUC2 mal mel gal2 CUP1 RPL37B-GFP::kanMX4 atg2Δ::hghNT1 | This study |
| SMY253 | MATα ura3-52 TOR1 (L2134M) atg8Δ::GFP-ATG8::hphNT1 atg11Δ::kanMX6 | TS184 (21) |
| SMY254 | MATα ura3-52 atg8Δ::GFP-ATG8::hphNT1 atg11Δ::kanMX6 | TS161 (21) |
Immunoblotting
Immunoblot analyses were performed as described previously (25, 35). Samples corresponding to 0.5 A600 units of cells were separated by SDS-PAGE, followed by Western blotting. For Fig. 5, 5-ml aliquots of cell culture were collected by centrifugation at the indicated time points. One-tenth of each sample was separated as described above. Primary antibodies against GFP (Roche Applied Science), ALP, PGK (Life Technologies, Inc.-Novex), HA (3F10, Invitrogen), API, GAPDH, Adh1, and Kar2 (laboratory stock) were acquired from the indicated sources. Chemiluminescence was induced using the Femtoglow HRP substrate (Michigan Diagnostics). Images were acquired on an LAS-4000 instrument and processed using the MultiGauge software (Fujifilm Life Sciences).
Phosphatase treatment
Yeast cells were lysed with alkaline solution (0.25 N NaOH and 1% β-mercaptoethanol) for 10 min on ice. Following TCA precipitation, samples were washed once with 1 m Tris and resuspended in the phosphatase assay buffer (50 mm HEPES (pH 7.5), 100 mm NaCl, 2 mm DTT, 0.01% Brij 35, 1 mm MnCl2) and a commercial mixture of protease inhibitors (Roche Applied Science). These samples were incubated with λ-phosphatase for 30 min at 30 °C. Samples were boiled for 5 min prior to loading onto the gel.
Other procedures
Intracellular localization of proteins was examined using an inverted fluorescence microscope as described previously (25). Ultrastructural analysis of yeast cells was performed by Tokai-EMA (Japan) as described (25). Elemental analysis by ICP-MS was performed as described in Ref. 36. Measurement of cellular glucose concentration was performed using a glucose assay kit (F-kit d-glucose, J. K. International).
Author contributions
T. K. and Y. O. designed the experiments and wrote the manuscript; T. K., T. H., M. M., and M. S. performed the experiments.
Acknowledgments
We thank Dr. Tatsuya Maeda for providing strains and helpful discussions. We are grateful to members of the Ohsumi and Nakatogawa laboratories for stimulating discussions and critical comments on the manuscript. We also thank Dr. Alexander May for proofreading.
This work was supported in part by Grants-in-aid for Scientific Research 23000015 (to Y. O.) and 16H06375 (to Y. O.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
- SD
- synthetic dextrose
- PAS
- pre-autophagosomal structure
- ICP-MS
- inductively coupled plasma-mass spectrometry
- TPEN
- N,N,N,N-tetrakis-[2-pyridyl-methyl]ethylenediamine.
References
- 1. Klionsky D. J. (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 [DOI] [PubMed] [Google Scholar]
- 2. Ohsumi Y. (2014) Historical landmarks of autophagy research. Cell Res. 24, 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mizushima N. (2009) Physiological functions of autophagy. Curr. Top. Microbiol. Immunol. 335, 71–84 [DOI] [PubMed] [Google Scholar]
- 4. Rabinowitz J. D., and White E. (2010) Autophagy and metabolism. Science 330, 1344–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reggiori F., Komatsu M., Finley K., and Simonsen A. (2012) Autophagy: more than a nonselective pathway. Int. J. Cell Biol. 2012, 219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartwell L. H. (1974) Saccharomyces cerevisiae cell cycle. Bacteriol Rev. 38, 164–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Takeshige K., Baba M., Tsuboi S., Noda T., and Ohsumi Y. (1992) Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 119, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kamada Y., Funakoshi T., Shintani T., Nagano K., Ohsumi M., and Ohsumi Y. (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 150, 1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kawamata T., Kamada Y., Kabeya Y., Sekito T., and Ohsumi Y. (2008) Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol. Biol. Cell 19, 2039–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujioka Y., Suzuki S. W., Yamamoto H., Kondo-Kakuta C., Kimura Y., Hirano H., Akada R., Inagaki F., Ohsumi Y., and Noda N. N. (2014) Structural basis of starvation-induced assembly of the autophagy initiation complex. Nat. Struct. Mol. Biol. 21, 513–521 [DOI] [PubMed] [Google Scholar]
- 11. Yamamoto H., Fujioka Y., Suzuki S. W., Noshiro D., Suzuki H., Kondo-Kakuta C., Kimura Y., Hirano H., Ando T., Noda N. N., and Ohsumi Y. (2016) The intrinsically disordered protein Atg13 mediates supramolecular assembly of autophagy initiation complexes. Dev. Cell 38, 86–99 [DOI] [PubMed] [Google Scholar]
- 12. Sutter B. M., Wu X., Laxman S., and Tu B. P. (2013) Methionine inhibits autophagy and promotes growth by inducing the SAM-responsive methylation of PP2A. Cell 154, 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Regalla L. M., and Lyons T. J. (2006) in Zinc in Yeast: Mechanisms Involved in Homeostasis, Topics in Current Genetics (Tamas M. J., and Martinoia E., eds) Vol. 14, pp. 37–58, Springer, New York [Google Scholar]
- 14. Eide D. J. (2009) Homeostatic and adaptive responses to zinc deficiency in Saccharomyces cerevisiae. J. Biol. Chem. 284, 18565–18569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. North M., Steffen J., Loguinov A. V., Zimmerman G. R., Vulpe C. D., and Eide D. J. (2012) Genome-wide functional profiling identifies genes and processes important for zinc-limited growth of Saccharomyces cerevisiae. PLoS Genet. 8, e1002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Noda T., Matsuura A., Wada Y., and Ohsumi Y. (1995) Novel system for monitoring autophagy in the yeast Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 210, 126–132 [DOI] [PubMed] [Google Scholar]
- 17. Qiao W., Ellis C., Steffen J., Wu C. Y., and Eide D. J. (2009) Zinc status and vacuolar zinc transporters control alkaline phosphatase accumulation and activity in Saccharomyces cerevisiae. Mol. Microbiol. 72, 320–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki K., Kirisako T., Kamada Y., Mizushima N., Noda T., and Ohsumi Y. (2001) The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shintani T., Huang W. P., Stromhaug P. E., and Klionsky D. J. (2002) Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev. Cell 3, 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bird A. J., Gordon M., Eide D. J., and Winge D. R. (2006) Repression of ADH1 and ADH3 during zinc deficiency by Zap1-induced intergenic RNA transcripts. EMBO J. 25, 5726–5734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takahara T., and Maeda T. (2012) Transient sequestration of TORC1 into stress granules during heat stress. Mol. Cell 47, 242–252 [DOI] [PubMed] [Google Scholar]
- 22. Garreau de Loubresse N., Prokhorova I., Holtkamp W., Rodnina M. V., Yusupova G., and Yusupov M. (2014) Structural basis for the inhibition of the eukaryotic ribosome. Nature 513, 517–522 [DOI] [PubMed] [Google Scholar]
- 23. Ben-Shem A., Garreau de Loubresse N., Melnikov S., Jenner L., Yusupova G., and Yusupov M. (2011) The structure of the eukaryotic ribosome at 3.0 Å resolution. Science 334, 1524–1529 [DOI] [PubMed] [Google Scholar]
- 24. Kaneko Y., Hayashi N., Toh-e A., Banno I., and Oshima Y. (1987) Structural characteristics of the PHO8 gene encoding repressible alkaline phosphatase in Saccharomyces cerevisiae. Gene 58, 137–148 [DOI] [PubMed] [Google Scholar]
- 25. Huang H., Kawamata T., Horie T., Tsugawa H., Nakayama Y., Ohsumi Y., and Fukusaki E. (2015) Bulk RNA degradation by nitrogen starvation-induced autophagy in yeast. EMBO J. 34, 154–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ehrensberger K. M., and Bird A. J. (2011) Hammering out details: regulating metal levels in eukaryotes. Trends Biochem. Sci. 36, 524–531 [DOI] [PubMed] [Google Scholar]
- 27. Lynch C. J., Patson B. J., Goodman S. A., Trapolsi D., and Kimball S. R. (2001) Zinc stimulates the activity of the insulin- and nutrient-regulated protein kinase mTOR. Am. J. Physiol. Endocrinol Metab. 281, E25–E34 [DOI] [PubMed] [Google Scholar]
- 28. Kim S., Jung Y., Kim D., Koh H., and Chung J. (2000) Extracellular zinc activates p70 S6 kinase through the phosphatidylinositol 3-kinase signaling pathway. J. Biol. Chem. 275, 25979–25984 [DOI] [PubMed] [Google Scholar]
- 29. Lee S. J., Cho K. S., and Koh J. Y. (2009) Oxidative injury triggers autophagy in astrocytes: the role of endogenous zinc. Glia 57, 1351–1361 [DOI] [PubMed] [Google Scholar]
- 30. Hwang J. J., Kim H. N., Kim J., Cho D. H., Kim M. J., Kim Y. S., Kim Y., Park S. J., and Koh J. Y. (2010) Zinc(II) ion mediates tamoxifen-induced autophagy and cell death in MCF-7 breast cancer cell line. Biometals 23, 997–1013 [DOI] [PubMed] [Google Scholar]
- 31. Liuzzi J. P., and Yoo C. (2013) Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol. Trace Elem. Res. 156, 350–356 [DOI] [PubMed] [Google Scholar]
- 32. Liuzzi J. P., Guo L., Yoo C., and Stewart T. S. (2014) Zinc and autophagy. Biometals 27, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., and Schiebel E. (1999) Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15, 963–972 [DOI] [PubMed] [Google Scholar]
- 34. Janke C., Magiera M. M., Rathfelder N., Taxis C., Reber S., Maekawa H., Moreno-Borchart A., Doenges G., Schwob E., Schiebel E., and Knop M. (2004) A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962 [DOI] [PubMed] [Google Scholar]
- 35. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 36. Horie T., Kawamata T., Matsunami M., and Ohsumi Y. (2017) Recycling of iron via autophagy is critical for the transition from glycolytic to respiratory growth. J. Biol. Chem. 292, 8533–8543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zimbro M. J., and Power D. A., eds (2009) Difco & BBL Manual, Manual of Microbiological Culture Media, 2nd Ed., BD Diagnostics-Diagnostics Systems, Sparks, MD [Google Scholar]