

Abstract
In the fission yeast Schizosaccharomyces pombe, acquisition of exogenous heme is largely mediated by the cell membrane–associated Shu1. Here, we report that Str3, a member of the major facilitator superfamily of transporters, promotes cellular heme import. Using a strain that cannot synthesize heme de novo (hem1Δ) and lacks Shu1, we found that the heme-dependent growth deficit of this strain is rescued by hemin supplementation in the presence of Str3. Microscopic analyses of a hem1Δ shu1Δ str3Δ mutant strain in the presence of the heme analog zinc mesoporphyrin IX (ZnMP) revealed that ZnMP fails to accumulate within the mutant cells. In contrast, Str3-expressing hem1Δ shu1Δ cells could take up ZnMP at a 10-μm concentration. The yeast Saccharomyces cerevisiae cannot efficiently transport exogenously supplied hemin. However, heterologous expression of Str3 from S. pombe in S. cerevisiae resulted in ZnMP accumulation within S. cerevisiae cells. Moreover, hemin-agarose pulldown assays revealed that Str3 binds hemin. In contrast, an Str3 mutant in which Tyr and Ser residues of two putative heme-binding motifs (530YX3Y534 and 552SX4Y557) had been replaced with alanines exhibited a loss of affinity for hemin. Furthermore, this Str3 mutant failed to rescue the heme-dependent growth deficit of a hem1Δ shu1Δ str3Δ strain. Further analysis by absorbance spectroscopy disclosed that a predicted extracellular loop region in Str3 containing the two putative heme-binding motifs interacts with hemin, with a KD of 6.6 μm. Taken together, these results indicate that Str3 is a second cell-surface membrane protein for acquisition of exogenous heme in S. pombe.
Keywords: heme, membrane protein, yeast physiology, Schizosaccharomyces pombe, transporter, heme transport, major facilitator transporter, fission yeast
Introduction
Heme is a macrocycle molecule constituted of a protoporphyrin ring in which one atom of iron is coordinated at its center (1). The coordinated iron can adopt both oxidized (Fe3+) and reduced (Fe2+) states. The redox-active nature of iron makes heme a critical cofactor for a wide variety of enzymes, including cytochromes, globins, and catalases that are required in vital biochemical processes, such as respiration, oxygen transport, and disproportionation of hydrogen peroxide, respectively (2). Heme is also known to serve as a signaling molecule in cellular responses, such as antioxidant defense and regulation of the circadian clock (3, 4). Due to its critical physiological importance, a majority of organisms have evolved with different means to secure heme (5). In the case of heme prototrophs, these organisms possess an eight-step enzymatic pathway that involves anabolic enzymes that are located either in mitochondria or the cytoplasm, depending where their specific action takes place along the biosynthetic pathway. In the case of the heme biosynthesis pathway, proteins that are required for heme production have been identified in most organisms as well as the reactions they catalyze (1). A second means used by a number of organisms is to acquire heme from external environmental sources (6). As opposed to the well-characterized enzymes responsible for the synthesis of the heme molecule, knowledge of cellular components that are required for acquisition of exogenous heme is more limited and has only been investigated in a small number of organisms.
In Schizosaccharomyces pombe, heme biosynthesis and exogenous heme uptake represent two different ways to acquire heme (7). Heme biosynthesis involves eight enzymes encoded by the following genes: hem1+, hem2+, hem3+, ups1+, hem12+, hem13+, hem14+, and hem15+. The first enzyme is named Hem1 (δ-aminolevulinate acid synthase) and produces δ-aminolevulinate (ALA)3 by condensation of succinyl-CoA with glycine. ALA is then converted to porphobilinogen by a second enzyme (Hem2) of the biosynthetic pathway toward heme biosynthesis that involves six additional steps. In fission yeast, the hem1+ gene is essential because its disruption is lethal for the cells. A strategy to keep hem1Δ cells alive consists of adding exogenous ALA, allowing heme biosynthesis to resume at step 2 and then proceed further along the biosynthetic pathway until production of heme. A second way to ensure viability of hem1Δ cells is to supplement the mutant cells with exogenous hemin (heme chloride). In this case, hem1Δ cells are therefore forced to use their own heme uptake system. This experimental design (hem1Δ + hemin) selectively blocks heme biosynthesis, setting conditions to investigate the mechanisms of exogenous heme acquisition by cells.
Following this approach, studies have previously shown that S. pombe iron-starved cells produced Shu1, which is a small cell-surface heme-binding protein of 25 kDa (7). Shu1 is attached to the plasma membrane via a glycosylphophatidylinositol anchor (8). Absorbance spectroscopy and hemin-agarose pulldown experiments have demonstrated that Shu1 exhibits a constant of dissociation (KD) of 2.2 μm for hemin (7). Furthermore, in vivo functional growth assays have shown that the presence of Shu1 is required for assimilation of exogenous hemin by S. pombe hem1Δ cells (7).
Studies have shown that Shu1 is required for cellular internalization of the heme analog zinc mesoporphyrin IX (ZnMP) (7, 8). When heme biosynthesis is selectively blocked in hem1Δ mutant cells, ZnMP first accumulates into vacuoles and then within the cytosol (8). Consistent with this observation, results have shown that in response to elevated concentrations of hemin or ZnMP, Shu1 undergoes internalization from the cell surface to the vacuole (8). Disruption of the vacuolar Abc-type transporter Abc3 results in hem1Δ abc3Δ mutant cells being unable to grow in the presence of hemin as the sole iron source (8). Although the pathway whereby internalized heme or its analog ZnMP becomes available to the cells is still unclear, results have shown that Abc3 participates in the mobilization of stored heme or ZnMP from the vacuole to the cytosol (8).
Besides Shu1, Abc3 (heme assimilation), and the Fip1-Fio1 iron-dependent permease-oxidase complex (reductive iron uptake) (9), there are other S. pombe transport-related proteins that are regulated at the transcriptional level in response to changes in iron concentrations (10). For instance, Str1, Str2, and Str3 expression is induced under conditions of iron starvation and repressed under iron-replete conditions (11). Str1, Str2, and Str3 proteins are classified as members of the major facilitator superfamily (MFS) of transporters (12, 13). In the case of Str1, its heterologous expression in a Saccharomyces cerevisiae mutant strain that is deficient in high-affinity iron uptake systems results in assimilation of iron from ferrichrome (11). This result is consistent with the fact that S. pombe possesses the ability to produce ferrichrome (14) and with the plasma membrane localization of Str1 in S. pombe. In the case of Str2, its physiological role in S. pombe is still unclear, and the ORFeome global analysis of protein localization indicates that Str2 localizes to the membrane vacuole (15). Str3 displays the lowest sequence identity with Str1 and Str2 (11). Although Str3 localizes to the plasma membrane of cells (15), its heterologous expression in an iron uptake–deficient S. cerevisiae strain fails to restore growth in the presence of siderophores, including ferrichrome and ferroxiamine B (11). The biological function of Str3 remains therefore undetermined.
To further investigate the mechanisms whereby S. pombe cells acquire exogenous heme, cells lacking Hem1 and Shu1 were incubated using a range of concentrations of hemin. Although growth of hem1Δ shu1Δ cells was inhibited in the presence of 0.075 μm hemin, medium supplementation with 0.15 and 0.5 μm hemin restored the ability of hem1Δ shu1Δ cells to grow when hemin was the sole source of iron. The fact that hemin-dependent growth deficiency of hem1Δ shu1Δ cells can be overcome in a medium supplemented with high hemin concentrations (≥0.15 μm) indicated the presence of a Shu1-independent hemin uptake pathway. Here, we show that the presence of Str3 was required to suppress inhibited growth of hem1Δ shu1Δ cells in the presence of 0.15 μm exogenous hemin. As previously shown, hem1Δ cells expressing Shu1 were able to take up the heme analog ZnMP at a concentration of 2 μm. In contrast, deletion of shu1+ (shu1Δ) led to defects in the assimilation of ZnMP, unless Str3 was expressed in hem1Δ shu1Δ cells in the presence of ZnMP at a concentration of 10 μm. Inactivation of str3Δ in a hem1Δ shu1Δ strain abolished assimilation of ZnMP. Expression of an active S. pombe Str3 protein in S. cerevisiae led to cellular accumulation of ZnMP, whereas untransformed S. cerevisiae failed to take up ZnMP due to the absence of an endogenous heme transport system. Results showed that Str3 binds to hemin-agarose and exhibits an equilibrium dissociation constant (KD) value of 6.6 μm for hemin. Taken together, these results are consistent with the existence of an additional hemin acquisition pathway in S. pombe that involves the action of Str3.
Results
Effect of the shu1Δ deletion on the ability to assimilate hemin
Two strategies can be used to keep S. pombe hem1Δ cells alive. The first approach is to add ALA in cultures of hem1Δ cells (7). In this case, ALA serves as a substrate for the downstream enzyme Hem2 of the biosynthetic pathway toward heme biosynthesis. The second approach is to supplement hem1Δ cells with exogenous hemin (Fig. 1A) (7, 8). This latter approach (hem1Δ + hemin) makes sure that heme biosynthesis is selectively blocked, setting conditions to investigate the mechanisms by which external hemin is taken up by the cells. Here, hem1Δ cells that had been supplemented with 0.075, 0.15, or 0.5 μm hemin exhibited cell growth to an A600 of 2.7, 3.4, and 3.9, respectively, over a time period of 80 h (Fig. 1A). In the absence of hemin (and ALA), hem1Δ cells were unable to grow compared with hem1Δ cells that had been supplemented with exogenous hemin (Fig. 1A). As we have shown previously (7), disruption of the gene encoding the cell-surface protein Shu1 in hem1Δ cells resulted in a poor growth of these cells (hem1Δ shu1Δ) (A600 of 0.4 after 80 h) in the presence of 0.075 μm hemin (Fig. 1B). However, hem1Δ shu1Δ cells grown in the same medium supplemented with high concentrations of hemin (0.15 and 0.5 μm) exhibited a robust cell growth to an A600 of 3.4 and 4.2 that was similar to hemin-replete hem1Δ (shu1+) cells (Fig. 1, A and B). We therefore concluded that Shu1 is required for hemin acquisition when hemin is present at very low concentrations (0.075 μm), whereas its presence is dispensable under conditions of high hemin concentrations (0.15 and 0.5 μm).
Figure 1.
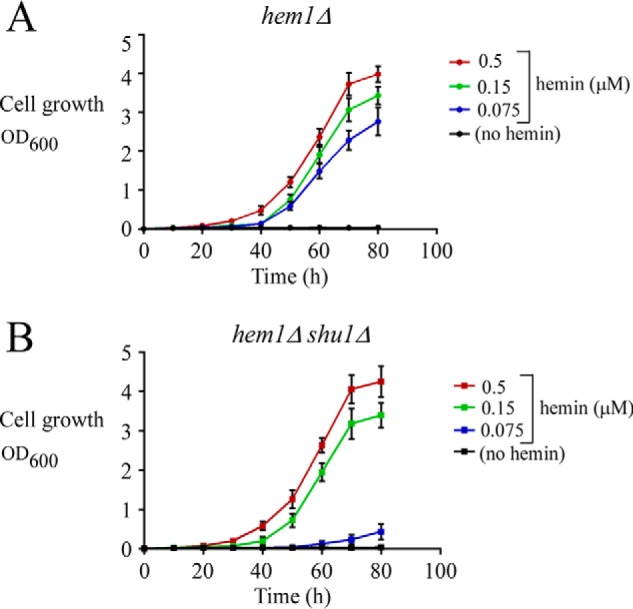
Deletion of shu1+ leads to poor growth in the presence of low hemin concentration (0.075 μm) but does not compromise cell growth with higher hemin concentrations (0.15 and 0.5 μm). A, growth of hem1Δ cells was assessed in ALA-free medium that was left untreated (no hemin; black) or supplemented with exogenous hemin. Hemin color codes are as follows. Blue, 0.075 μm; green, 0.15 μm; red, 0.5 μm. B, growth of hem1Δ shu1Δ cells was assessed under the same conditions as described for A. Values are represented as the averages ± S.D. (error bars) of a minimum of three independent experiments.
Str3 is an iron-regulated cell-surface protein that supports efficient hemin acquisition when hemin levels exceed 0.075 μm
Given the fact that hemin concentrations of 0.15 and 0.5 μm restored cell growth levels of hem1Δ shu1Δ mutant comparable with hem1Δ (shu1+) mutant, we sought to identify an additional protein that was involved in hemin acquisition. One possibility was that an additional protein involved in hemin acquisition was produced as a function of changes in iron concentrations. Its gene expression could therefore be under the control of Fep1, in a manner similar to Shu1 (7). Accordingly, its transcription would be repressed under high-iron conditions and derepressed under low-iron conditions. To further support hemin acquisition, one additional protein would have to be localized at the cell surface. In this connection, we noticed that the str3+ gene was immediately adjacent to shu1+ on chromosome I and possessed all of these characteristics (Fig. 2). Indeed, str3+ mRNA levels were repressed when cells were grown in the presence of iron. In contrast, str3+ mRNA levels were increased 5.0 ± 0.3-fold compared with basal levels observed in the untreated cells in the presence of the iron chelator 2,2′-dipyridyl (Dip) (Fig. 2B). Results showed that str3+ transcription was controlled by Fep1 because its disruption (fep1Δ) resulted in increased str3+ mRNA levels, which were unresponsive to iron for repression (Fig. 2B). In the absence of Fep1, str3+ was expressed 12.0 ± 0.5-fold compared with basal levels observed in WT untreated cells. Conversely, fep1Δ cells in which a WT fep1+ allele was reintegrated regained the ability to repress str3+ gene expression in response to iron (Fig. 2B).
Figure 2.

str3+ encodes a cell-surface protein that is biosynthetically regulated by cellular iron levels. A, schematic illustration of a genomic DNA region and location of the two neighbor str3+ and shu1+ genes. White arrows indicate the direction of gene transcription. A black double-headed arrow indicates the length of the intergenic region. B, WT (fep1+) and fep1Δ strains were either left untreated (−) or treated with Dip (250 μm) or FeCl3 (Fe; 100 μm) for 3 h. In the case of the fep1Δ strain, it was transformed with an empty vector or an integrative vector containing the WT fep1+ allele. Total RNA was prepared from each sample, and then str3+ and act1+ steady-state mRNA levels were analyzed by RNase protection assays. Results are representative of three independent experiments. C, hem1Δ shu1Δ str3Δ cells expressing str3+-GFP and shu1+-HA4 alleles were treated with Dip (250 μm) or FeCl3 (Fe; 100 μm), or they were left untreated for 3 h. Triton X-100–solubilized extracts were prepared and analyzed by immunoblotting using anti-GFP, anti-HA, or anti-α-tubulin antibodies. The positions of the molecular mass (M) of protein standards (in kDa) are indicated on each side. D, hem1Δ shu1Δ str3Δ cells expressing GFP-tagged Str3 were treated with Dip (250 μm) or FeCl3 (Fe; 100 μm) for 3 h. Cells were analyzed by fluorescence microscopy (right) for the presence of Str3-GFP. Nomarski optics (left) was used to ascertain cell morphology. E, a topological model of Str3 is shown. The gray boxes represent 12 predicted transmembrane domains, and their positions are indicated with numbers. The amino acid sequence numbers refer to the position relative to the first amino acid of Str3. The inset indicates the presence of YLREY and SLIREV amino acid residues that are located in the predicted extracellular loop 11 of Str3.
The iron-dependent regulated expression of str3+ prompted us to examine Str3-GFP protein levels in untreated cells and in cells incubated under conditions of low and high concentrations of iron. Functional str3+-GFP and shu1+-HA4 alleles expressed under the control of their own promoters were integrated into a hem1Δ shu1Δ str3Δ mutant strain. This strain was grown to mid-logarithmic phase and then incubated in ALA-free medium containing hemin (0.075 μm) and Dip (250 μm) or iron (100 μm) or in their respective absence. Results showed that the steady-state levels of Str3-GFP followed those of str3+ mRNA and were primarily detected in iron-starved cells (Fig. 2C). The Shu1-HA4 protein, which is known to be expressed under low-iron conditions (7), was used as a control in parallel experiments (Fig. 2C). Fluorescence microscopy analysis consistently showed Str3-GFP localization at the plasma membrane under conditions of iron starvation (Fig. 2D). In contrast, Str3-GFP fluorescence signal was lost when cells were incubated in the presence of iron (Fig. 2D).
Str3 is predicted to be a member of the MFS transporters (11). As commonly found in a large number of MFS proteins, a topological model of Str3 predicts the presence of 12 transmembrane spans (Fig. 2E). Transmembrane spans 1–6 form the first half of the protein, whereas transmembrane spans 7–12 constitute the second half of Str3. In the model, the two halves of Str3 are linked by the extended loop 6. This loop is predicted to be cytosolic and would allow a flexible movement between the N- and C-terminal halves of Str3. According to a predicted three-dimensional model by I-TASSER (16) and virtual docking simulation of heme to Str3, we observed the presence of a potential heme-binding pocket in the vicinity of extracellular loop 11. Furthermore, we found the presence of two putative heme-binding sequences, 530YX3Y534 and 552SX4Y557, in loop 11. Based on these observations and the fact that no experimental substrate was known for Str3, we created a hem1Δ shu1Δ str3Δ triple mutant strain to investigate whether Str3 may function in heme acquisition when the heme biosynthetic pathway was disrupted. First, hem1Δ shu1Δ str3Δ cells were incubated in the presence of ALA to verify that their growth was equivalent to hem1Δ, hem1Δ shu1Δ, and hem1Δ str3Δ strains (Fig. 3A). As a negative control, removal of ALA was lethal for a hem1Δ mutant strain (Fig. 3A). When hem1Δ and hem1Δ str3Δ cells (both strains expressing shu1+) were incubated in the absence of ALA and in the presence of 0.075 μm hemin, they exhibited a similar cell growth to A600 of 2.6 over a time period of 80 h (Fig. 3B). Under these conditions and over the same period of time, hem1Δ shu1Δ and hem1Δ shu1Δ str3Δ strains (both lacking shu1) exhibited poor growth (A600 of 0.33 and 0.29, respectively), which represented 8.0 ± 1.1-fold less growth compared with strains that expressed a functional shu1+ allele (Fig. 3B). This poor-growth phenotype was still observed when growth assays were performed using hem1Δ shu1Δ str3Δ cells containing an untagged str3+ or a GFP-tagged str3+ allele that had been reintegrated (Fig. 3B).
Figure 3.
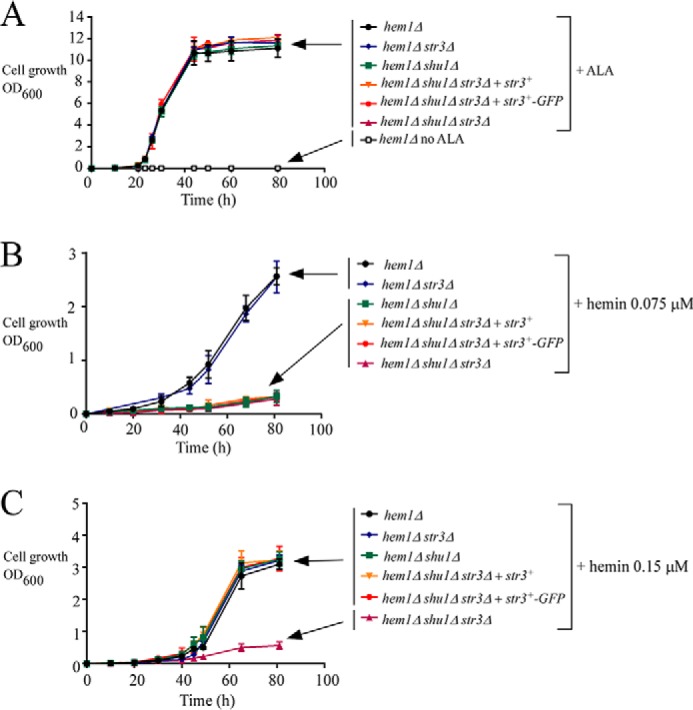
The str3+ gene is required for hemin acquisition by hem1Δ shu1Δ cells in ALA-free medium supplemented with 0.15 μm hemin. A, growth of the indicated yeast strains was assessed in YES medium that was left untreated (no ALA; open squares) or supplemented with exogenous ALA (200 μm). Strain color codes are as follows. Black, hem1Δ; blue, hem1Δ str3Δ; green, hem1Δ shu1Δ; orange, hem1Δ shu1Δ str3Δ expressing str3+; red, hem1Δ shu1Δ str3Δ expressing str3+-GFP; violet, hem1Δ shu1Δ str3Δ. B and C, growth of strains was assessed in the presence of 0.075 μm (B) or 0.15 μm (C) hemin but in the absence of ALA. Strain color codes are the same as described in A. Results are representative of three independent experiments. Values are represented as the averages ± S.D. (error bars).
We then investigated whether there was an effect of deletion of Str3 on hemin acquisition when hemin levels exceed 0.075 μm. Growth of hem1Δ shu1Δ (expressing endogenous str3+) and hem1Δ shu1Δ str3Δ cells was assessed in the presence of 0.15 μm hemin but in the absence of ALA. In the case of hem1Δ shu1Δ cells (expressing str3+), their ability to grow was restored in the presence of 0.15 μm hemin (A600 of 3.3 after 80 h), but not in the same medium supplemented with lower concentrations of hemin (0.075 μm) (A600 of 0.33 after 80 h) (Fig. 3, B and C). When growth assays were performed using hem1Δ shu1Δ str3Δ cells expressing a WT str3+ or a GFP-tagged str3+ allele in the presence of 0.15 μm hemin, results showed that these strains exhibited robust growth in a manner similar to hem1Δ shu1Δ cells expressing an endogenous str3+ (Fig. 3C). Taken together, these results revealed that Str3 supports hemin acquisition in the presence of 0.15 μm hemin, but it is not effective when hemin levels are as low as 0.075 μm.
Str3 is required for cellular assimilation of ZnMP in the absence of Shu1
To obtain further evidence that Str3 supported heme assimilation, we deleted the str3+ gene (str3Δ) in a strain lacking hem1+ and shu1+ (hem1Δ shu1Δ) and tested whether this triple disruption strain could accumulate fluorescent ZnMP compared with a hem1Δ shu1Δ strain (Fig. 4). The indicated strains were incubated in ALA-free medium containing Dip (250 μm) or FeCl3 (100 μm) for 3 h. Cells were then incubated for 90 min in the presence of two distinct concentrations of ZnMP (2 and 10 μm). The ZnMP fluorescent signal was mostly lost in hem1Δ shu1Δ str3Δ cells when they had been incubated in the presence of 2 and 10 μm ZnMP (Fig. 4, A and B). Interestingly, in the presence of 10 μm ZnMP, a high intracellular ZnMP fluorescence signal was detected in hem1Δ shu1Δ mutant cells expressing an endogenous str3+ allele or in hem1Δ shu1Δ str3Δ mutant cells expressing a WT str3+ allele that had been re-integrated (Fig. 4B). In the presence of 2 μm ZnMP, the above-mentioned mutant strains expressing str3+ failed to significantly accumulate ZnMP (Fig. 4A). In the case of hem1Δ and hem1Δ str3Δ cells that expressed an endogenous shu1+ allele, a strong intracellular ZnMP fluorescence signal was observed in the presence of both concentrations of ZnMP (Fig. 4). There was an absence of ZnMP fluorescent signal in hem1Δ cells when they had been incubated in the presence of high iron concentrations (100 μm) because expression of shu1+ and str3+ is repressed in iron-replete cells (7, 11). Taken together, these results indicated that the inability to take up ZnMP in a hem1Δ shu1Δ mutant strain can be overcome by Str3 when the medium is supplemented with 10 μm ZnMP. We therefore concluded that S. pombe possesses a Shu1-independent low-affinity heme uptake pathway that requires Str3.
Figure 4.

Deletion of shu1+ and str3+ leads to defects in the cellular assimilation of ZnMP, albeit in a different concentration threshold. The indicated strains were precultured in the presence of Dip (50 μm) or FeCl3 (100 μm) and ALA (200 μm). Cells were transferred in ALA-free medium containing Dip (250 μm) or FeCl3 (100 μm) for 3 h. In the final 90 min of treatment, ZnMP was added at the indicated concentration (2 μm (A) or 10 μm (B)). A hem1Δ shu1Δ str3Δ triple mutant strain in which a WT copy of the str3+ gene was reintegrated was cultured in an identical manner. Cells were analyzed by fluorescence microscopy for accumulation of fluorescent ZnMP (right). Nomarski optics (left) was used to examine cell morphology. Results of microscopy are representative of five independent experiments.
Heterologous expression of Str3 allows S. cerevisiae to acquire ZnMP
In contrast to S. pombe, the yeast S. cerevisiae is unable to acquire exogenous heme (2, 17). However, heterologous expression in S. cerevisiae of a heme uptake protein isolated from another organism confers the ability to S. cerevisiae to utilize heme or hemoglobin (18, 19). To validate the role of Str3 in the assimilation of exogenous ZnMP, we expressed the str3+ gene in an S. cerevisiae BY4741 hem1Δ mutant strain. In the presence of ALA, hem1Δ cells grew because ALA was used by Hem2, the second enzyme of the heme biosynthetic pathway. This observation suggested that the mutant cells generated sufficient quantities of heme for cellular needs of heme-dependent pathways. In contrast, there was a selective block of heme biosynthesis in the absence of ALA. This observation was used as a stepping stone to investigate the nature of proteins that may participate in exogenous acquisition of heme. Experimentally, the str3+ (S. pombe), str3+-GFP, and RBT5 (Candida albicans) (20) genes were placed under the control of the GPD gene promoter and then transformed in S. cerevisiae hem1Δ cells using a centromeric plasmid. The cell surface–anchored heme-binding protein Rbt5 was used as a control because it has been established that it confers the ability to utilize exogenous hemin in hem1Δ cells (18). To ascertain that Str3-GFP and Rbt5 were produced, crude membrane fractions were isolated by ultracentrifugation. Immunoblotting analysis showed that Str3-GFP, Rbt5, and Pma1 were associated with the membrane fractions (Fig. 5A). Plasma membrane protein Pma1 used as a control was detected in the membrane fraction (21). As expected, Str3-GFP, Rbt5, and Pma1 were not detected in the soluble fraction. In contrast, Pgk1, which is known to be found in the cytosolic fraction, was present in the supernatant fraction (Fig. 5A). Indirect immunofluorescence and direct fluorescence microscopy were performed to assess the cellular location of Rbt5 and Str3-GFP, respectively, heterologously expressed in S. cerevisiae. In the case of Rbt5, results showed that Rbt5-dependent fluorescence was primarily detected at the cell periphery (Fig. 5B). In the case of Str3-GFP, the GFP signal was predominantly localized at the cell surface (Fig. 5C). When S. cerevisiae hem1Δ cells were transformed with an empty plasmid or an untagged str3+, the fluorescence signal was absent (Fig. 5, B and C). Given the finding that Rbt5 and Str3-GFP were localized at S. cerevisiae cell surface, we investigated whether their presence led to cellular accumulation of fluorescent ZnMP. ALA was omitted from the culture medium to repress heme biosynthesis, and hem1Δ cells were preincubated for 3 h under conditions of iron starvation. Cells were then incubated for 90 min in the presence of ZnMP (50 μm). Results showed that S. cerevisiae hem1Δ cells expressing Rbt5, Str3, or Str3-GFP produced a ZnMP-associated fluorescence signal that was mainly located in the cytoplasm of cells (Fig. 5D). There was an absence of ZnMP fluorescent signal in cells transformed with an empty expression plasmid (Fig. 5D). Taken together, these results established the fact that heterologous expression of S. pombe Str3 in S. cerevisiae leads to cytoplasmic accumulation of the heme analog ZnMP.
Figure 5.
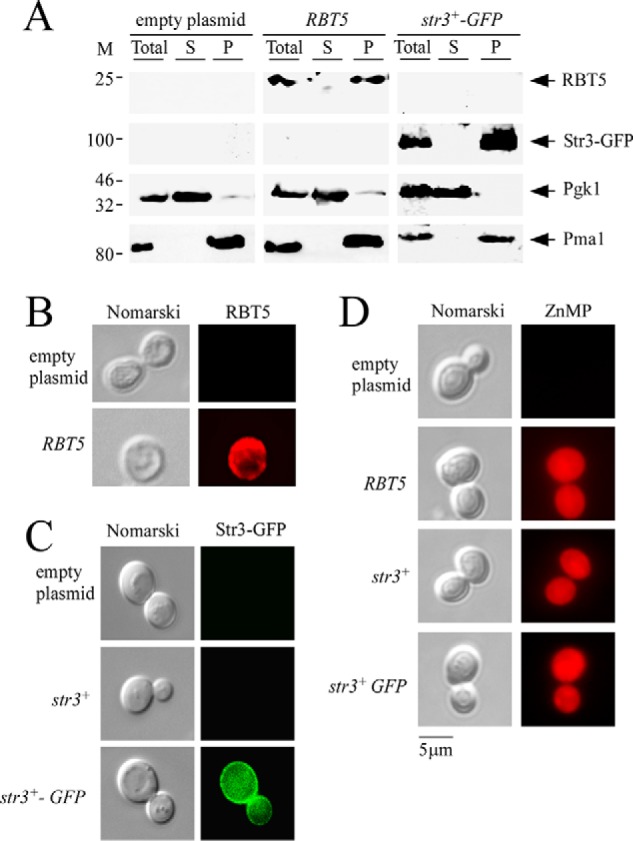
Expression of S. pombe Str3 in S. cerevisiae leads to cellular accumulation of ZnMP. A, an S. cerevisiae hem1Δ mutant strain was transformed with plasmid p415GPD alone (empty plasmid) or with plasmid p415GPDRBT5 or p415GPDstr3+-GFP. Cells were precultured in the presence of ALA (200 μm) and then transferred in an ALA-free medium in which they were incubated for 3 h. Total protein extracts (Total) from these cells were prepared and ultracentrifuged to obtain supernatant (S) and membrane protein pellet fraction (P). Supernatant and pellet fractions were resolved on SDS-polyacrylamide gels and analyzed for steady-state levels of Rbt5, Str3-GFP, Pgk1, and Pma1 by immunoblotting using anti-Rbt5, anti-GFP, anti-Pgk1, and anti-Pma1 antibodies. M, positions of molecular mass of protein standards (in kDa) are indicated on the left. B and C, hem1Δ cells expressing C. albicans Rbt5 (B) and S. pombe Str3 and Str3-GFP (C) proteins were analyzed by indirect immunofluorescence (Rbt5) and direct fluorescence (Str3-GFP) microscopy. An absence of fluorescence was observed in the case of cells harboring an empty plasmid or an untagged str3+ allele. D, S. cerevisiae hem1Δ cells expressing the indicated allele were precultured in the presence of ALA (200 μm). Cells were washed and incubated in ALA-free medium for 3 h. After a 90-min treatment with ZnMP (50 μm), cells were examined by fluorescence microscopy for accumulation of fluorescent ZnMP (right). Cell morphology was examined using Nomarski optics (left panels, B–D). Results of microscopy are representative of five independent experiments.
Str3 binds hemin
To further examine whether Str3 was able to bind heme, str3+-GFP, and shu1+-HA4, fusion alleles were co-expressed in a hem1Δ shu1Δ str3Δ triple mutant strain. Mid-logarithmic cells were washed to remove ALA and then treated with Dip (250 μm) or iron (100 μm) for 3 h. Total protein extracts were prepared, and cell membranes were isolated by ultracentrifugation. Membrane protein fractions were detergent-treated (Triton X-100) and then refractionated by ultracentrifugation. Solubilized Str3-GFP and Shu1-HA4 were mixed with hemin-agarose or agarose (control) beads. Immunoblotting analysis of proteins bound to hemin-agarose using anti-GFP and anti-HA antibodies revealed that both Str3-GFP and Shu1-HA4 isolated from iron-starved cells were detected in the bound fraction (Fig. 6A). In hemin-agarose pulldown assays, Shu1-HA4 served as a control of known heme-binding protein (7, 8). As an additional control for specificity of the resin, Str3-GFP and Shu1-HA4 were found in the unbound fraction (flow-through) when only agarose beads were used (Fig. 6A). Consistent with iron-dependent repression of str3+-GFP and shu1+-HA4 transcript levels (under the control of str3+ and shu1+ promoters, respectively), Str3-GFP and Shu1-HA4 were not detected in membrane fractions that had been prepared from iron-replete cells (Fig. 6A). Soluble PCNA was only detected in whole-cell extracts and was absent from the membrane protein fractions.
Figure 6.

Str3 binds hemin. A, hem1Δ shu1Δ str3Δ cells co-expressing GFP-tagged Str3 and HA4-tagged Shu1 were precultured in the presence of ALA (200 μm) and Dip (50 μm). Cells in the mid-exponential phase of growth were transferred to ALA-free medium and treated with Dip (250 μm) or FeCl3 (100 μm) for 3 h. Whole-cell extracts (Total) were prepared, and cell membranes were obtained by ultracentrifugation. Triton X-100–solubilized membrane proteins (input) were subjected to hemin pulldown assays using hemin-agarose or agarose alone. Unbound and bound protein fractions were analyzed by immunoblot assays using anti-GFP, anti-HA, and anti-PCNA antibodies. B, hem1Δ shu1Δ str3Δ cells expressing Str3-Y530A/Y534A/S552A/Y557A-GFP were analyzed for detection of a GFP-mediated signal by fluorescence microscopy (right). Before microscopic analysis, cells were incubated in the presence of Dip (250 μm) or FeCl3 (Fe; 100 μm) for 3 h. Nomarski optics was used to reveal cell morphology (left). C, hem1Δ shu1Δ str3Δ cells expressing GFP-tagged Str3-Y530A/Y534A/S552A/Y557A mutant protein were cultured under the same conditions as described for A. Protein fractionation, pulldown assays with hemin-agarose, and immunoblotting were carried out as indicated for A. M, positions of molecular mass of protein standards (in kDa) are indicated on the right. D, growth of the indicated strains was assessed in the presence of hemin (0.15 μm) but in the absence of ALA. Strain color codes were as follows: hem1Δ shu1Δ str3+ in black; hem1Δ shu1Δ str3Δ expressing str3+-GFP in orange; hem1Δ shu1Δ str3Δ with an empty plasmid in violet; hem1Δ shu1Δ str3Δ expressing str3-Y530A/Y534A/S552A/Y557A in green; and hem1Δ shu1Δ str3Δ expressing str3-Y530A/Y534A/S552A/Y557A-GFP in blue. Values are represented as the averages ± S.D. (error bars) of three independent experiments.
As mentioned above, two putative heme-binding motifs, 530YX3Y534 and 552SX4Y557, are present within the sequence of the predicted loop 11 of Str3. We therefore tested whether Tyr530, Tyr534, Ser552, and Tyr557 residues were involved in the ability of Str3 to interact with hemin. Our approach was to replace these residues (positions 530, 534, 552, and 557) with alanine residues. When expressed in hem1Δ shu1Δ str3Δ cells that had been washed to remove ALA and then incubated in the presence of Dip (250 μm, 3 h), the Str3-Y530A/Y534A/S552A/Y557A-GFP mutant protein was located at the cell surface, as observed in the case of WT GFP-tagged Str3 (Figs. 5 and 6B). In contrast, Str3-Y530A/Y534A/S552A/Y557A-GFP was not detected at the surface of iron-treated (100 μm, 3 h) cells. Membrane fractions collected by ultracentrifugation were subjected to Triton X-100 treatment, and the solubilized proteins were analyzed by hemin pulldown assays using hemin-agarose or agarose beads. Results showed that Str3-Y530A/Y534A/S552A/Y557A-GFP was largely detected in the unbound (flow-through) fraction (64 ± 5% of the preparation), consistent with a loss of affinity for hemin (Fig. 6C). A small but significant proportion of the mutant protein (36 ± 5% of the preparation) was still retained on hemin-agarose. In the case of Str3-Y530A/Y534A/S552A/Y557A-GFP, its presence in the unbound fraction (Fig. 6C, center) after hemin pulldown assays was markedly higher as compared with WT Str3-GFP, for which only minimal amounts (≥0.5 ± 0.05% of the preparation) of the protein were found in the unbound fraction (Fig. 6A, center). Str3-Y530A/Y534A/S552A/Y557A-GFP was exclusively found in the unbound fraction when agarose beads were used, and it was not detected in protein extracts that had been prepared from iron-replete cells as opposed to extracts prepared from iron-starved cells (Fig. 6C).
To investigate whether mutations of Tyr530, Tyr534, Ser552, and Tyr557 residues to Ala in Str3 affected the ability of cells to grow in the presence of exogenous hemin as a sole source of heme (no ALA; heme biosynthesis off), hem1Δ shu1Δ str3Δ mutant cells were transformed with integrated plasmids expressing the following alleles: str3+-GFP, str3-Y530A/Y534A/S552A/Y557A, str3-Y530A/Y534A/S552A/Y557A-GFP, and an empty plasmid as control. In the presence of hemin (0.15 μm), hem1Δ shu1Δ str3Δ cells expressing Str3-GFP grew to A600 of 3.6 over a time period of 80 h. This level of growth was comparable with that of hem1Δ shu1Δ mutant cells in which an endogenous str3+ gene had been expressed (A600 of 3.9 after 80 h) (Fig. 6D). In contrast, hem1Δ shu1Δ str3Δ cells expressing either Str3-Y530A/Y534A/S552A/Y557A or Str3-Y530A/Y534A/S552A/Y557A-GFP exhibited poor growth (A600 of 0.49 and 0.47, respectively, after 80 h) in a medium supplemented with hemin (0.15 μm) (Fig. 6D). Taken together, these results indicated that amino acid residues Tyr530, Tyr534, Ser552, and Tyr557 within the predicted extracellular loop between the eleventh and twelfth transmembrane regions of Str3 are required for its hemin-dependent transport function.
Str3 is an integral membrane protein
Cell-surface detection and a topological model of Str3 suggest that it is integrated into cellular membranes. To investigate this prediction, cell membranes were isolated by ultracentrifugation of whole-cell extracts of hem1Δ shu1Δ str3Δ cells expressing str3+-GFP under low-iron conditions. In parallel experiments, we used hem1Δ shu1Δ abc3Δ cells expressing abc3+-GFP under the same conditions because Abc3 is a control protein known to be integrated into cellular membranes (22). After a first ultracentrifugation, soluble and released peripheral membrane proteins present in the supernatants were left untreated and analyzed by immunoblot assays (Fig. 7, U1). In the case of the pellet fraction, it was resuspended and left untreated or was adjusted to 0.1 m Na2CO3 or 1% Triton X-100 and then refractionated through a second ultracentrifugation. Results showed that in the absence of any treatment, Str3-GFP and Abc3-GFP were detected only in the pellet fractions (Fig. 7, Buffer). An identical protein pattern was observed when the procedure had been performed in the presence of Na2CO3, which is known to selectively release nonintegral membrane proteins (with no effect on integral membrane proteins). Treatment of the pellet fraction with Triton X-100 induced the release of Str3-GFP and Abc3-GFP, which were detected in the supernatant fractions (Fig. 7, A and B). Taken together, these results indicated that Str3-GFP is an integral membrane protein, as characterized previously in the case of Abc3-GFP (22). As an additional control, soluble PCNA was detected only in the supernatant fraction (Fig. 7, A and B).
Figure 7.
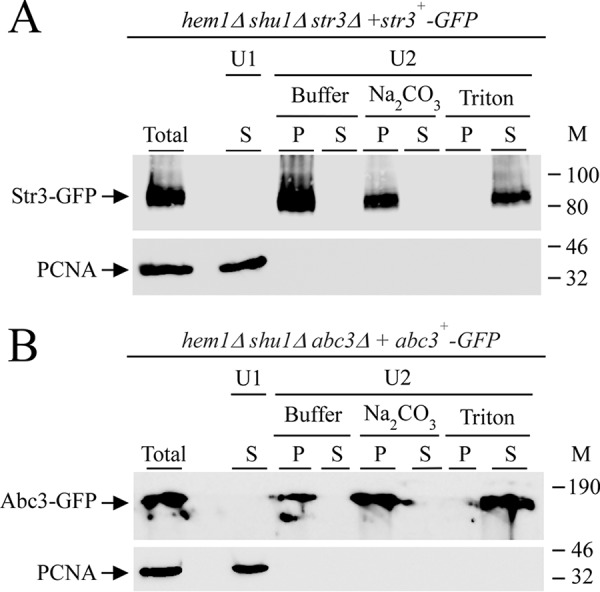
Str3 is an integral membrane-associated protein. hem1Δ shu1Δ str3Δ and hem1Δ shu1Δ abc3Δ mutant strains were transformed with str3+-GFP (A) and abc3+-GFP (used as a control; B) alleles, respectively. Transformed cells were precultured in the presence of Dip (50 μm) and ALA (200 μm). Cells were washed twice, and mid-logarithmic-phase cells were incubated in the presence of Dip (250 μm) for 3 h. Total extract preparation (Total) was subjected to a first ultracentrifugation (U1) at 100,000 × g. The supernatant (S; U1) was analyzed. The membrane-containing pellet fraction obtained from U1 was resuspended and kept untreated (buffer) or incubated in the presence of Na2CO3 (0.1 m) or Triton X-100 (1%) and then ultracentrifuged a second time at 100,000 × g (U2). Supernatant (S) and pellet (P) fractions were resolved on SDS-polyacrylamide gels and analyzed by immunoblotting using anti-GFP and anti-PCNA antibodies. M, positions of molecular mass of protein standards (in kDa) are indicated on the right. Str3-GFP, Abc3-GFP, and PCNA are indicated with arrows.
The predicted hydrophilic loop region 522–576 of Str3 binds hemin
The biophysical properties of Str3, which is an integral membrane protein that harbors 12 predicted hydrophobic membrane spans, prevent the full-length Str3 from being expressed as a soluble protein in Escherichia coli. However, when a portion of str3+ encoding the extracellular loop 11 corresponding to amino acid residues 522–576 was subcloned, the resulting polypeptide was highly soluble in E. coli. Of interest, Str3(522–576) contained two putative heme-binding motifs, 530YX3Y534 and 552SX4Y557 (Fig. 8A). We investigated the ability of purified, bacterially expressed Str3(522–576) to interact with hemin. In parallel experiments, a mutant version of Str3(522–576) in which Tyr530, Tyr534, Ser552, and Tyr557 had been substituted by Ala residues was generated and produced in E. coli. His6-tagged protein products (WT and mutant) were purified using two successive rounds of affinity chromatography on nickel-agarose beads. Results showed that WT Str3(522–576) was retained on hemin-agarose beads, with a trace amount of protein detected in the flow-through (unbound) fraction (Fig. 8B). In contrast, the mutant form of Str3(522–576) (harboring Y530A/Y534A/S552A/Y557A substitutions) was exclusively found in the flow-through (unbound) fraction (Fig. 8B).
Figure 8.

Str3(522–576) and hemin interact with one another. A, schematic representation of the Str3(522–576) region and its mutant derivative. B, purified WT Str3(522–576) (left) or Str3-Y530A/Y534A/S552A/Y557A)(522–576) mutant (right) (Input) was incubated with hemin-agarose beads. Unbound and bound purified WT or mutant Str3(522–576) was analyzed by immunblot assays using an anti-His6 antibody. C, differential spectral titration of WT Str3(522–576)-hemin interaction (left) using 5 μm hemin and increasing concentrations of Str3 (0–20 μm). Similar titration assays (right) were performed with increasing concentrations of Str3-Y530A/Y534A/S552A/Y557A(522–576) mutant (0–20 μm) and hemin (5 μm). D, hemin-binding curves for WT (left) and mutant (right) Str3(522–576) obtained by plotting changes in absorbance at the Soret peak as a function of Str3 concentrations. WT Str3(522–576) and hemin interacted with one another with a KD of 6.6 × 10−6 m.
Additional evidence that the amino acid region 522–576 of Str3 bound hemin was obtained by spectroscopy. Absorbance spectra of the addition of increasing quantities of purified Str3(522–576) (0–20 μm) to a fixed concentration of hemin (2 μm) were recorded. Absorption of hemin exhibited a typical Soret peak at 402 nm in the absence of Str3 (Fig. 8C). Results showed that the addition of Str3(522–576) caused a shift in the absorbance of hemin to shorter wavelengths (blue shift) corresponding to 394–397 nm (Fig. 8C). The absorption peak that was blue-shifted in the presence of Str3(522–576) decreased as a function of increasing concentrations of Str3 (Fig. 8C). In contrast, the blue shift was not observed in the case of the Str3-Y530A/Y534A/S552A/Y557A mutant. Furthermore, the intensity of the peak did not show significant changes as a function of increasing concentrations of Str3-Y530A/Y534A/S552A/Y557A (Fig. 8C). Analysis of the data yielded a value of 6.6 × 10−6 m for the constant of dissociation (KD) with respect to Str3(522–576). On the other hand, the lack of interaction between Str3-Y530A/Y534A/S552A/Y557A and hemin did not allow a KD value to be determined (Fig. 8D). Taken together, these results showed that the Str3(522–576) region interacts with hemin without the need for an additional protein partner.
Discussion
We have previously reported that S. pombe has the ability to utilize exogenous hemin as a heme source (7). Heme acquisition by S. pombe occurs through the activity of Shu1 in the presence of 0.075 μm hemin in the medium (7). Here, we showed that hem1Δ shu1Δ mutant cells were still able to acquire exogenous hemin at concentrations of 0.15 μm or higher, suggesting the presence of an additional mechanism for hemin acquisition. Genetic evidence for the role of Str3 as a hemin transporter was obtained by characterizing the consequence of its disruption in hem1Δ shu1Δ cells. Indeed, disruption of the str3+ gene (str3Δ) abrogated the ability of these cells to grow in the presence of 0.15 μm hemin.
In silico membrane topology analysis of Str3 predicted that the protein contained amino acid sequence signatures characteristic of members of the MFS transporters (13). Str3 has been predicted to possess 12 transmembrane spans that are connected by short hydrophilic loops, except in the case of loops 6 and 11, which are notably longer. Loop 6 has been predicted to be cytosolic and to interconnect the first six transmembrane spans with the second half of the protein, whereas loop 11 has been predicted to be extracellular and exposed to a potential ligand. In the case of the MFS-type iron-siderophore transporter, loop 11 contains the siderophore transporter domain, in which a highly conserved Tyr residue is present and required for uptake of siderophore into the cells (23). Although this highly conserved Tyr residue (position 571) was also found in Str3, loop 11 of Str3 was unique due to the presence of two putative heme-binding sequences, 530YX3Y534 and 552SX4Y557, that were absent in S. pombe Str1 and Str2 proteins. These two putative heme-binding sequences were reminiscent of near-iron transporter (NEAT) motifs that are found in several Gram-positive bacteria (24). Although the initial proposed role of NEAT domains was to bind siderophores for their delivery into Gram-positive cells, subsequent studies have shown that NEAT-containing proteins function to scavenge heme from hemoproteins and to uptake it through the bacterial cell surface for delivery into the cytosol (24–26). Several NEAT proteins contain a domain in which amino acid sequences are arranged in SX4Y and YX3Y configurations that are important for heme-binding function (24). In bacteria, the canonical SX4Y motif is generally found within a small and unique α-helix structure located at the N terminus of the NEAT domain. The remaining part of the domain is constituted of eight β-strands that form a conserved β-barrel fold. The YX3Y motif is found within the eighth β-strand located at the C terminus of the domain. In the case of loop 11 of Str3, its structural conformation is unknown. However, according to an in silico three-dimensional model by I-TASSER (16), a putative heme-binding pocket has been predicted and comprises the YX3Y and SX4Y motifs. In the current report, mutations of the YX3Y and SX4Y motifs in Str3 led cells expressing the str3-Y530A/Y534A/S552A/Y557A allele to be unable to rescue hem1Δ shu1Δ str3Δ cells to grow in the presence of hemin compared with hem1Δ shu1Δ str3Δ cells expressing a WT str3+ allele. Furthermore, pulldown assays using protein lysates prepared from cells expressing Str3-Y530A/Y534A/S552A/Y557A showed that substitutions of Tyr530, Tyr534, Ser552, and Tyr557 for the corresponding Ala residues markedly decreased Str3 binding to hemin-agarose. When the WT version of the loop 11 of Str3(522–576) was produced and purified from E. coli, it was retained on hemin-agarose beads using pulldown assays. In contrast, a mutant form of loop 11 containing Y530A/Y534A/S552A/Y557A substitutions failed to bind hemin-agarose beads, revealing that Tyr530, Tyr534, Ser552, and Tyr557 residues participated in hemin coordination in Str3. However, determination of whether one amino acid residue has a contribution to binding higher than the others or whether there is influence of additional amino acid residues flanking the motif or elsewhere will require additional studies. Based on an in silico-generated model, the YX3Y and SX4Y motifs located in loop 11 of Str3 may serve as a docking site to facilitate heme entry into the central cavity of the MFS transporter.
S. pombe Str3 is not the unique case of the MFS-type transporter that mediates heme transport. In humans, FLVCR1a, FLVCR1b, and FLVCR2 are MFS-type transporters (27–31). FLVCR1a is a plasma membrane heme exporter (30). FLVCR1b encodes a shorter isoform of FLVCR1a that localizes to the mitochondrial membrane (27). FLVCR1b possesses six hydrophobic transmembrane domains, and it is likely that FLVCR1b homodimerizes to form a full-length transporter with 12 transmembrane domains similar to FLVCR1a. It has been reported that FLVCR1b functions as a mitochondrial heme exporter (27). In the case of FLVCR2 expressed in mammalian cells and Xenopus laevis oocytes, it functions as an importer of heme (28). In cats, the cell-surface heme exporter FLVCR1a is recognized and bound by the feline leukemia virus subgroup C (FeLV-C) (32). Although loop 11 of FLVCR1a is shorter compared with that of Str3, FLVCR1a loop 11 is required for FeLV-C–FLVCR1 association (32). Upon binding of FLVCR1a by FeLV-C viruses, cellular heme efflux is blocked. The observation that FLVCR1a loop 11 functions as a docking site for FeLV-C is reminiscent of our findings that loop 11 of Str3 is involved in heme binding and may serve as an initial docking site for heme.
Structural studies of MFS transporters have shown that the 12 α-helical transmembrane spans are organized into two six-helix halves (12, 13). Substrate transport across the membrane probably occurs via a clamp-and-switch (also called rocker-switch) mechanism whereby the two halves of the transporter cycle through outward-opening (substrate loaded), occluded (substrate bound), and inward-opening (substrate released) conformations. At present, the nature of the amino acid residues of Str3 and FLVCR that coordinate heme in the occluded state of the transporters is unknown. However, in the case of FLVCR1a, an in silico sequence analysis of heme carrier proteins has suggested that His145, Tyr153, and His198 are likely to be involved in the heme transport process (30, 33). Pairwise sequence alignment between Str3 and FLVCR1a has revealed that Tyr153 is conserved in both proteins. Furthermore, the aromatic side chain of Tyr153 (found at position 160 in Str3) is predicted to be positioned in the center of the transporter and available for substrate coordination. Additional studies will be required to confirm the functional role of Tyr160 in Str3 as well as to identify additional amino acid residues that coordinate heme in the heme-bound occluded state of Str3.
S. pombe uses three distinct strategies for acquisition of iron from environmental sources. A first system involves the transport of reduced free iron ions through an oxidase-permease–based iron uptake machinery that consists of the Fio1 and Fip1 proteins, respectively (9). A second system takes up siderophore-bound iron through the ferrichrome transporter Str1 (11). A third system triggers assimilation of exogenous heme using the cell-surface proteins Shu1 and Str3. Interestingly, str3+ and shu1+ are gene neighbors on chromosome I of S. pombe, and their transcription is oriented in the same direction. In the case of Shu1, experiments using the heme analog ZnMP have shown that the prosthetic group first accumulates into vacuole and then within the cytoplasm (8). Furthermore, the same study has shown that mobilization of stored ZnMP from the vacuole for redistribution within the cytoplasm requires the vacuolar transporter Abc3 (8). Upon the addition of the iron chelator Dip, we observed that expression profiles of fio1+ (or fip1+) and str1+ were rapidly induced within 15–30 min of treatment. This step was followed by a sustained induction of fio1+ and str1+ mRNA levels for at least 4 h. In the case of shu1+, abc3+, and str3+, under the same conditions, their transcript levels were detected at times later than those of fio1+ and str1+. Furthermore, shu1+, abc3+, and str3+ mRNA levels were at their maximum levels only after 4 h in iron-starved cells. This observation suggests a common temporal regulation for shu1+, abc3+, and str3+ compared with fio1+ and str1+.
We have shown previously that Shu1 interacts with hemin with a KD of 2.2 μm (7). In the current study, results showed that loop 11 of Str3 exhibits a KD value of 6.6 μm for hemin. These results revealed that Str3 possesses a lower affinity for hemin than Shu1. Consistent with this observation, the hemin-dependent growth deficiency of a hem1Δ shu1Δ double mutant (expressing str3+) can only be rescued when exogenous hemin exceeded ≥2 times the concentration that is required to overcome hemin deficiency of a hem1Δ single mutant (expressing shu1+). When the abc3+ gene was deleted in a hem1Δ shu1Δ mutant strain, hem1Δ shu1Δ abc3Δ cells exhibited similar growth compared with hem1Δ shu1Δ cells in the presence of 0.15 μm hemin, revealing that Str3 function was unaffected by the absence of the vacuolar transporter Abc3. Together, the results revealed that S. pombe possesses two systems with different affinities to bring exogenous heme into the cell. As a saprophyte fungus, these two transport systems identified in S. pombe may strengthen its ability to acquire external heme from natural sources in its environment.
Experimental procedures
Yeast strains and growth media
Genotypes of S. pombe and S. cerevisiae strains used in this study are listed in Table 1. All S. pombe strains were maintained on yeast extract plus supplements medium (YES) containing 0.5% yeast extract, 3% glucose, and 225 mg/liter adenine, uracil, leucine, lysine, and histidine (34). In the case of cells lacking Hem1 (hem1Δ), they were supplemented with ALA (200 μm) under nonselective growth conditions. hem1Δ mutant cells could also be maintained alive in the absence of ALA by supplementing the medium with exogenous hemin at the indicated concentrations (0.075, 0.15, and 0.5 μm). In the presence of exogenous hemin, hem1Δ cells rely on their heme uptake machinery to sustain growth instead of using their own heme biosynthesis pathway. Strains used for DNA plasmid integration were cultured in Edinburgh minimal medium, in which specific amino acids were omitted as required to trigger chromosomal integration events. For monitoring cell growth, precultures of cells were carried out in YES medium containing 2,2′-dipyridyl (Dip) (50 μm) to chelate iron and, at the same time, to foster expression of iron starvation–inducible genes, including str3+ and shu1+. During precultures of cells, ALA (200 μm) was added to ensure biosynthesis of heme. Once precultures reached mid-logarithmic phase, cells were washed twice and then diluted 1000-fold in YES containing Dip (10 μm or the indicated concentration) and hemin (0.075, 0.15, or 0.5 μm) but in the absence of ALA (unless otherwise stated). Cell cultures were then initiated and monitored (A600) at each of the indicated times. S. cerevisiae hem1Δ cells were grown in standard minimal medium that was supplemented in ALA (200 μm), unless otherwise stated. Synthetic complete medium lacking leucine was used to transform S.cerevisiae cells with p415GPD (35) and its derivatives.
Table 1.
S. pombe and S. cerevisiae strains used in this study
| Strain | Genotype | Source or reference |
|---|---|---|
| FY435 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 | Ref. 43 |
| fep1Δ | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 fep1Δ::ura4+ | Ref. 43 |
| TMY1 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::KANr | Ref. 7 |
| TMY2 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::KANr | Ref. 7 |
| TMY17 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP str3Δ::KANr | This study |
| VNY13 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP str3Δ::KANr str3+:ade6+ | This study |
| VNY14 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP str3Δ::KANr str3+-GFP:ade6+ | This study |
| VNY15 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP str3Δ::KANr str3+-GFP:ade6+ shu1+-HA4:leu1+ | This study |
| VNY16 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP str3Δ::KANr str3+Y530A/Y534A/S552A/Y557A:ade6+ | This study |
| VNY17 | h+ his7-366 leu1-32 ura4-Δ18 ade6-M210 hem1Δ::loxP shu1Δ::loxP str3Δ::KANr str3+Y530A/Y534A/S552A/Y557-GFP:ade6+ | This study |
| BY4741 | MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 | Ref. 44 |
| VNY18 | MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 hem1Δ::KANr | This study |
| VNY19 | MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 hem1Δ::KANr p415-GPD | This study |
| VNY20 | MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 hem1Δ::KANr p415-GPD-RBT5 | This study |
| VNY21 | MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 hem1Δ::KANr p415-GPD-str3+ | This study |
| VNY20 | MATa his3Δ0 leu2Δ0 met15Δ0 ura3Δ0 hem1Δ::KANr p415-GPD-str3+-GFP | This study |
Plasmids
PCR amplification of the str3+ gene was performed using primers designed to generate XmaI and NotI restriction sites at the upstream and the downstream termini of the ORF, respectively. The PCR product was digested with XmaI and NotI and cloned into the corresponding sites of pBPade6+ (36). The resulting plasmid was named pBPade-str3+. Subsequently, the str3+ promoter region from position −785 upstream of the initiator codon of str3+ was isolated by PCR amplification and then inserted into pBPade-str3+ at the ApaI and XmaI sites. This pBPade-str3+ derivative was denoted pBPade-785str3+, and it allowed expression of str3+ under the control of its own promoter. The GFP coding sequence derived from pSF-GP1 (37) was isolated by PCR using primers designed to generate NotI and SacII sites at the 5′ and 3′ termini of the GFP gene. The NotI-SacII GFP-encoded DNA fragment was inserted in-frame with the 3′-terminal coding sequence of str3+, creating pBPade-785str3+-GFP. Plasmid pBPade-785str3+-GFP was used to introduce mutations in the coding sequence of str3+. Codons corresponding to Tyr530, Tyr534, Ser552, and Tyr557 were replaced by nucleotide triplets that encode alanine. These site-specific mutations were created by a PCR overlap extension method (38). The resulting plasmid containing the str3+ mutant allele was denoted pBPade-785str3-Y530A/Y534A/S552A/Y534A-GFP. A similar strategy was used to create an untagged str3+ mutant allele, and the plasmid was called pBPade-785str3-Y530A/Y534A/S552A/Y534A. The shu1+-HA4 gene was isolated from pBP-1317shu1+-HA4 (7) using the ApaI and SacII restriction enzymes. The purified DNA fragment was cloned into the corresponding sites of pJK148 (39). The resulting plasmid was denoted pJK-1317shu1+-HA4. The coding sequence corresponding to the str3+ gene was isolated by PCR and cloned into the BamHI-XhoI–cut p415GPD vector (35). This centromeric plasmid was named p415GPDstr3+. The str3+-GFP coding sequence was amplified from plasmid pBPade-785str3+-GFP using primers designed to generate XmaI and XhoI sites at each extremity of the PCR product. The DNA fragment was inserted into the corresponding sites of p415GPD, creating p415GPDstr3+-GFP. The RBT5 ORF was isolated by PCR from genomic DNA of the C. albicans SC5314 strain. The PCR-amplified fragment was digested with XmaI and XhoI and cloned into the corresponding sites of p415GPD. The resulting plasmid was denoted p415GPDRBT5. The C-terminal DNA coding region of Str3 (corresponding to residues 522–576) was amplified by PCR using primers designed to generate NcoI and XhoI sites at the 5′ and 3′ termini of the DNA fragment, respectively. The purified PCR product was subsequently cloned into the corresponding sites of pET28a, which resulted in the Str3 fragment (residues 522–576) fused downstream of and in-frame to the His6 tag coding region. The resulting plasmid, pET28a522Str3576, was used to introduce mutations in the coding sequence of str3+. Codons corresponding to Tyr530, Tyr534, Ser552, and Tyr557 were replaced with Ala residues. These site-specific mutations were created by a PCR overlap extension method (38). The new mutant plasmid was named pET28a522Str3576Y530A/Y534A/S552A/Y557A.
RNA isolation and analysis
Total RNA was extracted by a hot phenol method as described previously (40). Transcripts were analyzed using RNase protection assays as described previously (41). Plasmids pSKstr3+ (11) and pSKact1+ (42) were linearized with BamHI for subsequent labeling with the use of [α-32P]UTP and the T7 RNA polymerase. The resulting antisense RNA probes served to determine str3+ and act1+ steady-state mRNA levels. In the case of act1+ transcripts, they were probed as an internal control for normalization during quantification of RNase protection products.
Direct fluorescence and indirect immunofluorescence microscopy
Fluorescence and differential interference contrast images of cells were viewed with a Nikon Eclipse E800 epifluorescent microscope (Nikon, Melville, NY) equipped with a Hamamatsu ORCA-ER digital cooled camera (Hamamatsu, Bridgewater, NJ). Cells were subjected to microscopic analysis using ×1000 magnification and the following filters: 340–380 nm (blue), 465–495 nm (green), and 510–560 nm (red). For detection of fluorescent ZnMP accumulation in S. pombe cells, liquid cultures of the indicated strains were seeded to an A600 of 0.5. The cultures were then incubated in ALA-free medium containing Dip (250 μm) or FeCl3 (100 μm) for 3 h. Subsequently, ZnMP (2 or 10 μm) was added for 90 min. ZnMP accumulation was stopped by adding 5 volumes of ice-cold 5% BSA in PBS. After centrifugation, cells were resuspended in ice-cold 2% BSA in PBS and examined by fluorescence microscopy as described previously (7, 8). In the case of S. cerevisiae cells, a similar method was used for fluorescence microscopic visualization of ZnMP, except that the indicated strains were incubated in ALA-free standard minimal medium containing a higher concentration of ZnMP (50 μm) than used in S. pombe. To determine the cellular location of Rbt5 in S. cerevisiae cells, indirect immunofluorescence microscopy was performed using formaldehyde-fixed cells as described previously (36). Cells were spheroplasted using zymolase (30 mg/ml), β-mercaptoethanol (10 mm), and Triton X-100 (1%) as described previously (7). Spheroplasts were adsorbed on poly-l-lysine–coated (0.1%) multiwall slides as described previously (36). After a 30-min block with Tris-buffered saline (TBS) buffer (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% BSA, and 0.02% sodium azide), cells were incubated with a custom anti-Rbt5 antibody (Abcam, Burlingame, CA) diluted 1:250 in TBS. After an 18-h reaction, cells were washed with TBS and incubated for 90 min with tetramethylrhodamine-labeled goat anti-rabbit IgG (Invitrogen, T-2769) diluted 1:500 in TBS. Fields of cells shown in this study correspond to a minimum of five independent experiments.
Protein extraction and analysis
Preparation of cell lysates from S. pombe and S. cerevisiae was performed with glass beads using HEGN100 buffer, containing 20 mm HEPES, pH 7.9, 100 mm NaCl, 1 mm EDTA, 10% glycerol, 0.1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, 1 mm DTT, and a complete protease inhibitor mixture (Sigma-Aldrich, P8340). Cells were broken using a FastPrep-24 instrument (MP Biomedicals, Solon, OH). Cell lysates were fractionated using successive rounds of ultracentrifugation as described previously (8). Soluble and dissolved membrane proteins were resolved by electrophoresis on SDS-polyacrylamide gels and analyzed by immunoblot assays. For protein expression analysis, the following primary antibodies were used for immunodetection: monoclonal anti-GFP antibody B-2; monoclonal anti-HA antibody F-7 (Santa Cruz Biotechnology, Inc.); monoclonal anti-α-tubulin antibody B-5-1-2; monoclonal anti-PCNA antibody PC10 (Sigma-Aldrich); monoclonal anti-Pma1 antibody 40B7 (Thermo Fisher Scientific); monoclonal anti-Pgk1 antibody 22C5-D8 (Molecular Probes); monoclonal anti-His5 antibody (penta-His; Qiagen); and custom polyclonal anti-Rbt5 (Abcam). Following incubation with primary antibodies, membranes were washed and incubated with the appropriate horseradish peroxidase–conjugated secondary antibodies (Amersham Biosciences), developed with ECL reagents (Amersham Biosciences), and visualized by chemiluminescence. In the case of pulldown assays with hemin-agarose, proteins (∼100 μg) were incubated with 20 μl of hemin-agarose or agarose beads, and the suspensions were mixed end-over-end for 30 min at 25 °C. The beads were centrifuged, and unbound material was kept on ice. The beads were washed three times with 1 ml of buffer containing 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, and 1% Triton X-100. The beads were transferred to a fresh microtube before the last wash. Immunoprecipitates and unbound material were resuspended and mixed, respectively, with 50 μl of SDS loading buffer (100 mm Tris-HCl, pH 8.0, 1.4 mm β-mercaptoethanol, 1% SDS, 5 mm EDTA, 8 m urea) and heated for 30 min at 37 °C. Samples were resolved by electrophoresis on 10% SDS-polyacrylamide gels.
Purification of Str3(522–576) expressed in bacteria
E. coli Rosetta(DE3)pLysS cells harboring plasmid pET28a522Str3576 or pET28a522Str3576Y530A/Y534A/S552A/Y557A were grown to an A600 of 0.5. At this growth phase, pET28a-driven protein expression was induced with isopropyl-β-d-thiogalactopyranoside (0.4 mm) for 4 h at 37 °C in the presence of ethanol (2%). Cells were then broken up by sonication in buffer A (50 mm Tris-HCl, pH 7.5, 300 mm NaCl, 10% sucrose, 20 mm imidazole, 50 μg/ml lysozyme, and 1% Triton X-100) containing a mixture of protease inhibitors (P8340, Sigma- Aldrich). Protein extracts were incubated for 2 h at 4 °C with a suspension (2 ml) of nickel-nitrilotriacetic acid-agarose beads. Str3(522–576) or Str3Y530A/Y534A/S552A/Y557A(522–576) protein bound to the beads were eluted stepwise with buffer B (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10% glycerol) containing 100, 500, and 1000 mm imidazole. Samples (500 mm imidazole eluate fractions) containing Str3(522–576) or Str3Y530A/Y534A/S552A/Y557A(522–576) were dialyzed to remove imidazole (down to 5 mm) and processed for an additional purification on the same type of affinity resin.
Absorbance spectroscopy
A stock solution of hemin (5 mm) was prepared by dissolution in NaOH (0.1 m) as described previously (7). Fresh stock solution was diluted (1:1000), and hemin concentration was determined at 385 nm using an extinction coefficient of 58,400 liters mol−1 cm−1. Association of proteins with heme was determined by adding increasing amounts of a protein (purified Str3(522–576) or Str3Y530A/Y534A/S552A/Y557A(522–576); 0–20 μm) to hemin (2 μm) in 40% DMSO buffered with 20 mm HEPES (pH 7.4). Differences in absorption spectra over a range of 350–700 nm were recorded using a DU730 spectrophotometer (Beckman Coulter). Changes in absorbance at the Soret peak served to monitor formation of the protein-heme complex and determination of the dissociation constant (KD) as described previously (7). Data were analyzed using GraphPad Prism version 7 software.
Author contributions
V. N. planned, designed, and performed most of the experiments. T. M. planned and obtained several preliminary experimental results as well as performed numerous bioinformatics analyses. V. N., T. M., and S. L. analyzed data. V. N., T. M., and S. L. conceptualized research and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We are grateful to Dr. Gilles Dupuis for critical reading of the manuscript and for valuable comments.
This work was supported by Natural Sciences and Engineering Research Council of Canada (NSERC) Grant RGPIN-2015/2020-04878 (to S. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- ALA
- δ-aminolevulinate
- Dip
- 2,2′-dipyridyl
- GFP
- green fluorescent protein
- PCNA
- proliferating cell nuclear antigen
- TBS
- Tris-buffered saline
- YES
- yeast extract plus supplements
- ZnMP
- zinc (II) mesoporphyrin
- MFS
- major facilitator superfamily
- FeLV-C
- feline leukemia virus subgroup C
- NEAT
- near-iron transporter.
References
- 1. Severance S., and Hamza I. (2009) Trafficking of heme and porphyrins in metazoa. Chem. Rev. 109, 4596–4616 10.1021/cr9001116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hamza I., and Dailey H. A. (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 1823, 1617–1632 10.1016/j.bbamcr.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tsiftsoglou A. S., Tsamadou A. I., and Papadopoulou L. C. (2006) Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 111, 327–345 10.1016/j.pharmthera.2005.10.017 [DOI] [PubMed] [Google Scholar]
- 4. Yuan X., Rietzschel N., Kwon H., Walter Nuno A. B., Hanna D. A., Phillips J. D., Raven E. L., Reddi A. R., and Hamza I. (2016) Regulation of intracellular heme trafficking revealed by subcellular reporters. Proc. Natl. Acad. Sci. U.S.A. 113, E5144–E5152 10.1073/pnas.1609865113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reddi A. R., and Hamza I. (2016) Heme mobilization in animals: a metallolipid's journey. Acc. Chem. Res. 49, 1104–1110 10.1021/acs.accounts.5b00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Korolnek T., and Hamza I. (2014) Like iron in the blood of the people: the requirement for heme trafficking in iron metabolism. Front. Pharmacol. 5, 126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mourer T., Jacques J. F., Brault A., Bisaillon M., and Labbé S. (2015) Shu1 is a cell-surface protein involved in iron acquisition from heme in Schizosaccharomyces pombe. J. Biol. Chem. 290, 10176–10190 10.1074/jbc.M115.642058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mourer T., Normant V., and Labbé S. (2017) Heme assimilation in Schizosaccharomyces pombe requires cell-surface-anchored protein Shu1 and vacuolar transporter Abc3. J. Biol. Chem. 292, 4898–4912 10.1074/jbc.M117.776807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Askwith C., and Kaplan J. (1997) An oxidase-permease-based iron transport system in Schizosaccharomyces pombe and its expression in Saccharomyces cerevisiae. J. Biol. Chem. 272, 401–405 10.1074/jbc.272.1.401 [DOI] [PubMed] [Google Scholar]
- 10. Brault A., Mourer T., and Labbé S. (2015) Molecular basis of the regulation of iron homeostasis in fission and filamentous yeasts. IUBMB Life 67, 801–815 10.1002/iub.1441 [DOI] [PubMed] [Google Scholar]
- 11. Pelletier B., Beaudoin J., Philpott C. C., and Labbé S. (2003) Fep1 represses expression of the fission yeast Schizosaccharomyces pombe siderophore-iron transport system. Nucleic Acids Res. 31, 4332–4344 10.1093/nar/gkg647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Quistgaard E. M., Löw C., Guettou F., and Nordlund P. (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 10.1038/nrm.2015.25 [DOI] [PubMed] [Google Scholar]
- 13. Yan N. (2015) Structural biology of the major facilitator superfamily transporters. Annu. Rev. Biophys. 44, 257–283 10.1146/annurev-biophys-060414-033901 [DOI] [PubMed] [Google Scholar]
- 14. Schrettl M., Winkelmann G., and Haas H. (2004) Ferrichrome in Schizosaccharomyces pombe: an iron transport and iron storage compound. Biometals 17, 647–654 10.1007/s10534-004-1230-z [DOI] [PubMed] [Google Scholar]
- 15. Matsuyama A., Arai R., Yashiroda Y., Shirai A., Kamata A., Sekido S., Kobayashi Y., Hashimoto A., Hamamoto M., Hiraoka Y., Horinouchi S., and Yoshida M. (2006) ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 24, 841–847 10.1038/nbt1222 [DOI] [PubMed] [Google Scholar]
- 16. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 10.1038/nmeth.3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Protchenko O., Rodriguez-Suarez R., Androphy R., Bussey H., and Philpott C. C. (2006) A screen for genes of heme uptake identifies the FLC family required for import of FAD into the endoplasmic reticulum. J. Biol. Chem. 281, 21445–21457 10.1074/jbc.M512812200 [DOI] [PubMed] [Google Scholar]
- 18. Weissman Z., and Kornitzer D. (2004) A family of Candida cell surface haem-binding proteins involved in haemin and haemoglobin-iron utilization. Mol. Microbiol. 53, 1209–1220 10.1111/j.1365-2958.2004.04199.x [DOI] [PubMed] [Google Scholar]
- 19. Weissman Z., Shemer R., Conibear E., and Kornitzer D. (2008) An endocytic mechanism for haemoglobin-iron acquisition in Candida albicans. Mol. Microbiol. 69, 201–217 10.1111/j.1365-2958.2008.06277.x [DOI] [PubMed] [Google Scholar]
- 20. Kuznets G., Vigonsky E., Weissman Z., Lalli D., Gildor T., Kauffman S. J., Turano P., Becker J., Lewinson O., and Kornitzer D. (2014) A relay network of extracellular heme-binding proteins drives C. albicans iron acquisition from hemoglobin. PLoS Pathog. 10, e1004407 10.1371/journal.ppat.1004407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ambesi A., Miranda M., Petrov V. V., and Slayman C. W. (2000) Biogenesis and function of the yeast plasma-membrane H+-ATPase. J. Exp. Biol. 203, 155–160 [DOI] [PubMed] [Google Scholar]
- 22. Pouliot B., Jbel M., Mercier A., and Labbé S. (2010) abc3+ encodes an iron-regulated vacuolar ABC-type transporter in Schizosaccharomyces pombe. Eukaryot. Cell 9, 59–73 10.1128/EC.00262-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nevitt T., and Thiele D. J. (2011) Host iron withholding demands siderophore utilization for Candida glabrata to survive macrophage killing. PLoS Pathog. 7, e1001322 10.1371/journal.ppat.1001322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Honsa E. S., Maresso A. W., and Highlander S. K. (2014) Molecular and evolutionary analysis of NEAr-iron Transporter (NEAT) domains. PLoS One 9, e104794 10.1371/journal.pone.0104794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grigg J. C., Ukpabi G., Gaudin C. F., and Murphy M. E. (2010) Structural biology of heme binding in the Staphylococcus aureus Isd system. J. Inorg. Biochem. 104, 341–348 10.1016/j.jinorgbio.2009.09.012 [DOI] [PubMed] [Google Scholar]
- 26. Mazmanian S. K., Skaar E. P., Gaspar A. H., Humayun M., Gornicki P., Jelenska J., Joachmiak A., Missiakas D. M., and Schneewind O. (2003) Passage of heme-iron across the envelope of Staphylococcus aureus. Science 299, 906–909 10.1126/science.1081147 [DOI] [PubMed] [Google Scholar]
- 27. Chiabrando D., Marro S., Mercurio S., Giorgi C., Petrillo S., Vinchi F., Fiorito V., Fagoonee S., Camporeale A., Turco E., Merlo G. R., Silengo L., Altruda F., Pinton P., and Tolosano E. (2012) The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 122, 4569–4579 10.1172/JCI62422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Duffy S. P., Shing J., Saraon P., Berger L. C., Eiden M. V., Wilde A., and Tailor C. S. (2010) The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell. Biol. 30, 5318–5324 10.1128/MCB.00690-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keel S. B., Doty R. T., Yang Z., Quigley J. G., Chen J., Knoblaugh S., Kingsley P. D., De Domenico I., Vaughn M. B., Kaplan J., Palis J., and Abkowitz J. L. (2008) A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 319, 825–828 10.1126/science.1151133 [DOI] [PubMed] [Google Scholar]
- 30. Khan A. A., and Quigley J. G. (2013) Heme and FLVCR-related transporter families SLC48 and SLC49. Mol. Aspects Med. 34, 669–682 10.1016/j.mam.2012.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quigley J. G., Yang Z., Worthington M. T., Phillips J. D., Sabo K. M., Sabath D. E., Berg C. L., Sassa S., Wood B. L., and Abkowitz J. L. (2004) Identification of a human heme exporter that is essential for erythropoiesis. Cell 118, 757–766 10.1016/j.cell.2004.08.014 [DOI] [PubMed] [Google Scholar]
- 32. Brown J. K., Fung C., and Tailor C. S. (2006) Comprehensive mapping of receptor-functioning domains in feline leukemia virus subgroup C receptor FLVCR1. J. Virol. 80, 1742–1751 10.1128/JVI.80.4.1742-1751.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khan A. A., and Quigley J. G. (2011) Control of intracellular heme levels: heme transporters and heme oxygenases. Biochim. Biophys. Acta 1813, 668–682 10.1016/j.bbamcr.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sabatinos S. A., and Forsburg S. L. (2010) Molecular genetics of Schizosaccharomyces pombe. Methods Enzymol. 470, 759–795 10.1016/S0076-6879(10)70032-X [DOI] [PubMed] [Google Scholar]
- 35. Mumberg D., Müller R., and Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 10.1016/0378-1119(95)00037-7 [DOI] [PubMed] [Google Scholar]
- 36. Beaudoin J., Laliberté J., and Labbé S. (2006) Functional dissection of Ctr4 and Ctr5 amino-terminal regions reveals motifs with redundant roles in copper transport. Microbiology 152, 209–222 10.1099/mic.0.28392-0 [DOI] [PubMed] [Google Scholar]
- 37. Kim J., and Hirsch J. P. (1998) A nucleolar protein that affects mating efficiency in Saccharomyces cerevisiae by altering the morphological response to pheromone. Genetics 149, 795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ho S. N., Hunt H. D., Horton R. M., Pullen J. K., and Pease L. R. (1989) Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77, 51–59 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 39. Keeney J. B., and Boeke J. D. (1994) Efficient targeted integration at leu1–32 and ura4–294 in Schizosaccharomyces pombe. Genetics 136, 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen D., Toone W. M., Mata J., Lyne R., Burns G., Kivinen K., Brazma A., Jones N., and Bähler J. (2003) Global transcriptional responses of fission yeast to environmental stress. Mol. Biol. Cell 14, 214–229 10.1091/mbc.E02-08-0499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mercier A., Watt S., Bähler J., and Labbé S. (2008) Key function for the CCAAT-binding factor Php4 to regulate gene expression in response to iron deficiency in fission yeast. Eukaryot. Cell 7, 493–508 10.1128/EC.00446-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mercier A., Pelletier B., and Labbé S. (2006) A transcription factor cascade involving Fep1 and the CCAAT-binding factor Php4 regulates gene expression in response to iron deficiency in the fission yeast Schizosaccharomyces pombe. Eukaryot. Cell 5, 1866–1881 10.1128/EC.00199-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pelletier B., Beaudoin J., Mukai Y., and Labbé S. (2002) Fep1, an iron sensor regulating iron transporter gene expression in Schizosaccharomyces pombe. J. Biol. Chem. 277, 22950–22958 10.1074/jbc.M202682200 [DOI] [PubMed] [Google Scholar]
- 44. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., and Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 10.1002/(SICI)1097-0061(19980130)14:2%3C115::AID-YEA204%3E3.0.CO%3B2-2 [DOI] [PubMed] [Google Scholar]