

Abstract
Aminoacyl-tRNA synthetases (ARSs) are ubiquitously expressed enzymes implicated in several dominant and recessive disease phenotypes. The canonical function of ARSs is to couple an amino acid to a cognate transfer RNA (tRNA). We identified three novel disease-associated missense mutations in the alanyl-tRNA synthetase (AARS) gene in three families with dominant axonal Charcot–Marie–Tooth (CMT) disease. Two mutations (p.Arg326Trp and p.Glu337Lys) are located near a recurrent pathologic change in AARS, p.Arg329His. The third (p.Ser627Leu) is in the editing domain of the protein in which hitherto only mutations associated with recessive encephalopathies have been described. Yeast complementation assays demonstrated that two mutations (p.Ser627Leu and p.Arg326Trp) represent loss-of-function alleles, while the third (p.Glu337Lys) represents a hypermorphic allele. Further, aminoacylation assays confirmed that the third mutation (p.Glu337Lys) increases tRNA charging velocity. To test the effect of each mutation in the context of a vertebrate nervous system, we developed a zebrafish assay. Remarkably, all three mutations caused a pathological phenotype of neural abnormalities when expressed in zebrafish, while expression of the human wild-type messenger RNA (mRNA) did not. Our data indicate that not only functional null or hypomorphic alleles, but also hypermorphic AARS alleles can cause dominantly inherited axonal CMT disease.
Introduction
Aminoacyl-tRNA synthetases (ARS) are ubiquitously expressed enzymes with a basic role in protein synthesis. The canonical function of these enzymes is to catalyse the transfer of the cognate amino acid to their tRNAs. The human nuclear genome contains 37 ARS loci encoding the ARS enzymes in the cytoplasm and mitochondria. Over the last 2 decades, mutations in 31 ARS loci have been associated with a myriad of dominant and recessive disease phenotypes (1) with the vast majority of alleles causing a loss-of-function effect (2).
The sole dominant phenotype associated with ARS mutations is peripheral neuropathy, also known as Charcot–Marie–Tooth (CMT) disease. CMT is a hereditary disease affecting the peripheral nerves ultimately leading to loss of integrity and subsequent wasting of the distal muscles and varying degrees of sensory deficits. Foot deformities may be present as well. CMT is heterogeneous both at the genetic and clinical level. Many genes have been described to be associated with CMT and the number of genes is still expanding. Mutations in five cytoplasmic tRNA synthetases have also been described as the cause for CMT type 2, a subtype of hereditary neuropathy with a dominant mode of inheritance in which the axon is primarily affected. Specifically, mutations in GARS, YARS, alanyl-tRNA synthetase (AARS), HARS, and WARS have been reported to cause dominant or intermediate dominant CMT type 2 (3–7).
The mechanism through which ARS mutations cause dominant CMT disease is not fully known, and whether mutations act as a dominant-negative and/or have acquired a gain-of-function has been a matter of debate (1). Similarly, the exact role of complete (or partial) loss of the amino-acylation capacity is unclear, in contrast to most recessive mutations that have been described as loss-of-function mutations. The pathogenic mutations found in the AARS gene as well as those in the other CMT-associated tRNA synthetases are not clustered to a specific domain. Consequently, it is unknown whether a test for its canonical function such as an in vitro aminoacylation assay is the correct read-out for pathogenicity.
Remarkably, all CMT-causing ARS mutations give rise to missense changes or in-frame deletions whereas nonsense or frameshift mutations do not occur (1). Alternatively, one normal allele in addition to a null-allele might be enough for the production of sufficient amounts of the enzyme and thus not result in a clinical phenotype. However, as noted above, homozygosity or compound heterozygosity for loss-of-function mutations in tRNA synthetases have been reported but generally, they lead to a more severe clinical phenotype. Homozygosity for p.Tyr454Ser HARS gives rise to Usher syndrome type IIIb, with onset in early childhood resulting in blindness, deafness and periods of hallucinating fever (8). Compound heterozygosity for mutations in QARS lead to progressive microcephaly with onset in infancy with or without involvement of the cerebellum (9,10) or early onset epileptic encephalopathy (11). Similarly, loss-of-function mutations in AARS have been reported to cause autosomal recessive infantile epileptic encephalopathy with myelination defects (12,13). A spontaneously occurring sti/sticky (sti) mutation in the mouse Aars gene has also been identified which results in a recessive ataxia starting at 6 weeks of age and with concomitant Purkinje cell loss (14). The mutation causes a deficiency in the editing capacity of the enzyme for quality control, thus resulting in protein misfolding, formation of aggregates and consequent degeneration of the cells in which the mutant protein is expressed.
In this study we report three families each diagnosed with CMT type 2. A custom-made sequence capture containing all exons of known CMT-associated genes, followed by next generation sequencing, revealed a potentially pathogenic AARS mutation in each of these families with complete co-segregation with the disease. A complementation test in yeast showed that p.Ser627Leu and p.Arg326Trp AARS are loss-of-function alleles, but that p.Glu337Lys unexpectedly increases the function of the enzyme. Consistent with the yeast data, aminoacylation assays to test the enzyme kinetics of p.Glu337Lys demonstrated an increase in catalytic activity. However, because neither the yeast rescue experiments nor the aminoacylation assays assess the functional consequences of the variants in the context of a multi-cellular organism with a nervous system, we sought to set up an in vivo assay to monitor the pathogenicity of the mutations. Injection of human mutated AARS mRNAs into 1-cell stage Danio rerio embryos clearly caused toxicity while injection of the same amount of wild-type mRNA did not produce any significant effects. We conclude that these three novel missense mutations in the AARS gene are the cause of the disease in these families. Moreover, our data for the first time associate both hypomorphic and hypermorphic ARS alleles with CMT disease.
Results
Identification of novel missense mutations in the AARS gene
Although many genes have been implicated in CMT type 2, molecular diagnosis remains difficult due to the low frequency of occurrence with which each gene contributes to the CMT mutation spectrum (15). We collected 12 CMT2 families with unknown cause, having excluded mutations in genes most frequently implicated in CMT. Genomic DNA from one affected individual per family was used for a custom-made sequence capture that contained the exons of 45 CMT-associated genes followed by pyrosequencing. Per individual we found 218–300 variations with 1–9 variations affecting the encoded proteins. In family H9, only one potential pathogenic mutation was found in exon 14 of the AARS gene: c.1880 C > T; p.Ser627Leu (NM_001605) which was predicted to be pathogenic by all prediction programs available in the Alamut Visual 2.9.0 package (SIFT, Mutation Taster, Polyphen-2). In family H19, another missense variation was found in exon 8 of the same gene, c.1009G > A, p.Glu337Lys which was a predicted pathogenic mutation according to Mutation Taster and Polyphen-2. Both AARS mutations affect a conserved residue and are absent from public databases. Sanger sequence analysis was used to confirm the identified changes in each patient. Screening of routinely processed diagnostic samples yielded a third family, L21, with a potentially pathogenic mutation: c.976C > T, p.Arg326Trp that was detected only once in publicly available databases (0,000008/1 ExAc), also in exon 8 of the AARS gene. All three prediction programs used (SIFT, Polyphen-2 and Mutation Taster) assessed this variation as a pathogenic change. The three pedigrees are depicted in Figure 1.
Figure 1.
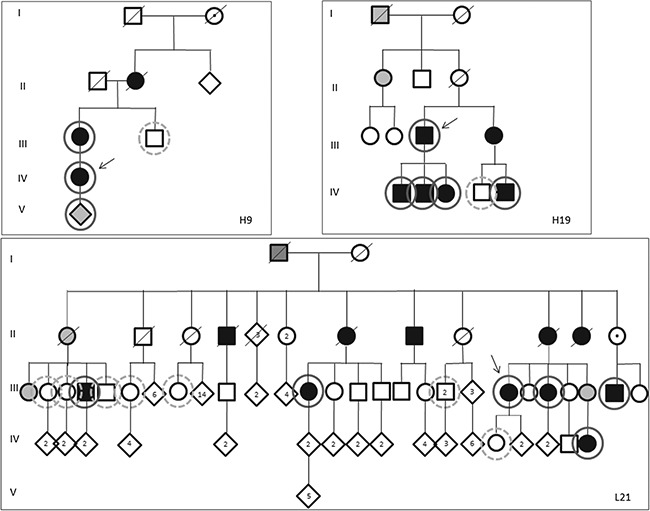
Pedigrees of families. Pedigrees of families with hereditary neuropathies as indicated in the figure. Tested individuals are marked by circles; solid line: tested positive for respective AARS mutation, dashed line: tested negative for AARS mutation. III.41,42: only one of the two individuals were tested. The mutations found were the following: c.1880C > T, p.Ser627Leu (H9), c.1009G > A, p.Glu337Lys (H19) and c.976GC > T, p.Arg326Trp (L21). Filled symbols indicate affected patients. Grey symbols: probably affected, said to be affected or no detailed neurologic examination. Open symbols: not affected. The family individuals are numbered consecutively from left to right. The index patient of each family is indicated by an arrow.
AARS sequence alterations lead to a sensorimotor polyneuropathy with axonal, and sometimes demyelinating, features
The details of clinical and electrophysiologic findings of available affected family members are given in Table 1. Family H09 is composed of two affected individuals (mother and daughter son).(individual IV-1 in pedigree; index patient). The former had distal weakness and wasting of legs and arms, and also distal sensory disturbances. The latter had calf hypertrophy and weakness of the anterior distal muscles.
Table 1.
Clinical and electrophysiologic findings of clinically affected persons of the families H9, H19 and L21
| ID in pedigree, gender | Age of onset (years) | Age at examination | Clinical features | EMG findings |
|---|---|---|---|---|
| H09-III-1, F | 30 years | 50 years | Distal atrophy and paresis arms (MRC grade 3) and legs (MRC 1–2). Weakness quadriceps MRC 4. Flat feet. Distal sensory disturbances arms and legs. Impaired joint position sense hallux, absent vibration sense of the legs Areflexia. |
Left ulnar MNCVa forearm 43 m/s (ADM)b; right ulnar MNCV forearm 41 m/s. No CMAPsc recordable of median (APB)d, peroneal (EDB)e and tibial (AHB)f nerves. No SNAPsg recordable of right median (2nd finger), left ulnar (5th finger) and right sural (lateral malleolus) nerves. Signs of denervation and reinnervation in predominantly distal leg and arm muscles.Conclusion: severe axonal sensorimotor polyneuropathy. |
| H09-IV, F (index) | unknown | 26 years | Calf hypertrophy. Paresis anterior tibial and peroneal muscles (MRC 4). Pes cavus. Decreased vibration sense hallux. Absent Achilles tendon jerks. |
Right median MNCV forearm 39 m/s, left ulnar MNCV forearm 52 m/s. No or very low CMAP amplitudes peroneal (EDB) both sides and left tibial (AHB) nerve. Decreased or no SNAP amplitudes right median, left ulnar and right sural with decreased SNCVsh. No denervation, signs of reinnervation in predominantly distal leg muscles.Conclusion: severe axonal sensorimotor polyneuropathy. |
| H19-III-3, M (index) | 7 years | 62 years | Atrophy lower legs. Paralysis anterior tibialis muscles and toe extensors, paresis MRC 4 calf muscles. Flat feet. Distal sensory disturbances of the legs. Decreased vibration sense of the feet. Impaired joint position hallux. Absent Achilles tendon jerks. | Right median MNCV (APB) forearm 34 m/s; left ulnar MNCV (ADM) forearm 40 m/s. No recordable CMAPs peroneal both sides, EDB) and tibial (AHB) nerves. No recordable SNAPs right median (2nd finger), left ulnar (5th finger) and right sural nerve (lateral malleolus). Signs of reinnervation in the predominantly distal leg and arm muscles.Conclusion: severe sensorimotor polyneuropathy with axonal and demyelinating features. |
| H19-III-4, F | 17 years | 65 years | Paralysis feet, otherwise no examination performed. | Median MNCVs forearm left 11 m/s and right 17 m/s, although with very low CMAP amplitudes (APB) both sides; Ulnar MNCV forearm both sides 40 m/s, upper arm left 32 m/s and right 35 m/s with relatively normal CMAP amplitudes (ADM). Very low to non-recordable SNAP amplitudes in arms and legs. Signs of de- and reinnervation in predominantly distal arm muscles (legs not assessed). Conclusion: severe sensorimotor polyneuropathy with axonal and demyelinating features. |
| H19-IV-1, M | Early childhood | 30 years | Atrophy and weakness MRC 4+ of distal leg muscles. Pes cavus and hammer toes. Slight sensory disturbances of the distal legs. Decreased Achilles tendon jerks. | Right median MNCV (APB) forearm 38 m/s; left ulnar MNCV (ADM) forearm 42 m/s. No CMAPs recordable left peroneal (EDB); decreased left CMAP amplitude of AHB muscle with decreased MNCV. No SNAPs recordable in arms and legs. No needle-EMG available. Conclusion: severe sensorimotor polyneuropathy with axonal and demyelinating features. |
| H19-IV-2, M | unknown | 26 years | Weakness (MRC 4) of the distal arm and leg muscles. No foot deformities. No sensory disturbances. Absent Achilles tendon jerks. Extensor plantar response on the left. | Right median MNCV (APB) forearm 40 m/s; left ulnar MNCV (ADQ)i forearm 39 m/s. No CMAPs recordable from the peroneal (both sides) and left tibial nerve. Very low to non-recordable SNAPs in the arms and legs. Signs of reinnervation in distal and proximal arm and leg muscles. Conclusion: severe sensorimotor axonal polyneuropathy. |
| H19-IV-3, F | unknown | 23 years | Data not available | Right median MNCV (APB) forearm 37 m/s; left ulnar MNCV (ADM) forearm 35 m/s; upper arm 50 m/s. No CMAPs peroneal (both sides) and left tibial nerve. Very low to non-recordable SNAPs in arms and legs. Signs of denervation in the distal leg muscles and signs of reinnervation in distal and proximal arm and leg muscles. Conclusion: severe sensorimotor polyneuropathy with axonal and demyelinating features. |
Table 1.
(Continued)
| ID in pedigree, gender | Age of onset (years) | Age at examination | Clinical features | EMG findings |
|---|---|---|---|---|
| H19-IV-5, M | 35 years | 38 years | Atrophy and weakness distal legs (MRC 4).Pes cavus, hammer toes and contractures Achilles tendons. Decreased vibration sense hallux. Absent Achilles tendon jerks, extensor plantar response on the right. | Right median MNCV (APB) forearm 39 m/s; left ulnar MNCV (ADQ) forearm 46 m/s. Very low to non-recordable CMAPs peroneal (EDB) both sides, left tibial (AHB)g nerve. Very low to non-recordable SNAPs of right median (2nd finger), left ulnar (5th finger) and right sural nerve (lateral malleolus). Signs of denervation and reinnervation in predominantly distal leg and arm muscles. Conclusion: severe sensorimotor axonal polyneuropathy. |
| L21 II.7 F | 60 years | 65 years | Bilateral paresis foot extensors (MRC 4). Pes cavus. Hypesthesia of left lower leg. Absent Achilles and knee tendon reflexes at both sides. | Near normal conduction studies of median and ulnar nerves. No recordable CMAPs and SNAPs in distal legs. Conclusion: severe sensorimotor axonal polyneuropathy. |
| L21 II.10 M | childhood | 62 years | Distal paresis (MRC 3-4). Hypotonic feet, steppage gait. No sensory complaints. Absent Achilles tendon jerks. | No data available |
| L21 II.13 F | 29 | 62 | Distal paresis. Atrophy of lower legs. Steppage gait. No sensory disturbances. Nystagmus. Reflexes arms: normal to brisk, absent Achilles tendon jerks, brisk knee tendon reflex. Originally diagnosed with multiple sclerosis. | Documented as severe sensorimotor axonal polyneuropathy. No recordings available. |
| L21II.4 M | 10 | 70 | Diffuse paresis of feet (MRC2-3). Steppage gait. Hypotonic feet. Absent Achilles tendon jerks. | No data available |
| L21 III.48 F | 30-40 | 50 | No paresis (MRC5). No sensory disturbances. Complaints of pain in legs and cramps in feet. Reflexes normal. | Right ulnar MNCV (ADM) forearm 58 m/s. Very low CMAP amplitudes right peroneal nerve (EDB) and low CMAP amplitudes left tibial nerve (AHB) with reduced MNCV. Non-recordable SNAP sural nerve (lateral malleolus) both sides and reduced SNAP amplitude right radial nerve (thumb). No signs of denervation or reinnervation in distal leg and arm muscles Conclusion: sensorimotor axonal polyneuropathy. |
| L21 III.46 F (index) | 35-40 | 42 | Distal paresis, specifically toe extensors. Foot drop, steppage gait, pes cavus.. No sensory disturbances. Decreased deep tendon reflexes. No Achilles tendon jerks (both sides) or knee jerk (right). | No recordable CMAPs in distal legs and very low SNAP amplitude sural nerve (lateral malleolus). Conclusion: severe sensorimotor axonal polyneuropathy. |
| L21 III.35 F | 44 | 49 | Diffuse paresis of psoas foot extensors MRC 4), toe extensors and extensors hallicus (MRC 3 (right), MRC 4 (left)). Pes cavus. No vibration sense toes. Absent Achilles tendon jerks, other reflexes: brisk. | Right ulnar MNCV (ADM) forearm 51 m/s. Non-recordable CMAPs right peroneal (EDB) and left tibial (AHB) nerves. Non-recordable SNAP left sural nerve (lateral malleolus). Signs of reinnervation in distal leg muscle. Conclusion: severe sensorimotor axonal polyneuropathy. |
The MRC grading is the Medical Research Council grading of muscle strength. Abbreviations used are: aMNCV = motor nerve conduction velocity; bADM = abductor digiti minimi muscle; cCMAP = compound muscle action potential; dAPB = abductor pollicis brevis muscle; eEDB = extensor digitorum brevis muscle; fAHB = abductor hallucis brevis muscle; gSNAP = sensory nerve action potential; hSNCV = sensory nerve conduction velocity; iADQ = abductor digiti quinti muscle.
Age of onset was in the third decade of life. Electromyography (EMG) demonstrated a severe sensorimotor axonal neuropathy.
Six affected members of family H19 showed atrophy and weakness of the distal legs, and in some patients the distal arm muscles were also affected, with variable (minor) distal sensory disturbances. Some patients had extensor plantar responses. Age of onset ranged from the first to third decade of life. EMG demonstrated a sensorimotor polyneuropathy with axonal, and in four out of six, including the index person (H19.III-3) also demyelinating features were found.
Eleven patients were affected in family L21, for seven patients more detailed information was available. Affected patients in family L21 showed variable weakness of the distal legs and no or minimal distal sensory disturbances. One patient (individual III.48) was asymptomatic upon clinical examination. Age of onset was in the first to third decade of life. EMG was consistent with a (severe) sensorimotor axonal neuropathy.
The AARS sequence alterations co-segregate with the disease in the respective families
After confirmation of the identified sequence variations, we analysed the remaining available family members for the presence of the mutation. For family H9, three additional family members could be assessed: one was not examined in detail due to young age but was suspected of carrying the disease, one was diagnosed as affected and one was unaffected. The mutation was indeed found in the affected mother of the index patient and her child, and was absent in an unaffected uncle. For family H19, four additional affected family members and one unaffected family member were available for analysis. Again, all affected persons carried the mutation while it was absent in the unaffected person. The third family, L21, is a large family. The index person is individual III.46 in the pedigree. Six affected and seven unaffected family members were tested and again, the mutation was present in all individuals with the disease and absent in unaffected family members. The clinical phenotype was highly variable, ranging from an asymptomatic obligate carrier to a patient who was wheelchair-bound.
All three AARS missense variants affect gene function in yeast complementation assays
To assess the pathogenicity of the identified AARS sequence alterations, we used a previously described yeast complementation assay to determine the functional consequences of each AARS variant (16,17). We compared the ability of wild-type and mutant human AARS to complement the loss of endogenous yeast ALA1 (the yeast ortholog of AARS). Complementation studies were performed in a previously validated haploid yeast strain deleted for the endogenous ALA1 gene and maintained viable with ALA1 on a URA3-bearing vector (16,17). Wild-type or mutant AARS expression constructs or empty control constructs were transformed into the above yeast strain and grown on 5-FOA, which selects for spontaneous loss of the URA3-bearing maintenance vector [Fig. 2A and B, (18)]. The empty plasmid did not support yeast growth at 30°C or 37°C confirming that ALA1 is an essential gene. Wild-type AARS supported some yeast growth at 30°C and more robust yeast growth at 37°C, which indicates that human AARS can complement the loss of the endogenous ALA1 locus, consistent with previous findings (19). The p.Arg326Trp AARS variant was unable to support any yeast growth at 30°C, suggesting that p.Arg326Trp represents a loss-of-function (i.e. functional null) allele. The p.Ser627Leu AARS variant demonstrated reduced yeast viability compared to wild-type AARS at 30°C and an even more dramatic reduction in viability at 37°C, consistent with a loss-of-function (i.e. hypomorphic) allele. The p.Glu337Lys AARS variant, however, unexpectedly showed increased yeast cell growth compared to wild-type AARS at both 30 and 37°C (Fig. 2A and B) indicating that this is a gain-of-function (i.e. hypermorphic allele). However, lysine is the wild-type amino acid at this residue in yeast (Fig. 2C), making it necessary to test the enzyme activity of this specific mutation to rule out the possibility of an artefact of the yeast assay (see below). Interestingly, the discovery of p.Glu337Lys as a hypermorphic allele provided us with a more robust assay to test the loss-of-function effects of p.Arg326Trp and p.Ser627Leu; we therefore tested the ability of each mutation to rescue yeast cell growth in cis with p.Glu337Lys. p.Arg326Trp in cis with p.Glu337Lys was unable to support any yeast cell growth (Fig. 2A and B), confirming that this is a functional null allele. In contrast, p.Ser627Leu in cis with p.Glu337Lys supported yeast growth similar to p.Glu337Lys alone (Fig. 2A and B), indicating that the reduced growth caused by p.Ser627Leu can be rescued.
Figure 2.
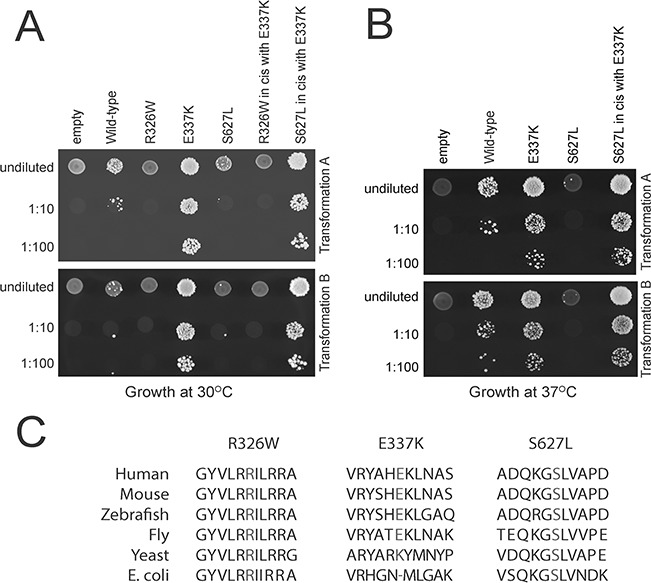
CMT-associated AARS mutations affect cellular growth in yeast. (A) Yeast lacking endogenous ALA1 (the yeast ortholog of AARS) were transformed with vectors containing wild-type AARS or mutant AARS or a vector without AARS insert (‘empty’). Transformation was performed twice (transformation A,B). Resulting cultures were plated undiluted or diluted (1:10 or 1:100) on media containing 5-FOA and grown at 30°C. (B) Cultures resulting from the same transformations as in A were plated undiluted or diluted on media containing 5-FOA and grown at 37°C. (C) Conservation of the affected amino acid residues. The position of each variant is shown along with flanking AARS amino acid residues from multiple, evolutionarily diverse species. The position of the affected residue is shown in red for each species.
p.Glu337Lys AARS demonstrates increased amino acid charging capacity
To further explore the hypermorphic effect of p.Glu337Lys AARS observed in yeast, we performed enzyme activity (aminoacylation) assays compared to the wild-type enzyme (Fig. 3). In steady-state assays under multiple turnover conditions, we showed that the mutant enzyme exhibited a 5-fold decrease in Km for tRNA relative to the wild-type enzyme (0.19 versus 1.13 μm), but a small decrease in the catalytic turnover number kcat (0.40 versus 0.61 s-1). These effects on kinetic parameters resulted in a nearly 4-fold increase in the catalytic efficiency of aminoacylation kcat/Km. This increase is consistent with the increased viability compared to wild-type AARS in the yeast complementation assay, supporting the notion that p.Glu337Lys is a hypermorphic allele.
Figure 3.
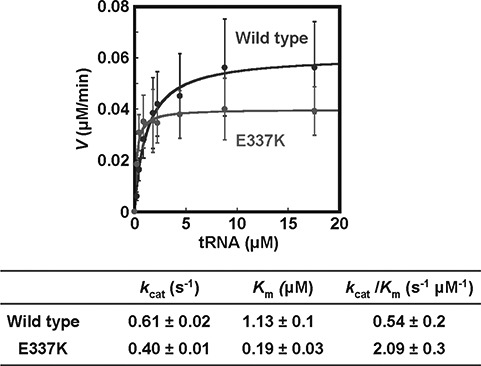
In vitro aminoacylation assays are suggestive of a gain-of-function mechanism for the p.p.Glu337Lys mutation. Steady state assays of aminoacylation of tRNAAla with 3H-alanine were performed at 37oC with each enzyme at 1.67 nm, and tRNAAla in varying concentrations. Error bars show standard deviations derived from averaging at least three independent sets of experiments.
Micro-injection of anti-sense splice morpholino oligonucleotides yields a CMT-associated phenotype in zebrafish
Yeast complementation and aminoacylation assays are limited in that they do not test the functional consequences of the AARS missense mutations in the context of a vertebrate nervous system. To address this, we developed a zebrafish assay. Previous research using injection into 1-cell stage embryos of Danio rerio demonstrated that targeting the start or splice site using anti-sense morpholino-oligonucleotides (MOs) directed against another CMT-associated gene, lrsam1, resulted in a clear morphologic phenotype in zebrafish (20). Therefore, we first assessed if two MOs directed against the translation start site of the zebrafish aars gene (ATG1-MO) or acceptor splice site of exon 14 (splice-MO) yielded a similar phenotype. Injection of an ATG-MO would rather mimic a recessive type of disease. Since a spontaneous recessive mutation was described in the sticky mouse (14), we expected a brain phenotype. However, except for a transient fuzziness in the brain in 15–40% of the ATG1-MO-injected embryos at 24 hpf, the majority otherwise developed normally over a wide range of concentrations as evaluated at 2–3 days post fertilization (dpf). In contrast, after optimization of the concentration used, the aars-splice MO yielded a similar phenotype as obtained for lrsam1 morphants (Table 2, Fig. 4). RT-PCR analysis of the targeted site in the aars-splice-MO-injected embryos demonstrated the presence of a larger reverse-transcriptase-polymerase-chain-reaction (RT-PCR) product, the amount of which increased upon use of a higher concentration of splice-MO, in addition to the wild-type RT-PCR product which had the expected size of 268 bp. (Fig. 5). Sequence analysis of the aberrantly spliced product showed that the larger product was 360 bp in size and that it also contained intron 13. The injected embryos may thus contain a truncated protein lacking the C-terminal and editing domain but with 12 additional amino-acids at the C-terminal tail, in addition to an excess of the wild-type product what resulted in a clearly aberrant morphology. To confirm that the morphology was at least partially based on neurologic abnormalities we performed a whole-mount immunohistochemical staining with znp-1, which detects the zebrafish synaptotagmin protein (Fig. 6). The segmented developmental pattern of motor neurons midline over each somite with normal branching not occurring before halfway is clearly less well organized as compared to that in wild-type embryos.
Table 2.
MO-injections in wild-type zebrafish (TL strain)
| Exp | conc.(um) | # injected | # survived | Abnormal fish (%) |
|---|---|---|---|---|
| 1 | 600 | 46 | 37 | 43% (13% severe) |
| 2 | 600 | 60 | 33 | 45% (15% severe) |
| 3 | 600 | 73 | 58 | 75% (53% severe) |
| 4 | 625 | 44 | 38 | 55% |
| 5 | 625 | 28 | 18 | 44% |
| 6 | 625 | 45 | 37 | 49% |
1 nl of splice-MO-dilution was injected with concentrations as indicated in the table. Separate columns indicate the number (#) of injected fish and surviving fish. Percentages of abnormal fish reflect the ratio of abnormal fish and total number of surviving fish.
Figure 4.
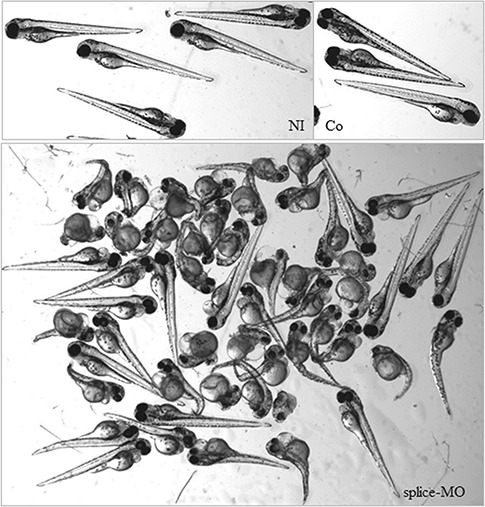
Morphology of zebrafish embryos at 72 hpf. Normal morphology was observed for control embryos at 72 hpf that were not injected (NI) or injected with control morpholino [GeneTools (Co)]. Splice-MO injected zebrafish showed abnormal morphology mostly consisting of shortened body axis, smaller eyes and curved bodies. Severe cases hardly developed a normal tail.
Figure 5.
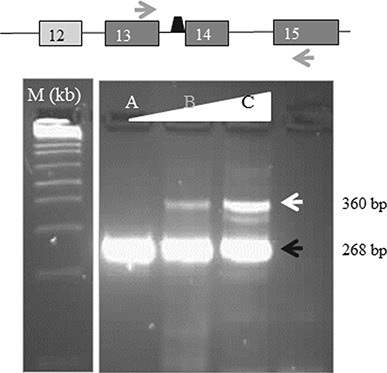
Injection of zebrafish embryos with splice-MO results in a concentration-dependant aberrant splicing. RT-PCR of splice MO-injected zebrafish with the position of the primers depicted by arrows and the position of the splice MO just preceding exon 14 of the zebrafish aars gene. Exons are indicated by numbered blocks. Sequence analysis of both products demonstrated that the expected wild-type product consisted of part of exon 13, exon 14 and part of exon 15 (black arrow, bottom panel; 268 bp) while the larger product consisted of part of exon 13, intron 13, exon 14 and part of exon 15 (white arrow, bottom panel; 360 bp). The bottom panel shows RT-PCR products of non-injected (A) or splice-MO injected zebrafish with 1 nl of 550 (B) and 650 um (C) solutions. As a molecular size marker (m) we used the 1 kb ladder (Invitrogen).
Figure 6.
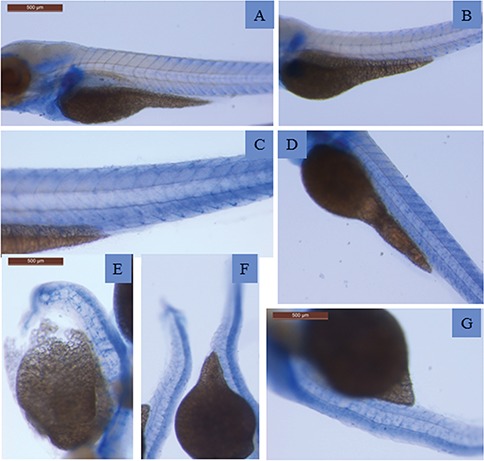
Whole mount immunohistochemical staining demonstrated aberrant patterns of synaptotagmin expression. Expression of synaptotagmin (blue staining) as detected by antibody znp-1 in wild-type zebrafish at 3 dpf (A) and in zebrafish injected at 1-cell stage with control MO (B,C) or with splice-MO (D–G) at 3 dpf. The immunostaining shows a less defined and increasingly irregular pattern as morphology grows worse.
Injection of human mutant AARS mRNAs leads to toxicity in zebrafish embryos
Based on the observed morphology of the splice-MO-injected zebrafish embryos and the fact that the identified AARS mutations lead to a dominant type of CMT, we reasoned that a direct injection of human mutant mRNA into 1-cell stage embryos would yield a similar phenotype as the splice-MO-injected embryos in cases of pathogenic mutations. We consequently introduced the patient mutations in the human AARS cDNA by in vitro mutagenesis and used in vitro transcription (IVT) to produce the wild-type and mutant mRNAs. Both wild-type and mutant constructs were checked by Sanger sequencing analysis. The quantity of the RNA was assessed using fluorometric measurement and equal amounts of mutant or wild-type mRNAs were injected into 1-cell stage embryos (Fig. 7, Fig. 8). Injection of AARS mutant mRNAs produced toxicity in the embryos while the same amount of wild-type mRNA did not result in an abnormal morphology (Fig. 8). The results were obtained using at least two independent IVT experiments. Injections were repeated and performed at several concentrations of the mRNAs (Table 3). To ensure that the identified differences were not due to large differences in translation efficiency, we analysed the level of human AARS in the injected zebrafish. The levels of expression were in the range of the endogenous zebrafish protein which was apparently recognized by this antibody (Fig. 7). Expression in the injected embryos is slightly increased compared to the non-injected controls with no marked differences between the mutants or the wild-type mRNA (Fig. 7D), suggesting that the differences in morphology in the injected embryos are not due to differences in expression. These results indicate that the mutations we identified are indeed pathogenic mutations.
Figure 7.
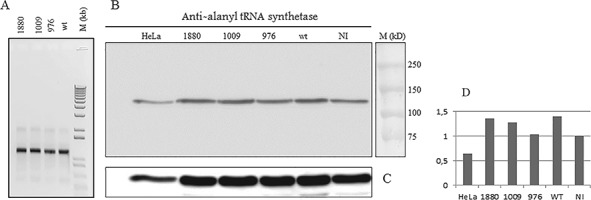
Western blot and RNA analysis of injected zebrafish embryos of 2 dpf shows no major changes in quantities of AARS mRNA and protein. Comparable quantities of AARS mRNA were obtained after IVT. For quality control, 500 ng of each RNA was run on an agarose gel (A). Analysis of AARS protein in zebrafish lysates of wild-type- (wt) or mutant-mRNA-injected or not injected controls (NI) is shown in (B). For loading control, anti-tubulin was used (C). A quantification of the expression of AARS protein corrected for loading differences expressed in arbitrary units as compared to the expression in non-injected controls (D). Molecular markers were dual precision protein marker (BioRad) and 1 kb ladder (Invitrogen).
Figure 8.
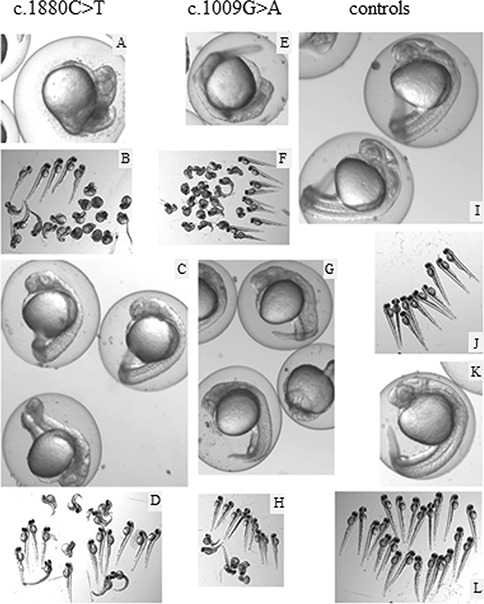
Zebrafish embryos injections of mutant mRNA (c.1009G > A, c.1880 C > T) lead to highly abnormal phenotypes. Embryos at 24 hpf (A,C,E,G,I,K) or 2 dpf (B,D,F,H,J,L) injected with 250 pg c.1880C > T, p.Ser627Leu (A–D), 200 pg c.1009G > A, p.Glu337Lys (E,F), 140 pg c.1009G > A and p.Glu337Lys (G,H) of mutant mRNAs or 475 pg wt mRNA (I,J) or non-injected controls (K,L). Wild-type mRNA injections were performed with 140–475 pg mRNA. The highest amount still did not affect the morphology of the embryos (K,L). At 2 dpf, a significant part of the surviving embryos injected with mutant mRNA is still affected.
Table 3.
Injections into 1-cell stage embryos of in vitro transcribed AARS constructs and non-injected controls
| Exp | Quantity | Construct | # | Abnormal at 1 dpf | Abnormal at 2 dpf |
|---|---|---|---|---|---|
| 1. | 316 pg | c.1880C > T | 24 | 10/20 blurry heads | 4/22 (18%) |
| 318 pg | c.1009G > A | 34 | all abnormal | 19/21 (90%) | |
| 308 pg | wt | 24 | 1/24 | 1/24 (4.2%) | |
| non-injected | - | 6 | 0/6 | 0/5 (0%) | |
| 2. | 475 pg | c.1880C > T | 23 | nearly all blurry heads | 12/23 (52%) |
| 475 pg | wt | 23 | 0 | 2/22 (9.1%) | |
| non-injected | - | 7 | 0 | 0 (0%) | |
| 3. | 250 pg | c.1009G > A | 33 | 27/30 | 20/23 (87%) |
| 250 pg | wt | 23 | 2 mild | 1/22 (4.5%) | |
| non-injected | - | 3 | 1 flatter head | 0/3 (0%) | |
| 4. | 200 pg | c.1009G > A | 33 | 27/30; 27% severe | 24/30 (80%); 57% severe |
| 140 pg | c.1009G > A | 15 | 11/14; 47% severe, 4 blurry heads | 7/14 (50%) | |
| 200 pg | wt | 38 | 1/38 | 0/34 (0%) | |
| non-injected | - | 10 | 0 | 0/10 (0%) | |
| 5. | 200 pg | c.1009G > A | 45 | All; 31% severe | 28/35 (80%); 46% severe |
| 140 pg | c.1009G > A | 36 | 7 severe | 19/33 (58%); 27% severe | |
| 200 pg | wt | 37 | 1–2 | 2/36 (5.5%) | |
| non-injected | - | 26 | 0 | 0/26 (0%) | |
| 6. | 500 ng | c.1009G > A | 37 | 18/36; 11% severe | 13/31 (42%) |
| 500 ng | c.1880C > T | 54 | 15/52 | 10/50 (20%) | |
| 500 ng | wt | 53 | 5/53 | 4/51 (7.8%) | |
| 500 ng | c.976 | 32 | 16/32 | 13/30 (43%) | |
| non-injected | - | 62 | 0 | 3/62 (4.8%) | |
| 7. | 600 ng | c.1009G > A | 23 | 7/17 | 6/16 (38%) |
| 600 ng | c.1880C > T | 34 | 8/24 | 12/23 (52%) | |
| 600 ng | c.976 | 17 | 7/14 | 5/13 (38%) | |
| 600 ng | wt | 26 | 4/25 | 3/21 (14%) | |
| non-injected | - | 18 | - | 0/17 (0%) | |
| 8. | 385 ng | c.976 | 39 | 13/39 | 12/39 (31%) |
| 385 ng | wt | 49 | 2/49 | 2/48 (4.2%) | |
| non-injected | - | 61 | 0 | 0/60 (0%) |
Experiments 1–5 were performed in wild-type zebrafish (TL), experiments 6–8 in the reporter strain. #: number of injected fish. Percentages of abnormal fish reflect the ratio of abnormal fish and total number of surviving fish.
Injection of mutant AARS in zebrafish leads to an altered neurologic phenotype
To evaluate if the injected embryos also displayed neurologic abnormalities we repeated the micro-injection experiments of mRNAs with each patient mutation in embryos of crossed reporter strains (olig2-DsRed/Olig2-DsRed x nkx2.2a-EGFP/+) (kindly provided by Bruce Appel and David Lyons). The reporter strain required a higher concentration of injected mRNAs to obtain similar results. We therefore raised the quantity of injected mRNAs to 500–600 pg. As visualized in Fig. 9, the morphology of the resulting embryos is clearly aberrant. A graphic representation of all injections in wild-type TL and reporter zebrafish is given in Fig. 10. Although some abnormalities were seen in the control-injected embryos of the reporter strain, a dramatic increase in the percentage of abnormal embryos was observed for all three mutations. The reporter strain allowed us to assess abnormalities during neural development. In Fig. 11 abnormal embryos show an irregular and less well defined pattern of nerves across each somite similar to the pattern we showed in a previous study with lrsam1 morphants (20) and equivalent to the znp-1 staining in the aars splice-morphants (Fig. 6). The degree of abnormalities was also reflected in a lower intensity of fluorescence in the early stages indicative of a reduced number of cells in the developing neural system or a reduced expression of the corresponding reporter genes (Supplementary Material, Fig. 1). Injection of mRNAs containing patient mutations occurring in the AARS gene into zebrafish embryos and evaluation of the resulting morphology can thus be used as an in vivo system to assess the pathogenicity of these mutations which is informative for both loss-of-function and gain-of-function alleles.
Figure 9.
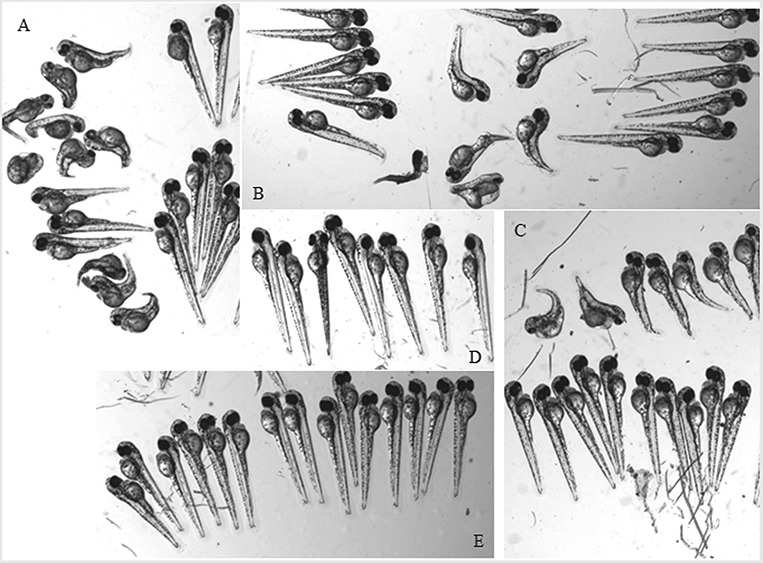
Injection of all three mutant mRNAs give aberrant morphologies after injection in embryos of zebrafish reporter lines while equal or larger amounts of wild-type mRNAs do not. Pictures were taken at 2 dpf after injection of 500 pg of mutant (A–C) mRNA or wt mRNA (E) in 1-cell stage embryos of reporter strains or of non-injected controls (D). Mutant injected mRNAs: c.1009G > A, p.Glu337Lys (A), c.1880C > T, p.Ser627Leu (B), c.976C > T, p.Arg326Trp (C).
Figure 10.
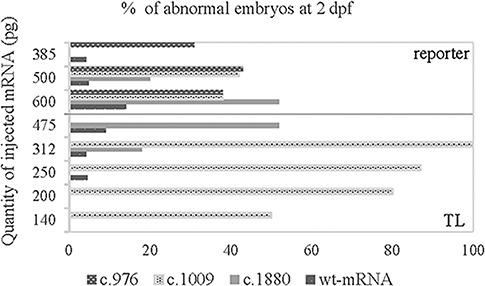
Increase of the number of abnormal embryos after injection of mutant mRNAs. Graphic representation of the percentage of abnormal embryos (number of abnormal embryos as compared to the total number of surviving embryos at 2 dpf) of injections with mutant or wild-type mRNA as indicated in the figure. The percentage of abnormal embryos of the non-injected embryos were not shown since no abnormalities were seen. Injections were performed in the wild-type strain (TL; below line) or reporter strain (above line).
Figure 11.
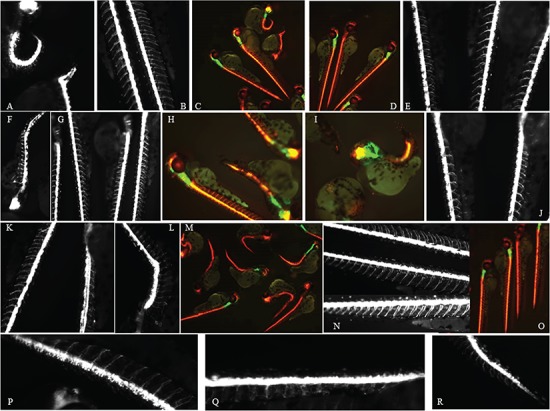
Aberrant neural development in mutant mRNA injected embryos. Red fluorescence is depicted as white (A,B,E,F,G,J,K,L,N,P,Q,R) or red (C,D,H,I,M,O) driven by the olig2 promoter in the reporter strain. The green fluorescence reflects expression driven by the nkx2.2a promoter. Pictures were taken at 2 dpf of embryos injected with mutant or wt-mRNA (D,E,J; 500 pg) or of non-injected controls (N,O). Mutant injected mRNAs: c.976C > T, p.Arg326Trp (A–C), c.1009G > A, p.Glu337Lys (F–I), c.1880C>T, p.Ser627Leu (P–R,K,L).
Discussion
In three families we identified the underlying genetic cause of CMT2 as a heterozygous missense mutation in the AARS gene. The genes encoding the tRNA synthetases are strongly conserved and all enzymes contain a catalytic domain. In the case of AARS, in addition to the N-terminal region with the catalytic site, an editing domain is present near the C-terminus, which prevents misincorporation of incorrect amino acids into proteins. This is followed by the conserved C-terminal domain, which is important for enzyme dimerization (21). For class II tRNA synthetases to which AARS belongs, the active form of the enzyme most probably consists of the loaded tRNA that interacts with two monomers via their active site, second additional domain (SAD)-domain which is assigned near the C-terminal part of the editing domain and the C-terminal end of the protein (http://www.uniprot.org). This architecture may attribute to the dominant nature of heterozygous missense mutations in AARS that are associated with CMT.
Two of the three mutations that we have identified were located in exon 8 affecting the middle helical part or the tRNA recognition domain of the protein. This region is also the site where a recurring dominant CMT-associated mutation (p.Arg329His) is located (5,16). Although outside of the catalytic domain, the recurring mutation was shown to result in a severely reduced aminoacylation activity (16) The identified exon 8 mutations in this study are only 3 and 8 amino acids apart from the position of the p.Arg329His mutation, suggesting that the two new mutations are likely loss-of-function. Indeed, we found that the p.Arg326Trp represents a true loss-of-function allele as assessed in the yeast complementation assay as was also reported for the p.Arg329His mutation. In contrast, the p.Glu337Lys mutation dramatically improved cell growth in yeast complementation assays and increased tRNA charging in aminoacylation assays upon comparison to the wild-type enzyme. This mutation may represent a gain-of-function, hypermorphic mutation, whose dominant neurotoxicity is associated with inappropriately increased enzyme kinetics.
The mutation in exon 14 of the AARS gene (p.Ser627Leu) represents a loss-of-function, hypomorphic allele since it showed reduced growth in the yeast complementation. Mutations in the editing domain have been associated with recessive disease (14,12,13). This is the first report of a dominant pathogenic mutation in this domain as the cause for CMT2, which raises questions regarding the (un)coupling of tRNA charging and editing in patients with CMT disease. Remarkably, the injection of the splice-MO into 1-cell stage embryos of Danio rerio, which is predicted to lead to a protein truncated at the C-terminal end lacking this domain, also created a dominant phenotype in zebrafish. The absence of a morphologic phenotype after injection of ATG MOs at 2–3 dpf could indicate that the remaining amount of protein was still sufficient and/or the ATG-MO was not functioning properly. For that reason, we also targeted the second ATG, which is located 25 amino acids downstream in the same reading frame but again, no overt major phenotype was observed at quantities lower than 8.5 ng per injection. A combination of these two gave the same results (data not shown). An alternative explanation is that duplications of part of the aars gene, present in the zebrafish genome may compensate for loss of the protein.
Despite the presence of the endogenous aars in the embryos, injections of the human mutant mRNA corresponding to the three patient mutations gave highly abnormal phenotypes in zebrafish suggesting that the mode of action is dominant-negative or a toxic gain of function. Although their canonical function in providing aminoacyl-tRNAs for protein synthesis is a shared characteristic of all tRNA synthetases is, other functions could attribute and/or be essential to the phenotype as well. Despite the fact that ARS enzymes are ubiquitously expressed and essential for every cell, only specific cells seem to be affected. This could imply that these enzymes have a cell-type-specific function in addition to their canonical function in protein synthesis and/or that the affected cell type is particularly vulnerable for these mutations. In fact, recent reports have attributed an essential role for the GARS enzyme, which is another ARS enzyme, the first one to be described as associated with CMT, in neddylation, where it acts as a chaperone for NEDD8-activated Ubc12 (22). Mutations in Gars were also found to cause aberrant interactions of the mutant proteins with Trk receptors essential for sensory neuron development (23), interfere with interactions between neuropilin and vascular endothelial growth factor (VEGF) (24), and inappropriately bind to HDAC6, leading to axonal mitochondrial transport defects (25). Haploinsufficiency of this specific ARS as a mechanism was ruled out previously, as mice carrying one normal allele of the Gars gene did not display a phenotype. In addition, Gars-mutant mice could not be rescued by reintroduction of an overexpressing wild-type allele (26) indicative of a gain-of-function mechanism. Splice-variants encoding ARS catalytic null variants have been described (27) with as yet unknown function. An alternative explanation is ectopic access or exclusion due to defects in trafficking or signalling as was recently suggested for gars in a Drosophila model (28).
In our zebrafish system, human mutations caused toxicity in embryos in the presence of a zebrafish wild-type allele suggestive of a dominant negative-mode of action but not excluding a toxic gain-of-function. In addition to AARS, mutations in GARS, YARS and HARS cause CMT2 and more recently, WARS mutations have also been described in CMT2 patients (4–7) CMT-associated mutations are not restricted to either one of the two classes of ARS enzymes nor to their association with a multimeric complex in mammalian cells (29). Despite the presence of different additional domains, all CMT-associated mutations lead to phenotypes in the nerve axons. We therefore propose a dominant-negative loss-of-function mechanism as one of the determinants for development of the disease. Another determinant is a toxic gain-of-function either as a prerequisite for the disease or as an additionally attributing factor to the phenotype. In this respect, it is remarkable that the splice morphants showed a similar phenotype in the fish as we observed for morphants of another CMT-associated gene, while the vast majority of the aars mRNA was normal, again suggestive of a gain-of-function mechanism.
While the vast majority of CMT-associated mutations in ARS enzymes lead to a change in aminoacylation capacity, gain-of-function mechanisms should still be addressed and a vertebrate in vivo functional read-out assay would be beneficial to provide essential information regarding the pathogenicity of the identified variations. Here, we report a novel in vivo model that, when coupled with established assays, revealed that functional null, hypomorphic and hypermorphic AARS alleles are all associated with dominant axonal CMT2 disease.
Patients, Materials and Methods
Patients and isolation of genomic DNA
Three four- to five-generation-families who were diagnosed with CMT2 were carefully examined by experienced neurologists. Pedigrees of the families are given in Fig. 1. All blood samples were obtained with patients’ consent. DNA of 24 patients of two to three generations was available for research.
Genomic DNA was isolated from blood samples using standard methods. It was used for amplification of specific exons of candidate genes prior to Sanger sequencing analysis and for the preparation of representative sequencing libraries.
Sequencing of candidate genes
For amplification of the exons of the candidate genes, M13-tagged specific exon-primers were used with 20 ng of genomic DNA and Hotfire polymerase (Solis Biodyne, Tartu, Estonia). Primers are available upon request. Polymerase chain reaction (PCR) was performed with a touchdown PCR program and a final annealing temperature of 55°C. Prior to Sanger sequencing using the Big Dye Terminator kit (Applied Biosystems, Thermofisher Scientific, Landsmeer, Netherlands), samples were treated with shrimp alkaline phosphatase (AP) and exonuclease I. Sequences were run on an ABI3730xl sequencer and analysed using the CodonCode Aligner software.
Targeted sequencing of CMT-associated genes
A CMT-specific custom-made capture (Nimblegen, Roche, Diagnostics, Almere, Netherlands) was developed for the following 45 CMT-associated genes: SEPT9, AARS, ARHGEF10, ATL1, BSCL2, CCT5, CTDP1, DCTN1, DNM2, EGR2, FAM134B, FGD4, FIG4, GAN, GARS, GDAP1, GJB1, HSN2, HSPB1, HSPB8, IGHMBP2, IKBKAP, LITAF, LMNA, LRSAM1,MFN2, MPZ, MTMR2, NDRG1, NEFL, NGFB, NTRK1, PLEKHG5, PMP22, PRPS1, RAB7A, SBF2, SETX, SH3TC2, SLC12A6, SOX10, SPTLC1, SPTLC2, TRPV4 and YARS, in addition to the exons of 24 genes of other neurologic disorders. For one capture, three samples were pooled, after which a sequencing library was prepared according to the manufacturer’s protocol (Newbler v.2.3, Nimblegen, Roche) prior to sequencing on a Titanium FLX pyrosequencer (Roche). Parts of PLEKHG5, SRGAP2, SBF2, SPTLC2, GAN, VAPB, SOX10, FAM134B, NTRK1, DNM2 and VCP with coverage lower than 10× were filled in using conventional Sanger sequencing of the respective amplified exons. All high-quality differences were visually checked using the Integrated Genome Viewer prior to confirmation using conventional sequencing analysis. Unreliable calls in homopolymer stretches were excluded for further analysis. Sequencing of the third family was part of the routine diagnostic procedure using the same capture and 1 μg of pre-amplified libraries of 6–14 patients, followed by sequencing with the miSeq V2 kit on an Illumina miSeq sequencer. Alignment and re-alignment were performed using HG19 and BWAMEM.0.7.8. For variant calling GATK-2.8-1-g932cd3a was used. Variants were callable when meeting the quality criteria we have for this procedure (i.e. coverage of at least 30× with both base and mapping quality ≥20).
Yeast complementation assays
Yeast complementation assays were performed as previously described in a validated haploid yeast strain in which the endogenous ALA1 locus is deleted and viability is maintained via a maintenance vector bearing wild-type ALA1 and URA3 (pRS316) (16,17); however, the AARS variants were modelled in the human AARS open-reading frame. Mutagenesis was performed using the Quickchange II XL Site-Directed Mutagenesis Kit, a sequence-validated wild-type AARS pDONR221 construct, and mutation-specific primers (primers available upon request). After transformation into Escherichia coli, individual colonies were sequenced to confirm the presence of the mutation and the absence of PCR-induced errors. Sequence-validated constructs for wild-type and mutant human AARS (p.Arg326Trp, p.Glu337Lys and p.Ser627Leu, alone or in cis with p.Glu337Lys) were cloned into pYY1 (30) using Gateway cloning technology (Invitrogen, Life Technologies, Carlsbad, CA, USA). The resulting colonies were analysed by restriction digestion with BsrGI to confirm proper recombination and the presence of an insert of appropriate size.
To assess the ability of wild-type and mutant alleles to support cellular growth, yeast cells were transformed with wild-type or mutant human AARS, or an empty vector control. pYY1 harbors the LEU2 gene, enabling selection for the presence of these constructs by growth in medium lacking leucine. Transformed yeast cells were selected for the presence of both constructs by growth on solid media lacking leucine and uracil. Two colonies per transformation were grown to saturation in 2 ml leu-ura- liquid medium at 30°C and 250 rpm for 48 h. Yeast cells from a 1 ml aliquot were collected by centrifugation at 10 000 rpm and re-suspended in 50 μl UltraPure RNase/DNase-free water. Undiluted cultures and dilutions of 1:10 and 1:100 were spotted on 0.1% 5-FOA complete solid medium (Teknova, Hollister, CA, USA) to select for cells that spontaneously lost the URA3-bearing maintenance vector (18). Yeast viability was assessed by visual inspection after 5 days of incubation at 30 or 37°C.
AARS aminoacylation assays
Wild-type and p.Glu337Lys human AARS were cloned in pET-DEST42 gateway vector (Life Technologies, Carlsbad, CA, USA) for expression in E. coli Rosetta 2 (DE3) with a C-terminal fusion to 6× His. Protein purification was performed with the Ni2+-affinity resin according to the manufacturer’s protocol (Novagen, Burlington, MA, USA). Following the T7 transcription, purification and annealing of human tRNAAla, steady-state aminoacylation assays were performed at 37oC as described (16,31), monitoring incorporation of 3H-Ala (specific activity of 16 500 dpm/pmole) into 3H-Ala-tRNAAla as acid precipitable counts on filter pads. Each reaction was initiated by addition of an AARS enzyme (1.67 nm) to a mixture of increasing concentrations of tRNAAla (0.2-18 μM). A quench factor of 4.9-fold was used to correct counts for to calculation of pmoles of 3H-Ala-tRNAAla. Data were fit to the Michaelis–Menten equation and an average of at least triplicate sets was presented with error bars showing standard deviation.
Zebrafish and injections
All zebrafish (D. rerio) procedures complied with standard animal care guidelines according to Dutch Animal Ethics Committee guidelines. Wild-type Tupfel long fin (TL) zebrafish outbred strains were crossed to generate embryos for morpholino-oligonucleotide (MO) injections or mRNA injections. Similarly, reporter strains were bred to obtain embryos for injection. MO antisense 25mers were designed (Genetools, Philomath, OR, USA) against the sequences surrounding the first two ATGs and the acceptor splice site of exon 14 (Genetools; ATG- MO:5′-AGATGAAGAGCCATAGATGATTTGC-3′; ATG2-MO: 5 ′-CGCAGTCAGTGAAGAGTCCATGCTT-3′; splice MO:5′- CTCCATCCTCTGAAACAGCCAAAAT-3′). One nl of MO in a range of 200–800 μM was injected into the yolk sac of 1-cell-stage embryos using fine glass needles and a microinjector (World Precision Instruments, Friedberg, Germany). Standard control MOs (GeneTools) and non-injected wild types were taken as controls. The developing embryos were incubated at 28°C. Morphology was monitored over time and images were taken using a Leica MZEBRAFISHLIII equipped with a Retiga 2000R Fast1394 Q imaging camera and the QCapture Pro program. Similarly, injections were performed with 1 nl of mRNAs (wild-type or mutant corresponding to 140–600 pg of mRNA). Injections into the reporter strain required at least double the amount that was used in the TL-strain.Wt-mRNA injections were matched accordingly.
Whole mount immunohistochemical staining of Zebrafish
After dechorionation, zebrafish were bleached for 3–4 min using a solution containing 5% formamide, 0.5 × saline sodium citrate and 9% H2O2. After permeabilization with 2.5 mg/ml trypsin in PBST for 3–4 min on ice, embryos were preincubated in blocking buffer prior to incubation with the primary znp1 antibody (ZIRC, Eugene, OR, USA) in a 1:100 dilution in blocking buffer at 4°C for 16–48 h under gentle agitation. Similarly, secondary antibody detections were performed overnight at 4°C in a 1:1000–2000 dilution (Southern Biotech, Birmington, AL, USA; AP-coupled goat anti-mouse IgG) followed by extensive washing prior to detection using the SK5300 (Vector Laboratories, Burlingham, CA, USA) AP detection kit. Images were captured using a Leica M125 stereomicroscope equipped with a DFC425 camera and LASv3.7 software.
IVT
Constructs with the complete coding parts of the human AARS wild-type gene (mammalian gene collection #3450606) or carrying a mutation (c.976C > T, c.1009G > A and c.1880C > T) were cloned (XbaI-EcoRI) into pCS2+ and 2.5 ug of the linearized clone was used for IVT (mMESSAGE mMACHINE kit Ambion, Fisher Scientific, Landsmeer, Netherlands) according to the manufacturers’ protocol. After purification, in vitro transcribed transcripts were checked for their integrity by agarose gel electrophoresis and quantified by fluorometric assays (Qubit, Fisher Scientific, Landsmeer, Netherlands).
Reverse Transcriptase-coupled PCR
Total RNA was isolated from snap-frozen injected embryos at 3 dpf or non-injected controls using Tripure (Roche). Total RNA (100–200 ng) was treated with RNase-free DNase prior to oligoVN-dT-primed first strand synthesis according to the manufacturer’s protocol (SuperscriptIII, Invitrogen), followed by PCR using gene specific-primers (sequences available upon request).
Western blot analysis
Zebrafish embryos were disrupted using an Eppendorf pestler, and lysed in RIPA buffer (50 mm Tris/HCl pH=7,6; 150 mm NaCl; 20 mm Ethylenediaminetetraacetic acid; 0,5% sodium deoxycholate; 1% Nonidet P40; 1% sodium dodecyl sulphate, SDS) supplemented with protease inhibitor cocktail (Roche). Following sonication (2 min in a bath sonicator), the samples were mechanically homogenized as much as possible. Protein concentrations were measured using the DC Protein Assay (Biorad, Veenendaal, Netherlands). Samples were heated for 5 min at 95°C after which the supernatants were loaded and analysed by polyacrylamide gel electrophoresis (SDS-PAGE 8%). Samples were transferred to a nitrocellulose membrane (Amersham GE Healthcare; Fisher Scientific, Landsmeer, Netherlands) by electroblotting. After preincubation in blocking buffer (Phosphate buffered saline, PBS supplied with 0,1% Tween20, 5% non-fat dry milk and 0,025% bovine serum albumin) membranes were incubated with anti-AARS in blocking buffer; α-tubulin was used as a loading control. Dilutions of the primary antibodies used were as follows: 1:5000 of anti-AARS (Abcam166915), and 1:1000 of anti-α-tubulin (Abcam 11304). As molecular size marker the Precision Plus Protein dual colour marker was used (Biorad). Subsequently, the blots were incubated with species-specific horseradish peroxidase-coupled secondary antibodies (Dako, Agilent Technologies, Amstelveen, Netherlands, 1:2000), and the resulting signals were visualized using the Lumi-lightPLUS western blotting substrate (Roche) and a LAS-3000 luminescent image analyser (Fuji Film).
Supplementary Material
Acknowledgements
We thank Dr Silver and Dr B Apple for generously providing embryos of the Tg (nkx2.2a-EGFP/wt) and Tg(DsRed:olig2/DsRed:olig2) reporter zebrafish lines. We also thank Dr M Kriek for supplying additional clinical information.
Conflict of Interest statement. None declared.
Funding
National Institute of General Medical Sciences (GM118647 to A.A.); NIH Medical Scientist Training Program Training Grant (GM007863 to M.K.); National Institute of General Medical Sciences (GM1081602, GM114343 to Y.M.H.).
References
- 1. Meyer-Schuman R. and Antonellis A. (2017) Emerging mechanisms of aminoacyl-tRNA synthetase mutations in recessive and dominant human disease. Hum. Mol. Genet., 26, R114–R127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oprescu S.N., Griffin L.B., Beg A.A. and Antonellis A. (2017) Predicting the pathogenicity of aminoacyl-tRNA synthetase mutations. Methods, 113, 139–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. et al. (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet., 72, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet., 38, 197–202. [DOI] [PubMed] [Google Scholar]
- 5. Latour P., Thauvin-Robinet C., Baudelet-Mery C., Soichot P., Cusin V., Faivre L., Locatelli M.C., Mayencon M., Sarcey A., Broussolle E. et al. (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet., 86, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vester A., Velez-Ruiz G., McLaughlin H.M., Program N.C.S., Lupski J.R., Talbot K., Vance J.M., Zuchner S., Roda R.H., Fischbeck K.H. et al. (2013) A loss-of-function variant in the human histidyl-tRNA synthetase (HARS) gene is neurotoxic in vivo. Hum. Mutat., 34, 191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tsai P.C., Soong B.W., Mademan I., Huang Y.H., Liu C.R., Hsiao C.T., Wu H.T., Liu T.T., Liu Y.T., Tseng Y.T. et al. (2017) A recurrent WARS mutation is a novel cause of autosomal dominant distal hereditary motor neuropathy. Brain, 140, 1252–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abbott J.A., Guth E., Kim C., Regan C., Siu V.M., Rupar C.A., Demeler B., Francklyn C.S. and Robey-Bond S.M. (2017) The Usher syndrome type IIIB Histidyl-tRNA synthetase mutation confers temperature sensitivity. Biochemistry, 56, 3619–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang X., Ling J., Barcia G., Jing L., Wu J., Barry B.J., Mochida G.H., Hill R.S., Weimer J.M., Stein Q. et al. (2014) Mutations in QARS, encoding glutaminyl-tRNA synthetase, cause progressive microcephaly, cerebral-cerebellar atrophy, and intractable seizures. Am. J. Hum. Genet., 94, 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salvarinova R., Ye C.X., Rossi A., Biancheri R., Roland E.H., Pavlidis P., Ross C.J., Tarailo-Graovac M., Wasserman W.W. and Karnebeek C.D. (2015) Expansion of the QARS deficiency phenotype with report of a family with isolated supratentorial brain abnormalities. Neurogenetics, 16, 145–149. [DOI] [PubMed] [Google Scholar]
- 11. Kodera H., Osaka H., Iai M., Aida N., Yamashita A., Tsurusaki Y., Nakashima M., Miyake N., Saitsu H. and Matsumoto N. (2015) Mutations in the glutaminyl-tRNA synthetase gene cause early-onset epileptic encephalopathy. J. Hum. Genet., 60, 97–101. [DOI] [PubMed] [Google Scholar]
- 12. Simons C., Griffin L.B., Helman G., Golas G., Pizzino A., Bloom M., Murphy J.L., Crawford J., Evans S.H., Topper S. et al. (2015) Loss-of-function alanyl-tRNA synthetase mutations cause an autosomal-recessive early-onset epileptic encephalopathy with persistent myelination defect. Am. J. Hum. Genet., 96, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakayama T., Wu J., Galvin-Parton P., Weiss J., Andriola M.R., Hill R.S., Vaughan D.J., El-Quessny M., Barry B.J., Partlow J.N. et al. (2017) Deficient activity of alanyl-tRNA synthetase underlies an autosomal recessive syndrome of progressive microcephaly, hypomyelination, and epileptic encephalopathy. Hum. Mutat., 38, 1348–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lee J.W., Beebe K., Nangle L.A., Jang J., Longo-Guess C.M., Cook S.A., Davisson M.T., Sundberg J.P., Schimmel P. and Ackerman S.L. (2006) Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature, 443, 50–55. [DOI] [PubMed] [Google Scholar]
- 15. Mathis S., Goizet C., Tazir M., Magdelaine C., Lia A.S., Magy L. and Vallat J.M. (2015) Charcot-Marie-Tooth diseases: an update and some new proposals for the classification. J. Med. Genet., 52, 681–690. [DOI] [PubMed] [Google Scholar]
- 16. McLaughlin H.M., Sakaguchi R., Giblin W., Program N.C.S., Wilson T.E., Biesecker L., Lupski J.R., Talbot K., Vance J.M., Zuchner S. et al. (2012) A recurrent loss-of-function alanyl-tRNA synthetase (AARS) mutation in patients with Charcot-Marie-Tooth disease type 2N (CMT2N). Hum. Mutat., 33, 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Motley W.W., Griffin L.B., Mademan I., Baets J., De Vriendt E., De Jonghe P., Antonellis A., Jordanova A. and Scherer S.S. (2015) A novel AARS mutation in a family with dominant myeloneuropathy. Neurology, 84, 2040–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boeke J.D., Trueheart J., Natsoulis G. and Fink G.R. (1987) 5-Fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol., 154, 164–175. [DOI] [PubMed] [Google Scholar]
- 19. Ripmaster T.L., Shiba K. and Schimmel P. (1995) Wide cross-species aminoacyl-tRNA synthetase replacement in vivo: yeast cytoplasmic alanine enzyme replaced by human polymyositis serum antigen. Proc. Natl. Acad. Sci. U S A, 92, 4932–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Weterman M.A., Sorrentino V., Kasher P.R., Jakobs M.E., Engelen B.G., Fluiter K., Wissel M.B., Sizarov A., Nurnberg G., Nurnberg P. et al. (2012) A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum. Mol. Genet., 21, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Naganuma M., Sekine S., Fukunaga R. and Yokoyama S. (2009) Unique protein architecture of alanyl-tRNA synthetase for aminoacylation, editing, and dimerization. Proc. Natl. Acad. Sci. U S A, 106, 8489–8494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mo Z., Zhang Q., Liu Z., Lauer J., Shi Y., Sun L., Griffin P.R. and Yang X.L. (2016) Neddylation requires glycyl-tRNA synthetase to protect activated E2. Nat. Struct. Mol. Biol., 23, 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sleigh J.N., Dawes J.M., West S.J., Wei N., Spaulding E.L., Gomez-Martin A., Zhang Q., Burgess R.W., Cader M.Z., Talbot K. et al. (2017) Trk receptor signaling and sensory neuron fate are perturbed in human neuropathy caused by Gars mutations. Proc. Natl. Acad. Sci. U S A, 114, E3324–E3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He W., Bai G., Zhou H., Wei N., White N.M., Lauer J., Liu H., Shi Y., Dumitru C.D., Lettieri K. et al. (2015) CMT2D neuropathy is linked to the neomorphic binding activity of glycyl-tRNA synthetase. Nature, 526, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benoy V., Van Helleputte L., Prior R., d'Ydewalle C., Haeck W., Geens N., Scheveneels W., Schevenels B., Cader M.Z., Talbot K. et al. (2018) HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain, 141, 673–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Motley W.W., Seburn K.L., Nawaz M.H., Miers K.E., Cheng J., Antonellis A., Green E.D., Talbot K., Yang X.L., Fischbeck K.H. et al. (2011) Charcot-Marie-Tooth-linked mutant GARS is toxic to peripheral neurons independent of wild-type GARS levels. PLoS Genet., 7, e1002399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lo W.S., Gardiner E., Xu Z., Lau C.F., Wang F., Zhou J.J., Mendlein J.D., Nangle L.A., Chiang K.P., Yang X.L. et al. (2014) Human tRNA synthetase catalytic nulls with diverse functions. Science, 345, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Grice S.J., Sleigh J.N., Motley W.W., Liu J.L., Burgess R.W., Talbot K. and Cader M.Z. (2015) Dominant, toxic gain-of-function mutations in gars lead to non-cell autonomous neuropathology. Hum. Mol. Genet., 24, 4397–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kellermann O., Tonetti H., Brevet A., Mirande M., Pailliez J.P. and Waller J.P. (1982) Macromolecular complexes from sheep and rabbit containing seven aminoacyl-tRNA synthetases I. species specificity of the polypeptide composition. J. Biol. Chem., 257, 11041–11048. [PubMed] [Google Scholar]
- 30. Chien C.I., Chen Y.W., Wu Y.H., Chang C.Y., Wang T.L. and Wang C.C. (2014) Functional substitution of a eukaryotic glycyl-tRNA synthetase with an evolutionarily unrelated bacterial cognate enzyme. PLoS One, 9, e94659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hou Y.M., Westhof E. and Giege R. (1993) An unusual RNA tertiary interaction has a role for the specific aminoacylation of a transfer RNA. Proc. Natl. Acad. Sci. U S A, 90, 6776–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.