

Abstract
A-type channels, encoded by the pore-forming α-subunits of the Kv4.x family, are particularly important in regulating membrane excitability in the CNS and the heart. Given the key role of modulation of A currents by kinases, we sought to investigate the protein structure–function relationships underlying the regulation of these currents by PKA. We have previously shown the existence of two PKA phosphorylation sites in the Kv4.2 sequence; therefore, we focused this study on the Kv4.2 primary subunit. In the present studies we made the surprising finding that PKA phosphorylation of the Kv4.2 α-subunit is necessary but not sufficient for channel modulation; channel modulation by PKA required the presence of an ancillary subunit, the K+ channel interacting protein (KChIP3). Therefore, these findings indicate a surprising complexity to kinase regulation of A currents, in that an interaction of two separate molecular events, α-subunit phosphorylation and the association of an ancillary subunit (KChIP3), are necessary for phosphorylation-dependent regulation of Kv4.2-encoded A channels by PKA. Overall, our studies indicate that PKA must of necessity act on a supramolecular complex of pore-forming α-subunits plus ancillary subunits to alter channel properties.
Keywords: KChIP, phosphorylation, neuromodulation, Kv4.2, shal-type, PKA, heart, neuron
Potassium currents, specifically A-type K+ currents, are known to regulate neuronal membrane excitability. The transient A-type K+ current is present in high densities in the dendrites of CA1 pyramidal neurons (Hoffman et al., 1997), where the neurons receive synaptic input. Rapid activation of these channels can limit the peak of back-propagating action potentials as well as modulate incoming synaptic information, exerting profound effects on hippocampal network communication (Hoffman et al., 1997). These A-type K+ currents are modulated by PKA activation (Hoffman and Johnston, 1998), and this modulation of A-current amplitude regulates the peak of back-propagating action potentials (Hoffman and Johnston, 1999).
The molecular components that make up this current are unknown; however, the shal-type channel-forming α-subunits (Kv4.1–Kv4.3) participate in transient A-type current characterized in neurons throughout the CNS. Specifically, Kv4.2 is a rapidly inactivating, voltage-gated K+ channel that activates at membrane potentials above −40 mV and is sensitive to 4-aminopyridine (Baldwin et al., 1991; Blair et al., 1991; Serodio et al., 1994, 1996). It is highly likely that Kv4.2 is a pore-forming subunit of A-type K+ channels in CA1 pyramidal-cell dendrites, because Kv4.2 is highly expressed in hippocampal pyramidal neuron soma and dendrites (Sheng et al., 1992;Maletic-Savatic et al., 1995; Tsaur et al., 1997); the pharmacological and kinetic properties of Kv4.2 expressed in oocytes are similar to the transient outward currents in dendrites (Blair et al., 1991; Serodio et al., 1994, 1996; Hoffman et al., 1997). In addition, Kv4.2 most likely plays a role in transient outward currents in other brain areas (Serodio and Rudy, 1998; Shibata et al., 1999), including the striatum and the basal forebrain (Song et al., 1998; Tkatch et al., 2000). The precise target of kinase modulation of hippocampal transient currents is not clear, although in previous studies we have shown that the cytoplasmic domains of Kv4.2 are phosphorylated by PKA (Anderson et al., 2002). In these previous studies we identified two PKA phosphorylation sites: Threonine 38 (T38) and Serine 552 (S552) in the cytoplasmic domains of Kv4.2 (Anderson et al., 2000). Given the presence of these two phosphorylation sites, and based on previous results in studies of other channels, a parsimonious hypothesis is that PKA regulation of Kv4.2 is mediated by direct phosphorylation of the α-subunit.
Voltage-dependent K+ channels are tetramers composed of homodimers or heteromers (within a family; i.e., shal) (Jan and Jan, 1992; Pongs, 1992; Chandy and Gutman, 1995). The Kv4-channel complex contains primary, pore-forming α-subunits but can also contain various β-subunits or interacting subunits. Auxiliary subunits are known to interact with channels containing principal subunits and contribute to the regulation of biophysical properties and the expression levels of K+ channels. One family of Kv4 interacting proteins, the K+channel interacting proteins (KChIPs), has been described recently (An et al., 2000). KChIPs act as chaperones for Kv4.2 and modulate the kinetic properties of Kv4 channels. Four subtypes of KChIPs (KChIP1, KChIP2, KChIP3, and KChIP4) have been described and are known to interact with the N-terminal of Kv4.2 or Kv4.3 (An et al., 2000;Morohashi et al., 2002). Interestingly, KChIP3 was first cloned as calsenilin, which interacts with presenilins (Buxbaum et al., 1998) and has also been described as donnstream regulatory element antagonist modulator (DREAM), a transcriptional repressor of the prodynorphin gene (Carrion et al., 1999). KChIP2 and KChIP3 are localized to the hippocampus (An et al., 2000); thus, the KChIPs are possible candidates for modulators of Kv4.2 in the hippocampus. In addition, KChIPs are also Ca2+ binding proteins, which have the potential for a role in activity-dependent plasticity.
In the present studies, we found that PKA regulation of Kv4.2-encoded currents required the presence of a KChIP subunit. Despite the ability of PKA to phosphorylate the Kv4.2 α-subunit in the absence of KChIP3, PKA was unable to alter channel biophysical properties by this mechanism alone. Thus, regulation of Kv4.2 by PKA appears to require that PKA act on a complex of pore-forming α-subunit plus an ancillary subunit. This reveals an unexpected complexity to the structure–function relationships for kinase regulation of membrane potassium channels.
MATERIALS AND METHODS
Functional expression in Xenopusoocytes.Oocytes were harvested from the ovarian lobe of femaleXenopus laevis frogs. Briefly, the frog was anesthetized by submersion in 0.15% tricaine. The ovarian lobe was then surgically removed and placed in Ca2+-free solution (in mm: 82.5 NaCl, 2.5 KCl, 1 MgCl2, and 5 HEPES). The frog was allowed to recover and placed back in the tank. The oocytes were digested in 1.6–2.4 mg/ml collagenase (Roche Diagnostics, Indianapolis, IN) for several hours. Digested oocytes were then incubated in ND-96, a solution containing (in mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.4, supplemented with pyruvic acid (2.5 mm) and gentamicin sulfate (50 mg/ml; Invitrogen, Grand Island, NY). After ∼24 hr, oocytes were injected with 30–50 ng of DNA using a Nanoject microinjector (Drummond Scientific Co., Broomall, PA) into the nucleus of stage V to stage VI oocytes. Currents were recorded after 2–3 d under two-electrode voltage-clamp using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA) at room temperature. Microelectrodes were pulled from filamented glass (1.5 × 0.86 mm; A-M Systems, Carlsborg, WA) filled with 3m KCl. The current electrode had a resistance of 0.30–0.50 MΩ, whereas the resistance of the voltage electrode ranged from 0.3 to 1.0 MΩ. Currents were leak-subtracted online using P/4 leak subtraction. Data were digitized at 2 kHz and stored using a Digidata 1200 (Axon Instruments). Current protocols used to obtain data include activation (hyperpolarization to −110 mV then depolarization to +40 mV for 400–800 msec and repeated in −5 mV intervals), inactivation (depolarization to 0 mV then hyperpolarization to −110 mV for 650 msec, changing this step by +5 mV intervals), and then depolarization to 0 mV. For recovery from inactivation for oocytes expressing Kv4.2 alone, the membrane was hyperpolarized to −110 mV from 20 mV for 5 msec (with subsequently longer hyperpolarizations increasing in increments of 50 msec), for a final hyperpolarization of 705 msec, then depolarized to 20 mV. Kv4.2 plus KChIP3 recovered from inactivation more quickly than Kv4.2 alone, so the initial hyperpolarization pulse was 2 msec, increasing in increments of 10 msec, for a final duration of hyperpolarization of 192 msec.
The chamber was continuously perfused with ND-96 with NaOH at a rate of 3–6 ml/min. Solution changes were achieved by a two-way valve system (General Valve, Fairfield, NJ). Forskolin (Sigma, St. Louis, MO) was dissolved in DMSO, stored in 50 mm aliquots at −20°C, and diluted to 50 μm when used. 8-Bromo (8-Br)-cAMP (Calbiochem, La Jolla, CA) was dissolved in DMSO at a concentration of 100 mm and then diluted to 100 μm when used. H-89 (Calbiochem) was dissolved in DMSO at a concentration of 10 mm and used at 10 μm. As a control, the effect of DMSO (0.1%) alone was tested on Kv4.2-expressing oocytes. This percentage of DMSO had no effect on Kv4.2 currents (n = 3).
Data were analyzed using Clampfit (Axon Instruments), Origin (Microcal Software Inc., Northampton, MA), and Prism (GraphPad Software, San Diego, CA) programs. Peak currents were obtained and conductance was determined using a reversal potential of −95 mV. Activation and inactivation curves were fitted with a Boltzman sigmoidal curve with the following equation: y = bottom + (top − bottom)/[1 + exp(V1/2 −x)/slope]). Inactivation time constants were fit in Clampex (Axon Instruments) with the Simplex method. Only currents that could be fit with a single exponential in control ND-96 were used and described here.
DNA preparation and site-directed mutagenesis. The original Kv4.2 and KChIP cDNA was provided by P. J. Pfaffinger. Both constructs are in a cytomegalovirus vector. Point mutations were made using the site-directed mutagenesis kit (Stratagene, La Jolla, CA). The primers used include 5′-GAGAGAAAAAGGGCCCAGGACGCTCTAATTGTG-3′ for the T38 site and 5′-GGAAGTCATAGAGGAGCCGTGCAAG- AACTC-3′ for the S552 site. The double-mutant was made by using the T38 primer on the S552A DNA. The KChIP3 double mutation was made using the primer 5′-GTGACAAAGGCGGCGGACGGCGCTCTT- CTG-3′. Mutations were confirmed by restriction enzyme digestion and DNA sequencing. In most cases, the entire Kv4.2 or KChIP3 region was sequenced to determine whether any other mutations existed.
Protein expression and purification. The KChIP3 protein was expressed in Escherichiacoli as glutathioneS-transferase (GST) fusion proteins using methods modified from Hakes and Dixon (1992). Plasmids containing the KChIP3 cDNAs were constructed using the GST-fusion vector pGEX-KN (Hakes and Dixon, 1992). A single colony of BL21(DE3)-pLysS cells transformed with the protein plasmid was grown in Luria broth (LB; 170 mm NaCl, pH 7.5, 1% tryptone, 0.5% yeast extract) containing 20 μg/ml carbenicillin and then used to seed a 500 ml culture. After growing to an optical density of 0.6–0.8 (A600) the culture was centrifuged (1000 ×g, 15 min, 4°C) (Beckman J2-21M; Beckman Instruments, Fullerton, CA). The cell pellet was resuspended in 500 ml of LB with carbenicillin. The bacteria was induced by incubation at room temperature with 200 μmisopropyl-β-d-thiogalactopyranoside for 4 hr and harvested by centrifugation.
The cells were resuspended and incubated in Tris buffer 1 (50 mm Tris-HCl, pH 8.0, 2 mm EDTA, 10 mg/ml pepstatin, 10 μg/ml leupeptin, and 100 μm PMSF) containing 10 mm β-mercaptoethanol and 100 μg/ml lysozyme (Sigma) for 15 min at 30°C. After solubilization with 1.5%N-laurylsarcosine, the lysate was incubated with 20 μg/ml DNase I (Roche Diagnostics) and 10 mmMgCl2. The lysate was then centrifuged (1000 × g, 15 min, 4°C; Sorvall RT 6000B; Kenoro Laboratory Products, Newton, CT) and adjusted to a 2% Triton X-100 concentration.
The GST-fusion protein was purified using glutathione affinity absorption. Glutathione agarose beads were washed, resuspended in Tris buffer 1, and then incubated with the lysate for 1 hr at 4°C. The beads were washed three times with Tris buffer 1 by repeat centrifugation (100 × g, 5 min, 4°C; Sorvall). After the final wash, the bead preparation was resuspended in Tris buffer 2 (20 mm Tris-HCl, pH 7.5, 0.5 mm EDTA, 0.5 mm EGTA, 1 mmNa4P2O7, 10 μg/ml aprotinin, and 10 μg/ml leupeptin).
Phosphorylation of KChIP3. KChIP GST-fusion proteins were incubated at 37°C in reaction mixtures (25 μl) containing 70 ng of the catalytic subunit of PKA, Tris buffer 2, and ATP mix 1 (100 μm ATP, 100 mmMgCl2, and 10 μCi [γ-32P]ATP). Reactions were stopped by boiling for 5 min with sample buffer (30 mmTris-HCl, pH 6.8, 200 mm DTT, 40% glycerol, 8% SDS, and 0.04 mg/ml bromophenol blue). The GST-fusion proteins were separated by SDS-PAGE (10%) and visualized with Coomassie blue staining. Phosphopeptides were identified by autoradiography. As a control, parallel reactions are performed for GST with and without the kinases and ATP mix 1. Kinase phosphorylation was also performed.
Phosphopeptide mapping. Phosphorylation reactions were performed as described above, with some modifications; a preparative scale (reaction volume, 300–400 μl) of the reaction was made. The incubation period was adjusted, based on the time course of kinase phosphorylation of the fusion proteins. The phosphorylated GST-fusion protein was separated by SDS-PAGE (10%). After Coomassie blue staining of the gels, the bands corresponding to the KChIP fusion proteins were excised, and an in-gel digestion with trypsin was performed as described previously (Frangioni and Neel, 1993), with minor modifications. After extraction from the gel, the peptides were separated using reverse-phase HPLC with absorption monitoring at 214, 254, and 280 nm. Counts per minute in each HPLC fraction were measured as Cherenkov radiation. Phosphopeptides identified as HPLC fractions containing high radioactivity were applied to Sequelon arylamine membranes (Millipore, Bedford, MA) essentially as described by the manufacturer. After drying, the membrane was rinsed two times sequentially with 10 ml of methanol, then with water, and finally with 5 ml 10% trifluoroacetic acid and 50% acetonitrile in water. The membrane was air-dried, cut into pieces with a scalpel, inserted in a BLOTT cartridge, and sequenced in an Applied Biosystems Model 477A Protein Sequencer with an inline 120 A phenylthiohydantoin (PTH) Analyzer (Applied Biosystems, Foster City, CA) using optimized cycles. Instead of butyl chloride, 90% methanol-containing phosphoric acid (15 μl/100 ml) was used to extract the cleaved amino acids. After conversion, 50% of the sample was transferred to the HPLC for PTH-amino acid identification; the other 50% was collected in the instrument fraction collector for the determination of radioactivity by scintillation counting.
Expression in COS-7cells and Western blotting. The FuGene 6 Transfection Reagent (Roche Diagnostics) was used for COS-7 cell transfections with plasmid DNAs of Kv4.2 and/or KCHIP3 (1:1 ratio, 0.1–2.0 μg/μl). Transfected cells were grown on 35 mm plates to a 2 × 105 cell density. The cells were then harvested and centrifuged. The cell pellet was resuspended in 10% SDS with 100 mm DTT, 10 μg/ml pepstatin, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 100 μm phenylmethylsulfonyl fluoride. Sample buffer was then added, and the samples were loaded on an SDS-PAGE gel (10%) for Western blotting. Two antibodies were used for the Western blots, as follows: (1) the phosphoselective C-terminal antibody (Anderson et al., 2000) and (2) the general C-terminal antibody (not phosphoselective). Immunoreactivity was measured using densitometry (NIH Image). Densitometry data were analyzed with a paired Student'st test.
RESULTS
PKA is a potential critical regulator of Kv4.2-encoded channels in the CNS and the heart. We had previously identified two sites in the Kv4.2 protein cytoplasmic domains (amino acids T38 and S552) that are phosphorylated by PKA. To test the hypothesis that direct phosphorylation of Kv4.2 by PKA causes a functional modulation of current, we expressed the primary subunit of Kv4.2 alone inXenopus oocytes and manipulated PKA activity in the cell. Forskolin (50 μm) was bath-applied to activate PKA. Bath application of forskolin (15 min) had no significant effect on the activation kinetics of Kv4.2 alone (n = 7), as measured by the V1/2 of control (−9 ± 2 mV) and V1/2 in forskolin (−6 ± 2 mV) (p = 0.13) and slope (20 ± 1 and 21 ± 1) (p = 0.15), respectively (Fig. 1). Similarly, bath application of 8-Br-cAMP (n = 6; data not shown) had no significant effect on Kv4.2 when expressed alone in oocytes. The V1/2 of the control was −10 ± 2 versus −8 ± 2 (p = 0.12) in 8-Br-cAMP; the slope was 20 ± 2 in the control and 20 ± 2 in 8-Br-cAMP (p = 0.92). In addition, there was no effect on steady-state inactivation or on the time of recovery from inactivation. Indeed, this concentration of forskolin has been shown to activate PKA in oocytes (Schorderet-Slatkine and Baulieu, 1982), and we have shown that PKA is capable of phosphorylating the Kv4.2 α-subunit at both the C- and N-terminals of Kv4.2 in COS cells (see Fig. 8) and hippocampal slices using phosphospecific antibodies (Anderson et al., 2000). Therefore, to our surprise, when Kv4.2 was expressed in Xenopus oocytes, the activation of PKA with forskolin or cAMP analog had no significant effect on the kinetics of the K+current.
Fig. 1.

PKA activation does not modulate transient outward current when Kv4.2 is expressed alone. A, Activation curve of Kv4.2 in the control (squares) and in forskolin (50 μm; triangles). B, Mean current–voltage plot of the peak amplitude of the current versus test voltage for the control and forskolin for all cells tested (n = 7). C, Bar graph of the meanV1/2 of each cell fitted individually (Boltzman sigmoidal). The V1/2 in control was −9 ± 1.7 mV, which did not differ significantly from the −5.7 ± 2.3 mV in forskolin. D, Raw current trace of depolarization to +20 mV in control (black) and forskolin (gray) (forskolin current is scaled up to the amplitude of the control current because forskolin caused a small decrease in amplitude). E, Scatterplot of the time constant of inactivation of the current evoked with a depolarization to +20 mV fitted with a single exponential (from peak to 230 msec) in the control (triangles) and in forskolin (diamonds). F, Raw current trace showing the current in forskolin scaled up to the control amplitude to illustrate the inactivation time constant further. Forskolin does not significantly alter the time constant of inactivation of Kv4.2 expressed alone.
Fig. 8.

Mutation of S552A blocks the immunoreactivity for the C-terminal phosphospecific antibody. A, Western blot of a COS cell homogenate showing immunoreactivity for the PKA C-terminal (CT) phosphospecific site (Phospho-site) (top) and total Kv4.2 (bottom). The phosphospecific antibody recognizes only Kv4.2 plus KChIP3 and not the S552A mutation. B, COS cells were transfected with Kv4.2 alone and treated with DMSO or forskolin (50 μm). Western blots of cellular homogenates show that forskolin caused phosphorylation of the C-terminal site, as evidenced by the increase in immunoreactivity for the PKA C-terminal-specific antibody (top) and no change in total protein (bottom).
Coexpression of Kv4.2 and KChIP3
The KChIPs are a family of interacting proteins that have been shown recently to interact with the Kv4 family of primary subunits (An et al., 2000). Interaction of the KChIPs and Kv4.2 causes various changes in the kinetics of Kv4.2. It has been shown previously that coexpression of the KChIPs with Kv4.2 caused an increase in current density, an increase in the rate of recovery from inactivation, an increase in the time constant of inactivation, and a shift of activation and inactivation curves in Chinese hamster ovary cells and oocytes (An et al., 2000; Decher et al., 2001; Beck et al., 2002). Might KChIP be an ancillary subunit necessary for PKA modulation of Kv4.2? Given our surprising finding described above indicating a lack of PKA regulation of Kv4.2 α-subunit alone, we set out to test this hypothesis. As a first step, we assessed the effect of KChIP3 on Kv4.2 channel biophysical properties in our experimental system (Fig.2). In our preparation, coexpression of KChIP3 with Kv4.2 shifted the V1/2activation toward the left 11 mV in the hyperpolarizing direction (−23 ± 1.0 mV; slope, 16 ± 0.3; n = 13) compared with Kv4.2 alone (−12 ± 1 mV; slope, 19 ± 1;n = 11), and shifted inactivation +15 mV (−63 ± 1 mV; slope, −4 ± 0.1 vs −78 ± 1 mV; slope, −6 ± 0.2) and strikingly increased the rate of recovery from inactivation at −110 mV (τ = 70 ± 3 msec for Kv4.2 alone and 12 ± 1 msec for Kv4.2 plus KChIP3) (Fig. 2A–D). The time constant of inactivation, fitted with one exponential, was increased from 20 ± 1 msec in Kv4.2 alone and 49 ± 3 msec in Kv4.2 plus KChIP3. Overall, these altered properties attributable to coexpression with KChIP3 made the properties of the K+current more similar to native A currents observed in neurons and cardiac myocytes.
Fig. 2.

Modulation of Kv4.2 current by KChIP3.A, Representative example of transient outward current recorded from an oocyte expressing only Kv4.2. B, Example of transient outward current recorded from an oocyte when KChIP3 is coexpressed with Kv4.2. C, Steady-state activation and inactivation curves for Kv4.2 alone (squares) and Kv4.2 plus KChIP3 (triangles). D, Plot of the time of recovery from inactivation for Kv4.2 expressed alone (squares) and with KChIP3 (triangles). KChIP3 speeds the recovery from inactivation (see Table 1).
Moreover, KChIP3 coexpression rescued the PKA regulation of Kv4.2. In contrast to what was observed with Kv4.2 alone, activation of PKA with forskolin caused a significant shift in the activation curve of Kv4.2 coexpressed with KChIP3 (Fig.3A). The activation curve of Kv4.2 with KChIP3 was shifted to the right by 6 mV toward more depolarized membrane potentials from −23 ± 1 to −17 ± 2 mV (n = 13; p = 0.005) (Fig. 3). The slope was not significantly affected: 16 ± 0.3 compared with 17 ± 0.6 in forskolin (p = 0.3). The amplitude of the current decreased by 17 ± 2% compared with the control amplitude in the presence of forskolin (at +20 mV) (Fig.3D). This effect of forskolin was reversible (Fig.3B). No significant effect was seen on steady-state inactivation (the V1/2 of inactivation was −62 ± 2 mV in control and −63 ± 2 mV in forskolin) or on recovery from inactivation (Fig. 3D).
Fig. 3.

KChIP3 coexpression is required for modulation of the current by PKA. A, Activation curve of Kv4.2 plus KChIP3 current in the control (squares) and forskolin (triangles). B, The current evoked from depolarizing pulses to +20, −5, −30, and −60 mV in the control (black, left), 15 min after the start of forskolin application (gray,middle), and 15 min after washout (black,right). C, Mean current–voltage plot of the peak amplitude of the current versus test voltage for the control and forskolin for all cells tested (n = 13).D, Time of recovery from inactivation for Kv4.2 plus KChIP3 in control and in forskolin. Forskolin does not significantly alter the recovery from inactivation.
Coexpression with KChIP3 also revealed an effect of forskolin on channel-inactivation properties (Fig. 4). The inactivation rate (at +20 mV) was best fitted with a single exponential in a control solution in 10 of 13 oocytes (50 ± 3 msec). However, after forskolin application, a double exponential was necessary to fit the inactivation rate in 7 of 10 oocytes, with a fast time constant of 13 ± 3 msec. The slow time constant increased to 74 ± 7 msec (Fig. 4). An increase in channel inactivation rate is a possible mechanism for the decreased amplitude and change in the number of channels open at certain potentials. Regardless, these observations are consistent with an interaction of KChIP3 and PKA phosphorylation (specifically, that KChIP3 is necessary for PKA regulation of Kv4.2).
Fig. 4.

Forskolin modulates the inactivation kinetics of Kv4.2 plus KChIP3. A, The time constant of inactivation is fitted with a single exponential in the control and a double exponential in forskolin. In a representative example of the current evoked by a depolarization to +20 mV, the forskolin current is scaled up to the control amplitude to compare the decay phases. Only currents that could be fitted by a single exponential in the control were evaluated. B, Scatterplot showing the single exponential in the control (squares) and the fast and slow time constant in forskolin (fsk) (triangles).
The control analog for forskolin, 1,9-dideoxyforskolin (50 μm) was also tested (Fig.5A). Dideoxyforskolin had no effect on the activation curve andV1/2 (p = 0.1;n = 3). A reduction of the time constant of inactivation (65 ± 5 to 45 ± 2 msec) was observed, but dideoxyforskolin did not cause the current to be fitted by a double exponential (Fig. 5A, inset).
Fig. 5.

The effect of forskolin is not mimicked by 1,9-dideoxyforskolin and is mimicked by 8-Br-cAMP and blocked by H-89, the PKA inhibitor. A, Activation curve of Kv4.2 plus KChIP3 in the presence of 1,9-dideoxyforskolin (Ddfsk; 50 μm). Dideoxyforskolin did not cause a significant shift in the activation curve (inset): Example of currents evoked by a depolarizing pulse to +20 mV in control (black) and dideoxyforskolin (gray) showing the effect of dideoxyforskolin on the current. The dideoxyforskolin current is scaled up to the size of the control. Dideoxyforskolin did not cause the inactivation time constant to be fitted by two exponentials, but it did have a small effect on amplitude and on the time constant of inactivation.B, Activation curve of Kv4.2 plus KChIP3 treated with 8-Br-cAMP (100 μm). C, The effect of forskolin on the activation curve is blocked by H-89 (10 μm). D, Raw current traces showing the effect of forskolin in H-89. H-89 (10 μm) blocked forskolin-induced changes in the time constant.
The forskolin-induced shift of the activation curve is mimicked by 8-Br-cAMP (100 μm) (Fig. 5B). 8-Br-cAMP caused a rightward shift of the activation curve (6 mV) from −23 ± 1 to −17 ± 1 mV (n = 5; p = 0.003), and application of 8-Br-cAMP decreased the peak amplitude of the current at +20 mV by 14 ± 4%. The slope of the activation curve was not significantly affected (15 ± 0.3 vs 15 ± 0.4). These observations strongly support the interpretation that the effects of forskolin are attributable to the elevation of the cAMP levels.
The effect of forskolin was blocked by the PKA inhibitor H-89 (10 μm) (Fig. 5C,D). H-89 alone caused a small (not significant) shift in V1/2(−25 ± 2 mV in the control vs −23 ± 1 mV in H-89;n = 4) (Fig. 5D). In the presence of H-89 (with a 10 min preincubation), there was no significant forskolin-induced shift in the voltage dependence of activation (−23 ± 1 mV in H-89 vs −24 ± 1 mV in forskolin;p = 0.55). In addition, the slopes of the activation curves were not significantly different. H-89 also blocked the effect of forskolin on the time constant of inactivation (at +20 mV, −49.3 ± 9 msec in H-89 vs 49 ± 6 msec in H-89 plus forskolin) (Fig. 5F). Thus, we found that the effects of forskolin were mimicked by 8-Br-cAMP and blocked by H-89, a specific PKA inhibitor, strongly indicating that these effects are indeed attributable to PKA activation. These findings suggest that phosphorylation of some component of the Kv4.2/KChIP protein complex is necessary for modulation of the channel properties.
Mutation of phosphorylation sites
The effects of phosphorylation on the Kv4.2/KChIP complex could be attributable to phosphorylation of the α-subunit, phosphorylation of KChIP, or both. As mentioned, we have shown previously that the N- and C-terminal cytoplasmic domains of Kv4.2 are phosphorylated at T38 and S552, respectively (Anderson et al., 2000). Therefore, we first tested whether phosphorylation of Kv4.2 at these sites is necessary for PKA modulation of Kv4.2/KChIP. Thus, we mutated T38 and S552 to an alanine to block phosphorylation at these sites (see Materials and Methods). However, when expressed without KChIP3, each mutant had kinetic characteristics similar to the wild type expressed alone (Table1), and the time constant of inactivation was modestly but significantly decreased in the N-terminal mutant and increased in the C-terminal mutant (Table 1). In addition, the mutations did not significantly affect the interaction of Kv4.2 with KChIP3, because the kinetics (including increased current density and recovery from inactivation) was similar to wild type plus KChIP3 (Table 1). These data indicate that the mutation of T38 or S552 to alanine does not grossly alter the biophysical properties of the channels or the interaction with KChIP3.
Table 1.
Effects of KChIP3 on biophysical parameters of wild-type Kv4.2 and PKA site-point mutants
| − KChIP3 | + KChIP3 | |||||
|---|---|---|---|---|---|---|
| Wild type | S552A | T38A | Wild type | S552A | T38A | |
| V1/2activation (mV) | −15 ± 1 | −17 ± 1 | −15 ± 1 | −23 ± 1 | −20 ± 1 | −23 ± 2 |
| n = 11 | n = 11 | n = 6 | n = 13 | n = 9 | n = 8 | |
| V1/2 inactivation (mV) | −78 ± 1 | −76 ± 1 | −75 ± 1 | −63 ± 1 | −63 ± 1 | −67 ± 1 |
| Inactivation time constant (ms) | 20 ± 1 | 26 ± 2* | 16 ± 1* | 49 ± 3 | 80 ± 111-160 | 42 ± 1 |
| Recovery from inactivation (msec) from −110 mV | 73 ± 3 | 71 ± 3 | 70 ± 4 | 12 ± 1 | 14 ± 5 | 11 ± 2 |
p < 0.05.
F1-160: p < 0.01.
T38A
Each mutant was then coexpressed with KChIP3. When the T38A mutant was coexpressed with KChIP3, forskolin caused a shift in the activation curve similar to that seen with wild type (Fig.6). The controlV1/2 was −23 ± 2 mV compared with −16 ± 1.5 mV in forskolin (p < 0.05; n = 7); there was no change in the slope of the activation curve (17.5 ± 0.85 in the control vs 17.4 ± 0.2 in forskolin). The amplitude was decreased by 28 ± 5% (at 20 mV) in the presence of forskolin (Fig. 6B).
Fig. 6.

Phosphorylation of the PKA N-terminal site is not required for modulation of the current by PKA. The N-terminal PKA phosphorylation site (T38) was mutated to an alanine.A, Activation curve of T38A plus KChIP3 current in the control (squares) and forskolin (triangles). B, Mean current–voltage plot of the peak amplitude of the current versus the test voltage for the control and forskolin for all cells tested (n = 7). C, A representative example of the current evoked by depolarizing pulses in control (black) and forskolin (gray). D, top, Example of current evoked by a depolarization to +20 mV in control (black) and forskolin (gray). Forskolin current is scaled up to the control amplitude to compare the decay phases. Forskolin caused an increase in the rate of inactivation.D, bottom, Scatterplot showing that the time constant of inactivation is fitted with a single exponential in the control and a double exponential in forskolin (Fsk). Only currents that could be fitted by a single exponential in the control were evaluated. Currents from five cells were fitted with a single exponential in control. For all five, the decay was fitted with a double exponential in forskolin. E, Time of recovery from inactivation for Kv4.2 plus KChIP3 in the control and in forskolin. Forskolin does not significantly alter the recovery from inactivation.
The time constant of inactivation in cells expressing the T38A plus KChIP3 mutant was affected by forskolin in a manner similar to wild type. The decay of inactivation at +20 mV was fitted with a single exponential in the control (42 ± 1 msec; n = 5); in forskolin all cells were fitted with a double exponential as well, with a fast time constant of 11 ± 1 msec and the slower time constant increased to 77 ± 5 msec (Fig. 6D). The recovery from inactivation was not affected by forskolin (Fig.6E). Overall, these data suggest that PKA phosphorylation of T38 is not involved in PKA regulation of the biophysical properties of the Kv4.2/KChIP3 complex.
S552A
In contrast, phosphorylation at S552 in the complex was essential for channel modulation. Thus, mutation of the S552 site to an alanine completely blocked forskolin-induced regulation of the properties of the Kv4.2/KChIP complex. Figure 7 shows the lack of effect of forskolin on the S552A mutant coexpressed with KChIP3. The V1/2 of S552A plus KChIP3 in the control and forskolin was not significantly different (p = 0.3). TheV1/2 for control was −20 ± 1 mV and −19 ± 1 mV in forskolin (n = 8). Also, there was no effect on the slope (19.4 ± 0.5 in the control vs 20 ± 0.3 in forskolin). The amplitude was decreased by only 7 ± 5% in forskolin (at 20 mV), compared with 17 ± 2% in the control and 28 ± 5% in the T38A mutant (Fig. 7B).
Fig. 7.

Phosphorylation of the PKA C-terminal site is required for modulation of the current by PKA.A, The C-terminal PKA phosphorylation site (S552) was mutated to an alanine. The activation curve of S552A plus KChIP3 current in the control (squares) and forskolin (triangles) is shown. B, Mean current–voltage plot of the peak amplitude of the current versus test voltage for control and forskolin for all cells tested (n = 9). C, The current evoked from depolarizing pulses in control (black) and forskolin (gray). Forskolin did not affect the current.D, top, An example of the current evoked by a depolarization to +20 mV in the control and in forskolin. Forskolin did not have an effect on the time constant of inactivation in the S552A mutant. D, bottom, The time constant of inactivation of the current from nine cells is fitted with a single exponential in control. Forskolin (Fsk) forced a double exponential in only three cells. E, Time of recovery from inactivation for Kv4.2 plus KChIP3 in the control and in forskolin. Forskolin does not significantly alter the recovery from inactivation.
In addition, the time constant of inactivation in cells expressing S552A was significantly longer (80 ± 11 msec; p = 0.003) than in wild type (48.7 ± 3.3 msec) or T38A (41.7 ± 1 msec) when coexpressed with KChIP3. After forskolin application, only three of eight decays at +20 mV were fitted with double exponentials. The fast time constant was 20 ± 3.0 msec (n = 3), and the long time constant was 102 ± 6 msec (n= 8) (Fig. 7D). These data suggest that phosphorylation of the S552 site is necessary for the effect of PKA activation on Kv4.2 coexpressed with KChIP3.
We wished to confirm that the mutation of S552 to an alanine did indeed block PKA phosphorylation of the C-terminal site. Therefore, we expressed wild-type Kv4.2 and the S552A mutant ± KChIP3 in the COS cell expression system. Western blots on cells expressing Kv4.2 plus KChIP3 probed with a selective antibody directed against the 552 phosphorylation site revealed immunoreactivity for the C-terminal site in cells expressing Kv4.2 plus KChIP3 but not in cells expressing S552A plus KChIP3 (n = 3) (Fig.8), suggesting that site S552 was not phosphorylated in the mutant. In our previous studies we observed that the Kv4.2 subunit when expressed by itself is a substrate for PKA (Anderson et al., 2000). As a control for the present studies, we confirmed this observation (Fig. 8B). These data show that although the Kv4.2 α-subunit by itself is phosphorylated at S552, KChIP3 coexpression is necessary for modulation of the current by phosphorylation.
In an additional series of control experiments, we confirmed that the double mutation (T38A, S552A) also showed no modulation by forskolin (data not shown). The V1/2 of T38AS552A plus KChIP3 in control and forskolin was not significantly different (n = 9; p = 0.3). TheV1/2 for control was −21 ± 2 and −18 ± 2 mV in forskolin. Also, there was no effect on the slope (17 ± 0.6 vs 16.1 ± 0.6 in forskolin). The time constant of inactivation of the double mutant plus KChIP3 was 57 ± 4 msec (n = 7). After forskolin application, only two of seven decays were better fitted with two exponentials, the fast time constant was 9.2 ± 2, and the slower time constant was 103 ± 10.6 for all seven cells (data not shown).
KChIP3 phosphorylation
Thus, phosphorylation at S552 appears to be essential for PKA regulation of the Kv4.2/KChIP complex, although it is clearly not sufficient to cause modulation of the Kv4.2 α-subunit alone. We wondered whether PKA phosphorylation of KChIP3 might be a missing component necessary for PKA modulation of Kv4.2. One prediction of this hypothesis is that PKA should phosphorylate KChIP3. We used in vitro phosphorylation, peptide mapping, and direct amino acid sequencing and found that indeed KChIP3 is a PKA substrate (Fig.9). KChIP3 is phosphorylated by PKA at serine 14 (S14), with a minor site at serine 11 (S11) (Fig. 9). Although other PKA phosphorylation sites may exist within KChIP3, we used this information to make site-directed mutants. We mutated the major site (S14) site to an alanine to determine whether phosphorylation of this site is necessary for PKA modulation of the Kv4.2/KChIP complex.
Fig. 9.
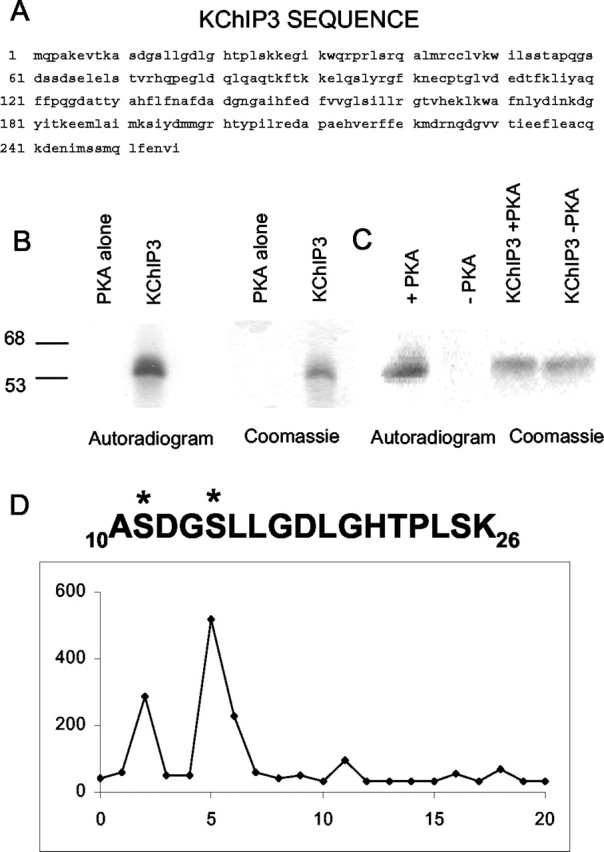
KChIP3 is phosphorylated by PKA. A, KChIP3 protein sequence. Inspection of the KChIP3 sequence reveals several consensus sites for PKA phosphorylation. KChIP3 was expressed as a GST-fusion protein in bacteria. The purified GST-fusion protein was reacted with PKA and γ-32P in vitro.B, Reaction products were separated by SDS-PAGE and stained with Coomassie blue (right). Autoradiography was performed to analyze the efficacy of KChIP3 as a substrate for PKA.C, Control experiment showing 32P incorporation in KChIP3 in the presence of PKA; however, there was no32P incorporation when PKA was not included in the experiment. D, The peptide was digested with trypsin, and two fragments were determined to contain radioactivity. One of the sequences determined by automated amino acid sequencing corresponded to amino acids 10–26 in the KChIP3 sequence. S14 was found to be a major site and S11 a minor site, as indicated by asterisks above serines.
Mutation of S14 in KChIP3 to an alanine did have a significant effect (relative to wild-type KChIP3) on theV1/2 of Kv4.2 plus KChIP (−18 ± 1 vs −23 ± 1 mV for −Kv4.2 plus wild-type KChIP;p = 0.003) and recovery from inactivation properties, but it had no effect on steady-state inactivation when the mutant KChIP3 was coexpressed with Kv4.2. Mutation of the S14 site in KChIP3 did not block the effect of PKA activation on the modulation of the properties of the Kv4.2/KChIP complex (Fig.10). TheV1/2 activation was −18 ± 1 mV in control and −14 ± 1 mV in forskolin (n = 10;p < 0.05). The mean time constant of inactivation at +20 mV was 52.2 ± 2.3 msec. After forskolin application, inactivation was fitted with a double exponential (12 ± 1 and 81.4 ± 5.5 msec) in all 10 cells.
Fig. 10.

Phosphorylation of the PKA site S14 in the KChIP3 sequence is not required for modulation of the current by PKA.A, The PKA phosphorylation site (S14) of KChIP3 was mutated to an alanine. The activation curve of wild type plus S14A KChIP3 current in control (squares) and forskolin (triangles) is shown. B, Examples of current evoked from depolarizing pulses in control (black) and forskolin (gray).C, The time constant of inactivation is fitted with a single exponential in the control and a double exponential in forskolin (Fsk). The currents from six cells were fitted by a single exponential in the control. Forskolin caused a double exponential decay in all six cells. The inset shows an example of the current; the forskolin current is scaled up to the control amplitude to compare the decay phases. D, Time of recovery from inactivation for Kv4.2 plus KChIP3 in the control and in forskolin. Forskolin does not significantly alter the recovery from inactivation.
We sought to determine whether phosphorylation of both S11 and S14 within the KChIP3 sequence is necessary for the modulation of the current mediated by Kv4.2 (Fig. 11). Therefore, we mutated both S11 and S14 to alanines. Kv4.2 coexpressed with S11S14A KChIP3 showed no significant change compared with wild type on activation (V1/2 = −24 ± 2 mV; n = 9). However, this KChIP3 mutant did show a significant shift in the time constant of inactivation (68 ± 2.5 msec; p = 0.002). The double KChIP3 mutant still demonstrated PKA modulation of channel properties. Forskolin application caused a significant (p < 0.01) shift in the V1/2 to −20 ± 1.5 mV (n = 9) (Fig. 11). Similar to wild type, forskolin application forced a double exponential fitted with a fast time constant of 11 ± 2 msec and a slow time constant of 111 ± 10 msec in all cells tested (n = 9). In both KChIP mutants, the S14A and S11AS14A, the amplitude was reduced by forskolin by a larger amount than in the control or the T38A mutant. The amplitude was reduced by 33 ± 2% in forskolin relative to control in the S14 mutant; forskolin caused a greater reduction in amplitude of the current evoked by a depolarization to +20 mV with the S11S14A mutant also, decreasing the amplitude by 39 ± 4%. This amount of current reduction is significantly more than that seen in the Kv4.2 primary subunit mutants; therefore, additional investigation is warranted to determine the mechanisms of this effect. In addition to these effects, forskolin also significantly slowed the recovery from inactivation. This suggests that the PKA phosphorylation sites in KChIP3 sites may play a role in maintaining the speed of recovery from inactivation, but that the overall PKA phosphorylation of sites S11 and S14 in KChIP3 is not necessary for PKA modulation of the ion-channel complex.
Fig. 11.

Phosphorylation of PKA sites S14 and S11 in the KChIP3 sequence is not required for modulation of the current by PKA. A, The PKA phosphorylation sites (S11 and S14) of KChIP3 were mutated to alanines. The activation curve of wild type plus S11S14A KChIP3 current in the control (squares) and forskolin (triangles) is shown. B, Mean current–voltage plot of the peak amplitude of the current versus test voltage for the control and forskolin for all cells tested (n = 9).C, Examples of the current evoked from depolarizing pulses in control (black) and forskolin (gray). D, The time constant of inactivation is fitted with a single exponential in the control and a double exponential in forskolin (Fsk). The currents from nine cells were fitted by a single exponential in the control. Forskolin caused a double exponential decay in all nine cells. Theinset shows an example of the current; the forskolin current is scaled up to the control amplitude to compare the decay phases. E, Time of recovery from inactivation for Kv4.2 plus KChIP3 in the control and in forskolin. Forskolin significantly alters the recovery from inactivation.
DISCUSSION
Our data indicate that two distinct molecular events are necessary for PKA regulation of the Kv4.2 pore-forming α-subunit. First, the α-subunit must be phosphorylated at S552. In addition, for this phosphorylation event to be efficacious, an ancillary subunit (KChIP3 in our experiments) must also be present. This indicates that the Kv4.2 pore-forming subunit and the KChIP ancillary subunit are operating as a functional supermolecular complex to allow modulation by phosphorylation. This indicates a surprising complexity to the protein structure–function relationships for A-current modulation by PKA. Direct phosphorylation of the pore-forming α-subunit is necessary but not sufficient for modulation of channel biophysical properties; additional allosteric effects by an ancillary subunit appear to be necessary.
This complexity of A-current regulation by PKA is further compounded by the likelihood that PKA phosphorylates additional sites in the complex. Thus, T38 in Kv4.2 and S11 and S14 in KChIP3 likely can be phosphorylated by PKA. The roles of these additional phosphorylation events remain mysterious. However, there are intriguing possibilities. We have found previously that the T38-phosphorylated form of Kv4.2 is differentially localized from the S552 variant, suggesting perhaps that T38 phosphorylation may participate in channel subcellular distribution (Varga et al., 2000). Other studies have also shown a role for post-translational modification in the regulation of channel localization (Ma and Jan, 2002). PKA phosphorylation of KChIP3 also has interesting potential roles as well, because KChIP3 can function not only as a Kv4.2 ancillary subunit but also as a calcium sensor and as a transcriptional regulator. KChIPs have four EF-hand-like domains and bind calcium ions (An et al., 2000). The function of these Ca2+ binding domains in the regulation of Kv4.2 is unknown; however, a Ca2+dependence of transient K+ currents has been reported previously (Bourque, 1988; Fisher and Bourque, 1998; Song et al., 1998). Future studies are necessary to determine whether PKA phosphorylation of KChIP regulates these functions.
In our studies, we observed that Kv4.2 is modulated by coexpression with KChIP3. This is similar to what has been reported previously and makes the Kv4.x-encoded channel behave much more like a native A current in heterologous expression systems. Kv4.x currents in expression systems are slightly different from that reported in the native brain (Rudy et al., 1988; Chabala et al., 1993; Serodio et al., 1994, 1996), and low-molecular-weight molecules extracted from the brain can correct this difference when coexpressed with Kv4.x α-subunits (Rudy et al., 1988; Chabala et al., 1993; Serodio et al., 1994, 1996). Indeed, a recent study shows that KChIP1 coexpression is necessary to see the effects of arachidonic acid, similar to what is seen in the native system (Holmqvist et al., 2001). We also found that coexpression with a KChIP3 ancillary subunit was necessary to reconstitute an additional native-channel function: modulation by PKA.
How might phosphorylation of the C-terminal lead to changes in the voltage dependence of activation? Phosphorylation of cytosolic domains is known to regulate K+-channel function (Levitan, 1994; Jonas and Kaczmarek, 1996), including effects on voltage-dependent activation (Murakoshi et al., 1997). Moreover, PKC phosphorylation of Kv3.4 has been shown to remove fast inactivation, converting a rapidly inactivating K+current to a noninactivating one (Covarrubias et al., 1994). We suggest that phosphorylation actually increases the rate of inactivation. Indeed, Kv4.1 has been shown to inactivate via interactions of the C and N terminals (Jerng and Covarrubias, 1997; Jerng et al., 1999). Therefore, we suggest that an interaction of the N-terminal (through binding to KChIP) and the C-terminal phosphorylation is involved in PKA modulation of Kv4.2.
It is interesting to consider the possibility that other Kv4.2 ancillary subunits might be able to substitute for KChIP. In particular, β-subunits have been shown to alter channel properties (Rettig et al., 1994; Heinemann et al., 1995; Leicher et al., 1998), act as chaperone proteins that promote and/or stabilize the cell-surface expression of α-subunits (Levin et al., 1996; Shi et al., 1996; Nagaya and Papazian, 1997; Trimmer, 1998; E. K. Yang et al., 2001), or transduce phosphorylation-dependent alterations in K+ channel properties (Jin et al., 2002), similar to what we have observed in this study. These subunits lack putative transmembrane domains and potential glycosylation sites or leader sequences, suggesting that they are cytoplasmic proteins (Scott et al., 1994). It will be interesting in the future to determine whether β-subunits also have in common with KChIPs the capacity to confer PKA modulation.
Kv4.2 is also a substrate for the MAP kinase (MAPK) extracellular signal-regulated kinase (ERK); ERK regulation of the dendritic transient K+ currents plays an important role in the neuromodulation of hippocampal pyramidal neurons (Watanabe et al., 2002; Yuan et al., 2002). It will be interesting to see whether ERK modulation of Kv4.2 depends on the presence of an ancillary subunit, as does PKA modulation. Moreover, because both PKA and ERK can directly phosphorylate the Kv4.2 α-subunit, it is intriguing to consider that there might be functional interactions of these two molecular events. It has been shown previously that activation of PKA or PKC caused a shift in the activation curve of transient outward K+ currents in hippocampal dendrites (Hoffman and Johnston, 1998). PKA activation has been shown to couple to ERK activation in the hippocampus (Roberson et al., 1999). This effect of PKA and PKC activation on the transient outward K+ current is blocked by the ERK/MAPK inhibitor U0126 (Yuan, 2002). In addition, glial cell line-derived neurotrophic factor and 8-Br-cAMP have been shown to increase cell excitability via reduction of A-type K+currents in midbrain dopaminergic neurons, an effect that is mediated by ERK/MAPK (F. Yang et al., 2001). Because U0126 blocked the effect of PKA activation in the endogenous system, we would predict that phosphorylation of one (or several) of the previously characterized ERK/MAPK sites (Adams et al., 2000) may also be necessary for the PKA-dependent modulation of Kv4.2 or Kv4.3 channels in vivo. This would be one specific example of the general hypothesis that there are functional interactions of the various phosphorylation sites identified on Kv4.2. This complexity of regulation would allow Kv4.2 (plus interacting subunits) to serve as a novel locus for coincidence detection of various signaling mechanisms.
One interesting issue is whether other Kv4 family ion channels are similarly regulated as part of a supramolecular complex. Indeed, dopaminergic neurons express Kv4.3 (Serodio and Rudy, 1998; Liss et al., 2001), which is highly homologous to Kv4.2. Although the PKA or ERK phosphorylation sites on Kv4.3 have not been mapped, several consensus sequences for both kinases exist within the Kv4.3 sequence; it is highly likely that either kinase can phosphorylate Kv4.3. Specifically, in the context of the present findings it appears that the S552 site is conserved in Kv4.3, so similar mechanisms to what we have observed with Kv4.2 may apply to Kv4.3 as well.
What functional roles might the PKA modulation of Kv4.2 play in the intact cell? Transient A-type K+ current exists at high density in hippocampal pyramidal cell dendrites; these currents play a major role in pyramidal neuron excitability (Hoffman et al., 1997). Recent work by Quirk et al. (2001) has shown a correlation between learning behavior and back-propagating spike amplitude, which is subject to regulation by hippocampal A currents (Hoffman et al., 1997), thus suggesting that modulation of these currents plays a role in learning and memory. These A-type currents are modulated by kinase activation, and this modulation has been shown to regulate the amplitude of back-propagating action potentials, which can have dramatic effects on the induction of long-term potentiation and information processing (Hoffman and Johnston, 1998, 1999). Although the molecular subunits of these currents are unknown, the primary subunit protein, Kv4.2, is an excellent candidate, because it exists in the pyramidal cell dendrites and exhibits properties similar to the native current. The Kv4 family of K+ channels has been shown recently to interact with the KChIP family of accessory subunits in vitro, and they are likely an important component of the K+-channel complex. Modulation of the properties of these channels (e.g., voltage dependence, inactivation rate, number of channels, and distribution) represents a powerful potential site for the regulation of pyramidal neuron excitability in long-term potentiation. Thus, these transient outward currents (most likely composed of Kv4.2 α-subunits and other α-subunits or interacting proteins) appear to be ideally suited, both in terms of their biophysical properties and subcellular localization, for contributing significantly to the regulation of pyramidal neuron excitability and synaptic responsiveness.
In summary, we have demonstrated a novel mechanism for ion-channel regulation by kinases. We show that interaction of the Kv4.2 α-subunit with an interacting subunit (KChIP3) is necessary for modulation by PKA phosphorylation. These data indicate a surprising complexity to the structure–function relationships for the regulation of ion channels by phosphorylation.
Footnotes
This work was supported by grants from the National Institute of Mental Health and the National Alliance for Research on Schizophrenia and Depression (L.A.S., J.D.S.) and from the National Institute of Neurological Disorders and Stroke (J.D.S., A.E.A.). We thank Dr. Dan Johnston for helpful discussions.
Correspondence should be addressed to Dr. J. David Sweatt, Division of Neuroscience, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. E-mail: jsweatt@bcm.tmc.edu.
REFERENCES
- 1.Adams JP, Anderson AE, Varga AW, Dineley KT, Cook RG, Pfaffinger PJ, Sweatt JD. The A-type potassium channel Kv4.2 is a substrate for the mitogen-activated protein kinase ERK. J Neurochem. 2000;75:2277–2287. doi: 10.1046/j.1471-4159.2000.0752277.x. [DOI] [PubMed] [Google Scholar]
- 2.An WF, Bowlby MR, Betty M, Cao J, Ling HP, Mendoza G, Hinson JW, Mattsson KI, Strassle BW, Trimmer JS, Rhodes KJ. Modulation of A-type potassium channels by a family of calcium sensors. Nature. 2000;403:553–556. doi: 10.1038/35000592. [DOI] [PubMed] [Google Scholar]
- 3.Anderson AE, Adams JP, Qian Y, Cook RG, Pfaffinger PJ, Sweatt JD. Kv4.2 phosphorylation by cyclic AMP-dependent protein kinase. J Biol Chem. 2000;275:5337–5346. doi: 10.1074/jbc.275.8.5337. [DOI] [PubMed] [Google Scholar]
- 4.Baldwin TJ, Tsaur M-L, Lopez GA, Jan YN, Jan LY. Characterization of a mammalian cDNA for an inactivating voltage-sensitive K+ channel. Neuron. 1991;7:471–483. doi: 10.1016/0896-6273(91)90299-f. [DOI] [PubMed] [Google Scholar]
- 5.Beck EJ, Bowlby M, An WF, Rhodes KJ, Covarrubias M. Remodelling inactivation gating of Kv4 channels by KChIP1, a small-molecular-weight calcium-binding protein. J Physiol (Lond) 2002;538:691–706. doi: 10.1113/jphysiol.2001.013127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blair TA, Roberds SL, Tamkun MM, Hartshorne RP. Functional characterization of RK5, a voltage-gated K+ channel cloned from the rat cardiovascular system. FEBS Lett. 1991;295:211–213. doi: 10.1016/0014-5793(91)81420-d. [DOI] [PubMed] [Google Scholar]
- 7.Bourque CW. Transient calcium-dependent potassium current in magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol (Lond) 1988;397:331–347. doi: 10.1113/jphysiol.1988.sp017004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buxbaum JD, Choi EK, Luo Y, Lilliehook C, Crowley AC, Merriam DE, Wasco W. Calsenilin: a calcium-binding protein that interacts with the presenilins and regulates the levels of a presenilin fragment. Nat Med. 1998;4:1177–1181. doi: 10.1038/2673. [DOI] [PubMed] [Google Scholar]
- 9.Carrion AM, Link WA, Ledo F, Mellstrom B, Naranjo JR. DREAM is a Ca2+-regulated transcriptional repressor. Nature. 1999;398:80–84. doi: 10.1038/18044. [DOI] [PubMed] [Google Scholar]
- 10.Chabala LD, Bakry N, Covarrubias M. Low molecular weight poly(A)+ mRNA species encode factors that modulate gating of a non-Shaker A-type K+ channel. J Gen Physiol. 1993;102:713–728. doi: 10.1085/jgp.102.4.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chandy K, Gutman G. Voltage-gated potassium channel genes. In: North R, editor. Handbook of receptors and channels: ligand and voltage-gated ion channels. CRC; Boca Raton, FL: 1995. [Google Scholar]
- 12.Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decher N, Uyguner O, Scherer CR, Karaman B, Yuksel-Apak M, Busch AE, Steinmeyer K, Wollnik B. hKChIP2 is a functional modifier of hKv4.3 potassium channels: cloning and expression of a short hKChIP2 splice variant. Cardiovasc Res. 2001;52:255–264. doi: 10.1016/s0008-6363(01)00374-1. [DOI] [PubMed] [Google Scholar]
- 14.Fisher TE, Bourque CW. Properties of the transient K+ current in acutely isolated supraoptic neurons from adult rat. Adv Exp Med Biol. 1998;449:97–106. doi: 10.1007/978-1-4615-4871-3_9. [DOI] [PubMed] [Google Scholar]
- 15.Frangioni JV, Neel BG. Use of general purpose mammalian expression vector for studying intracellular protein targeting: identification of critical residues in the nuclear lamin A/C nuclear localization signal. J Cell Sci. 1993;105:481–488. doi: 10.1242/jcs.105.2.481. [DOI] [PubMed] [Google Scholar]
- 16.Hakes DJ, Dixon JE. New vectors for high level expression of recombinant proteins in bacteria. Anal Biochem. 1992;202:293–298. doi: 10.1016/0003-2697(92)90108-j. [DOI] [PubMed] [Google Scholar]
- 17.Heinemann SH, Rettig J, Wunder F, Pongs O. Molecular and functional characterization of a rat brain Kvβ3 potassium channel subunit. FEBS Lett. 1995;377:383–389. doi: 10.1016/0014-5793(95)01377-6. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman DA, Johnston D. Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci. 1998;18:3521–3528. doi: 10.1523/JNEUROSCI.18-10-03521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoffman DA, Johnston D. Neurotransmitter modulation of dendritic action potentials. J Neurophysiol. 1999;81:408–411. doi: 10.1152/jn.1999.81.1.408. [DOI] [PubMed] [Google Scholar]
- 20.Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- 21.Holmqvist MH, Cao J, Knoppers MH, Jurman ME, Distefano PS, Rhodes KJ, Xie Y, An WF. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J Neurosci. 2001;21:4154–4161. doi: 10.1523/JNEUROSCI.21-12-04154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jan L, Jan Y. Structural elements involved in specific K+ channel functions. Annu Rev Physiol. 1992;54:537–555. doi: 10.1146/annurev.ph.54.030192.002541. [DOI] [PubMed] [Google Scholar]
- 23.Jerng HH, Covarrubias M. K+channel inactivation mediated by the concerted action of the cytoplasmic N- and C-terminal domains. Biophys J. 1997;72:163–174. doi: 10.1016/S0006-3495(97)78655-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jerng HH, Shahidullah M, Covarrubias M. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J Gen Physiol. 1999;113:641–660. doi: 10.1085/jgp.113.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin P, Weiger TM, Wu Y, Levitan IB. Phosphorylation-dependent functional coupling of hSlo calcium-dependent potassium channel and its hβ4 subunit. J Biol Chem. 2002;277:10014–10020. doi: 10.1074/jbc.M107682200. [DOI] [PubMed] [Google Scholar]
- 26.Jonas EA, Kaczmarek LK. Regulation of potassium channels by protein kinases. Curr Opin Neurobiol. 1996;6:318–323. doi: 10.1016/s0959-4388(96)80114-0. [DOI] [PubMed] [Google Scholar]
- 27.Leicher T, Bahring R, Isbrandt D, Pongs O. Coexpression of the KCNA3B gene product with Kv1.5 leads to a novel A-type potassium channel. J Biol Chem. 1998;273:35095–35101. doi: 10.1074/jbc.273.52.35095. [DOI] [PubMed] [Google Scholar]
- 28.Levin G, Chikvashvili D, Singer-Lahat D, Peretz T, Thornhill WB, Lotan I. Phosphorylation of a K+ channel α subunit modulates the inactivation conferred by a β subunit. Involvement of cytoskeleton. J Biol Chem. 1996;271:29321–29328. doi: 10.1074/jbc.271.46.29321. [DOI] [PubMed] [Google Scholar]
- 29.Levitan IB. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu Rev Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 30.Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J. 2001;20:5715–5724. doi: 10.1093/emboj/20.20.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma D, Jan LY. ER transport signals and trafficking of potassium channels and receptors. Curr Opin Neurobiol. 2002;12:287–292. doi: 10.1016/s0959-4388(02)00319-7. [DOI] [PubMed] [Google Scholar]
- 32.Maletic-Savatic M, Lenn NJ, Trimmer JS. Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci. 1995;15:3840–3851. doi: 10.1523/JNEUROSCI.15-05-03840.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morohashi Y, Hatano N, Ohya S, Takikawa R, Watabiki T, Takasugi N, Imaizumi Y, Tomita T, Iwatsubo T. Molecular cloning and characterization of CALP/KChIP4, a novel EF-hand protein interacting with presenilin 2 and voltage-gated potassium channel subunit Kv4. J Biol Chem. 2002;277:14965–14975. doi: 10.1074/jbc.M200897200. [DOI] [PubMed] [Google Scholar]
- 34.Murakoshi H, Shi G, Scannevin RH, Trimmer JS. Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol. 1997;52:821–828. doi: 10.1124/mol.52.5.821. [DOI] [PubMed] [Google Scholar]
- 35.Nagaya N, Papazian DM. Potassium channel α and β subunits assemble in the endoplasmic reticulum. J Biol Chem. 1997;272:3022–3027. doi: 10.1074/jbc.272.5.3022. [DOI] [PubMed] [Google Scholar]
- 36.Pongs O. Molecular biology of voltage-dependent potassium channels. Physiol Rev. 1992;72:S69–S88. doi: 10.1152/physrev.1992.72.suppl_4.S69. [DOI] [PubMed] [Google Scholar]
- 37.Quirk MC, Blum KI, Wilson MA. Experience-dependent changes in extracellular spike amplitude may reflect regulation of dendritic action potential back-propagation in rat hippocampal pyramidal cells. J Neurosci. 2001;21:240–248. doi: 10.1523/JNEUROSCI.21-01-00240.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rettig J, Heinemann SH, Wunder F, Lorra C, Parcej DN, Dolly JO, Pongs O. Inactivation properties of voltage-gated K+ channels altered by presence of β-subunit. Nature. 1994;369:289–294. doi: 10.1038/369289a0. [DOI] [PubMed] [Google Scholar]
- 39.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rudy B, Hoger JH, Lester HA, Davidson N. At least two mRNA species contribute to the properties of rat brain A-type potassium channels expressed in Xenopus oocytes. Neuron. 1988;1:649–658. doi: 10.1016/0896-6273(88)90164-x. [DOI] [PubMed] [Google Scholar]
- 41.Schorderet-Slatkine S, Baulieu EE. Forskolin increases cAMP and inhibits progesterone induced meiosis reinitiation in Xenopus laevis oocytes. Endocrinology. 1982;111:1385–1387. doi: 10.1210/endo-111-4-1385. [DOI] [PubMed] [Google Scholar]
- 42.Scott VE, Rettig J, Parcej DN, Keen JN, Findlay JB, Pongs O, Dolly JO. Primary structure of a β subunit of α-dendrotoxin-sensitive K+ channels from bovine brain. Proc Natl Acad Sci USA. 1994;91:1637–1641. doi: 10.1073/pnas.91.5.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serodio P, Rudy B. Differential expression of Kv4 K+ channel subunits mediating subthreshold transient K+ (A-type) currents in rat brain. J Neurophysiol. 1998;79:1081–1091. doi: 10.1152/jn.1998.79.2.1081. [DOI] [PubMed] [Google Scholar]
- 44.Serodio P, Kentros C, Rudy B. Identification of molecular components of A-type channels activating at subthreshold potentials. J Neurophysiol. 1994;72:1516–1529. doi: 10.1152/jn.1994.72.4.1516. [DOI] [PubMed] [Google Scholar]
- 45.Serodio P, Vega-Saenz de Miera E, Rudy B. Cloning of a novel component of A-type K+ channels operating at subthreshold potentials with unique expression in heart and brain. J Neurophysiol. 1996;75:2174–2179. doi: 10.1152/jn.1996.75.5.2174. [DOI] [PubMed] [Google Scholar]
- 46.Sheng M, Tsaur M-L, Jan YN, Jan LY. Subcellular segregation of two A-type K+ channel proteins in rat central neurons. Neuron. 1992;9:271–284. doi: 10.1016/0896-6273(92)90166-b. [DOI] [PubMed] [Google Scholar]
- 47.Shi G, Nakahira K, Hammond S, Rhodes KJ, Schechter LE, Trimmer JS. β subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron. 1996;16:843–852. doi: 10.1016/s0896-6273(00)80104-x. [DOI] [PubMed] [Google Scholar]
- 48.Shibata R, Wakazono Y, Nakahira K, Trimmer JS, Ikenaka K. Expression of Kv3.1 and Kv4.2 genes in developing cerebellar granule cells. Dev Neurosci. 1999;21:87–93. doi: 10.1159/000017370. [DOI] [PubMed] [Google Scholar]
- 49.Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ. Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci. 1998;18:3124–3137. doi: 10.1523/JNEUROSCI.18-09-03124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tkatch T, Baranauskas G, Surmeier DJ. Kv4.2 mRNA abundance and A-type K+ current amplitude are linearly related in basal ganglia and basal forebrain neurons. J Neurosci. 2000;20:579–588. doi: 10.1523/JNEUROSCI.20-02-00579.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trimmer JS. Regulation of ion channel expression by cytoplasmic subunits. Curr Opin Neurobiol. 1998;8:370–374. doi: 10.1016/s0959-4388(98)80063-9. [DOI] [PubMed] [Google Scholar]
- 52.Tsaur ML, Chou CC, Shih YH, Wang HL. Cloning, expression and CNS distribution of Kv4.3, an A-type K+ channel α subunit. FEBS Lett. 1997;400:215–220. doi: 10.1016/s0014-5793(96)01388-9. [DOI] [PubMed] [Google Scholar]
- 53.Varga AW, Anderson AE, Adams JP, Vogel H, Sweatt JD. Input-specific immunolocalization of differentially phosphorylated Kv4.2 in the mouse brain. Learn Mem. 2000;7:321–332. doi: 10.1101/lm.35300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Watanabe S, Hoffman DA, Migliore M, Johnston D. Dendritic K+channels contribute to spike-timing dependent long-term potentiation in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2002;99:8366–8371. doi: 10.1073/pnas.122210599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang EK, Alvira MR, Levitan ES, Takimoto K. Kvβsubunits increase expression of Kv4.3 channels by interacting with their C termini. J Biol Chem. 2001;276:4839–4844. doi: 10.1074/jbc.M004768200. [DOI] [PubMed] [Google Scholar]
- 56.Yang F, Feng L, Zheng F, Johnson SW, Du J, Shen L, Wu CP, Lu B. GDNF acutely modulates excitability and A-type K+ channels in midbrain dopaminergic neurons. Nat Neurosci. 2001;4:1071–1078. doi: 10.1038/nn734. [DOI] [PubMed] [Google Scholar]
- 57.Yuan LLA, JP, Swank M, Sweatt JD, Johnston D. Protein kinase modulation of dendritic K+ channels in hippocampus involves a mitogen-activated protein kinase pathway. J Neurosci. 2002;22:4860–4868. doi: 10.1523/JNEUROSCI.22-12-04860.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]