

Abstract
Neuronal nitric oxide synthase (nNOS) generates superoxide, particularly at sub-optimal l-arginine (L-Arg) substrate concentrations. Heat shock protein 90 (Hsp90) was reported to inhibit superoxide generation from nNOS protein. However, commercially available Hsp90 product from bovine brain tissues with unspecified Hsp90α and Hsp90β contents and an undefined Hsp90 protein oligomeric state was utilized. These two Hsp90s can have opposite effect on superoxide production by NOS. Importantly, emerging evidence indicates that nNOS splice variants are involved in different biological functions by functioning distinctly in redox signaling. In the present work, purified recombinant human Hsp90α in its native dimeric state was used in electron paramagnetic resonance (EPR) spin trapping experiments to study the effects of Hsp90α on superoxide generation from nNOS splice variants nNOSµ and nNOSα. Human Hsp90α was found to significantly increase superoxide generation from nNOSµ and nNOSα proteins under l-Arg-depleted conditions and Hsp90α influenced superoxide production by nNOSµ and nNOSα at varying degrees. Imidazole suppressed the spin adduct signal, indicating that superoxide was produced at the heme site of nNOS in the presence of Hsp90α, whereas l-Arg repletion diminished superoxide production by the nNOS-Hsp90α. Moreover, NADPH consumption rate values exhibited a similar trend/difference as a function of Hsp90α and l-Arg. Together, these EPR spin trapping and NADPH oxidation kinetics results demonstrated noticeable Hsp90α-induced increases in superoxide production by nNOS and a distinguishable effect of Hsp90α on nNOSµ and nNOSα proteins.
Keywords: nitric oxide synthase, superoxide, heat shock protein 90, EPR spin trapping, heme
Graphic Abstract
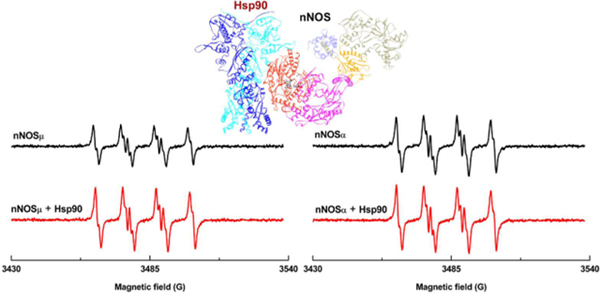
Previous studies showed that heat shock protein (Hsp) 90 binds to heme domain of nitric oxide synthase (NOS). Electron paramagnetic resonance (EPR) spin trapping results herein indicated that dimeric human Hsp90α increases superoxide generation from neuronal NOS (nNOS) heme, and that the enhancement effect differs between nNOSµ and nNOSα isoforms.
Introduction
Nitric oxide synthase (NOS) is a family of flavohemoproteins responsible for biosynthesis of nitric oxide (NO) in vertebrates [1]. There are three mammalian NOS isoforms with distinct functions in various tissues: neuronal, endothelial and inducible NOS (nNOS, eNOS and iNOS, respectively). Structurally, each subunit of the homodimeric NOS protein is comprised of an N-terminal heme and tetrahydrobiopterin (H4B)-containing oxygenase domain and a C-terminal reductase domain that comprises the FMN, FAD and NADPH-binding (sub)domains. The oxygenase and reductase domains are joined by a linker that binds a calcium-sensing protein, calmodulin (CaM). The NOS enzyme catalyzes oxidation of l-arginine (L-Arg) to NO, which requires CaM-enabled electron transport across the NOS domains [2].
In addition to NO, NOS generates superoxide (O2•−) free radical [3, 4]. This phenomenon is termed NOS uncoupling since O2•− generation mainly occurs when NOS is not coupled with its substrate l-Arg or cofactor H4B [5, 6]. It is of current interest to elucidate the mechanism controlling NOS’s O2•− production [7] because: a) excessive O2•− plays a key role in generating reactive oxygen species (ROS) and reactive nitrogen species (RNS) including hydrogen peroxide and peroxynitrite, and b) low level O2•− production emerges as an important redox signaling molecule [8]. Early reports showed that nNOS produces O2•− under l-Arg depletion condition [9, 10]. Further studies demonstrated that O2•− is a product of all three NOS isoforms [3, 11] and that NOS also generates some O2•− in the presence of l-Arg at physiologically relevant concentration [11, 12]. These studies relied on a) unambiguous measurement of free radicals by EPR spin trapping [13] and b) purified active NOS proteins [3, 4, 13]. Direct detection of O2•− is difficult in aqueous solution at room temperature due to its short lifetime. To address this, an electron paramagnetic resonance (EPR)-silent spin trapping reagent can be used to react with O2•−, forming a more stable EPR-active spin adduct that can then be analyzed by EPR spectroscopy. Zweier, Vasquez-Vivar, Rosen and their co-workers have presented elegant studies of O2•− production by the NOS isoforms. These extensive studies have provided important insights into molecular aspects of NOS regulation, such as the site of O2•− production in the NOS isoforms and the role of l-Arg and H4B in NOS coupling [4, 14]. In general, O2•− production by eNOS is mainly controlled by H4B levels [5], while O2•− generation from nNOS is more l-Arg-dependent [12]. These results provide valuable mechanistical information to explain the in vitro and in vivo phenomena where NOS uncoupling contributes to the onset of pathological events [15, 16].
NOS is regulated in vivo by interacting with other proteins in response to various stimuli. For example, heat shock protein 90 (Hsp90) is a major modulator of NOS. In vitro cellular studies of nNOS regulation by Hsp90 have demonstrated important roles of Hsp90 in nNOS protein quality control [17] and heme insertion into the NOS enzyme [18]. Besides these chaperone functions, Hsp90 protein was found to affect NOS’s ability to produce O2•−, with most of the previous studies focusing on eNOS [19–22]. Compared to the much-studied eNOS-Hsp90 system, to the best of our knowledge, there is only one report, which focused on the effect of Hsp90 on the nNOS O2•− production [23]. Their EPR-spin trapping studies of nNOS-Hsp90 complex used commercially available Hsp90 proteins isolated from bovine brain tissue [23]; recombinant Hsp90 protein was not accessible at that time. The cytosolic Hsp90 protein in tissues mainly exist in its β form, except in the brain tissue [24]: porcine brain tissue contains ~ 6 times more abundant Hsp90α than the β form. In other words, it is very likely that the isolated Hsp90 product used in the previous study [23] is a mixture of Hsp90α and Hsp90β forms. Not until recently has it been realized that these two Hsp90 forms can have opposite effects on O2•− production by eNOS [25]. Furthermore, the native form of Hsp90α protein is dimeric, while Hsp90 readily aggregates during purification [26]. Of note, the oligomeric state of the commercial Hsp90 proteins is not defined in their data sheets. Therefore, the previous studies using the commercially available Hsp90 product at that time [23] may not faithfully reproduce its native dimeric state. These Hsp90 oligomeric and isoform issues are major and may have complicated the previous EPR spin trapping studies [23], raising possibilities of alternative interpretations.
One of the other important processes in regulating expression and function of nNOS is its translational splice variants [27–30]. This is particularly important for tissue-specific regulation of nNOS. For example, nNOSµ in skeletal muscle is slightly larger in its size than nNOSα in brain, due to alternative splicing that adds a 102-base pair insert between exons 16 and 17 [29]; nNOSµ contains 34 additional residues in an autoregulatory (AR) insert within the FMN domain, compared to nNOSα (Figure 1). Both nNOSα and nNOSµ are full-length functional proteins that have similar NO production activity [31], but differ in O2•− production under substrate-depleted conditions [32]. The involvement of nNOS splice variants in different biological functions [33] by functioning distinctly in redox signaling [27, 34, 35] has just begun to be explored. For example, deviations in nNOS splice variant signaling in skeletal muscle have been established in pathologies of several neuromuscular diseases [27], but the underlying biochemical mechanisms remain largely unclear. Taken together, it is timely to unambiguously investigate the effect of specific Hsp90 form on O2•− production by nNOS splice variants, as the first necessary step to comprehend redox signaling pathways in tissues.
Figure 1.
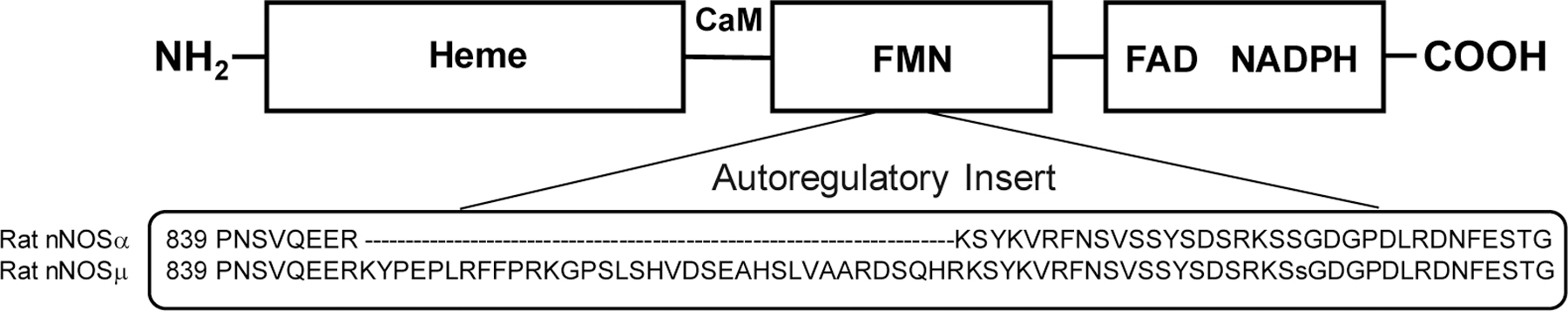
Schematic view of dimeric heme-containing oxygenase domain and FMN-, FAD- and NADPH-binding (sub)domains in nNOS holoprotein. The two nNOS splice variants, nNOSα and nNOSµ, are both full-length proteins, which only differ in the number of amino acids in the autoregulatory insert within the FMN domain: nNOSµ contains extra 34 residues, compared to nNOSα. CaM binds to the linker connecting the FMN and heme domains, while Hsp90 associates with the heme domain.
To address this unanswered important question in the field, herein we utilized dimeric recombinant human Hsp90α purified in our laboratory, to overcome the abovementioned limitations in commercial Hsp90 product. Parallel experiments with rat nNOSµ and nNOSα proteins were conducted to compare the Hsp90 effect on their O2•− production and to also ensure that the observed results are substantiated. The EPR spin trapping experiments were further complemented with NADPH oxidation rate assays, since NADPH is the electron source for NOS-mediated reactions. Of interest are the findings that Hsp90α enhances O2•− production by nNOS proteins in the absence of l-Arg, and that Hsp90α differentially influences O2•− generation from the nNOSµ and nNOSα proteins. This work should inspire future mechanistic and biological studies of the role of specific Hsp90 isoforms in regulation of nNOS splice variants.
Materials and Methods
Materials.
Superoxide dismutase (SOD) was purchased from MP Biomedicals, USA. High purity (> 99%) 5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide (BMPO) was obtained from Enzo Life Sciences. Diethylenetriaminepentaacetic acid (DTPA; > 99%), a metal chelator, was purchased from Sigma-Aldrich, USA.
Protein expression and purification.
Rat nNOSα and nNOSµ plasmids were supplied by Dr. Linda Roman, a collaborator and co-author in previous studies [31, 36]. Recombinant rat nNOSα and nNOSµ proteins were overexpressed and purified as reported [31], with a few modifications [37]. The nNOS protein was purified in the presence of l-Arg and H4B, and l-Arg was then removed from the isolated protein by 3-cycle buffer-exchanging into the l-Arg-free buffer. The human Hsp90α plasmid on a vector of pET15b containing a N-terminal His-tag is a generous gift from Dr. Daniel Gewirth. Recombinant human Hsp90α protein was prepared as recently reported in an electron transfer kinetic study [37], and native PAGE analysis was used to probe the oligomeric states of the Hsp90 elution from a Mono Q column [37]. The dimeric Hsp90 fractions were then pooled and concentrated for the EPR spin trapping experiments. CaM was prepared by the method of Zhang and Vogel [38].
Spin trapping O2•− by X-band EPR.
The generation of O2•− from purified nNOS was monitored using EPR spin trapping technique as reported in the literature [39] with slight modification. The concentrated nNOS, CaM and Hsp90α proteins were incubated in the presence of 0.5 mM CaCl2 at room temperature for 15 minutes. The protein mixture was then added into a reaction system containing 0.5 mM CaCl2 and 20 mM BMPO (a nitrone spin trap) in a phosphate buffer (50 mM, pH 7.4, 100 µM DTPA and 100 µM EDTA). The final concentration of nNOS and Hsp90 were 20 nM and 3 µM, respectively, if added. 3 µM Hsp90 was used here to ensure binding of Hsp90 to nNOS since the Hsp90-NOS binding affinity is modest, with reported value of 200–500 nM [40]. A saturated amount of CaM (1 µM) was added into all the samples in which nNOS was present. The concentration of l-Arg, H4B, and imidazole in the reaction system were 100 µM, 10 µM, and 5 mM, respectively, if added. Upon addition of NADPH (150 µM final concentration) to trigger the reaction, the sample (400 µL total volume) was immediately transferred into a custom-made gas permeable Teflon tubing (Zeus Industries, Raritan, NJ), which was folded four times and inserted into a quartz EPR tube open at each end. The EPR tube was inserted within the cavity of a Bruker EleXsys E500 X-band EPR spectrometer (Bruker Biospin, Billerica, MA) and the spectra of BMPO-•OOH, spin-trapped O2•−, were acquired after spectrometer tuning at room temperature.
The EPR spectrum was acquired with a scan time of 20 s, and 3 scans were obtained and averaged to produce significant signal-to-noise ratio using the following instrument settings: microwave frequency, 9.8 GHz; modulation frequency, 100 kHz; center magnetic field, 3509 G; microwave power, 21 mW; modulation amplitude, 1.0 G; scan range, 110 G; time constant, 10 ms; receiver gain, 60 dB. The X-band EPR spectrometer was upgraded with a digital console upgrade and new 10” magnet for higher resolution, greater sensitivity and improved usability and stability. The EPR spectra were collected at 10, 15 and 20 minutes after initiating the reaction, and the data were stored using the Bruker Software Xepr (Billerica, MA) or converted to an ASCII file for further processing.
NADPH consumption assay.
Rate of NADPH oxidation by nNOS was determined in a pH 7.4 buffer containing 50 mM Tris, 100 mM NaCl, and 200 µM CaCl2. NADPH solution was freshly prepared daily prior to the measurements, and the final concentration in the assay solution is 100 µM. Concentrated nNOS, CaM and Hsp90α were first incubated in the presence of 0.2 mM CaCl2 at room temperature for 15 min, if added. The reaction was then triggered by adding the concentrated protein sample into the assay solution, with final concentrations of 20 nM nNOS and 1 µM CaM. The final concentrations of l-Arg and Hsp90α protein, if added, were 100 µM and 3 µM, respectively. The rate of NADPH consumption was determined using extinction coefficients of 6.2 mM−1 cm−1 at 340 nm [31].
Spectrophotometric Methods.
CO difference spectra were performed as described [41]. The molar protein concentrations for nNOS variants were determined based on heme content via reduced CO difference spectra, where ε=100 mM−1cm−1 for ΔA445–470, and thus reflect only active enzyme.
Results and Discussion
We first prepared recombinant nNOS and Hsp90 proteins for the EPR spin trapping and NADPH consumption studies. The purified nNOS variants are full-length protein, evidenced by the SDS-PAGE image in Figure S1 in Supporting Information. The NO production rates of the purified nNOSµ and nNOSα are 63 ± 4 and 72 ± 7 min−1, respectively, which are consistent with the reported values [31]. This confirms that the nNOS proteins used in this work are active. The absorption spectra of the purified nNOSµ and nNOSα proteins are similar [31]. The heme center is primarily in the high spin state after treatment with H4B and l-Arg during purification, as indicated by the Soret maximum near 396 nm (Figure S2). The CO difference spectrum (inset of Figure S2) has the characteristic peak of Fe(II)−CO at 446 nm, showing that the nNOS heme iron center is in the native heme-thiolate complex. To study O2•− production by nNOS under l-Arg-depleted conditions, l-Arg was removed from the purified nNOS protein by 3-cycle of buffer exchange into l-Arg-free buffer. Removal of l-Arg was clearly evidenced by the shift of Soret peak (Figure S3).
The native form of Hsp90α protein is dimeric, while Hsp90 easily aggregates during purification [26]. We have optimized the preparation protocol to reduce the aggregation and isolate the dimeric Hsp90α protein fractions, and native PAGE images indicated that the isolated dimeric Hsp90 was > 85 % purity [37]. Our purified dimeric Hsp90α protein set the stage for us to explicitly investigate effect of specific Hsp90 isoform on O2•− generation from nNOS.
For EPR spectroscopy studies, the BMPO nitrone was selected for spin trapping O2•− because it forms a much more stable radical adduct with O2•− than the commonly used spin trap 5,5-Dimethyl-1-pyrroline N-oxide, DMPO (BMPO-•OOH t1/2 = 23 minutes; DMPO-•OOH t1/2 = 45 seconds) and unlike with DMPO, the BMPO-•OOH adduct does not decay readily into a hydroxyl adduct [42]. The BMPO blank sample did not give any EPR signal, while the spin-adduct BMPO-•OOH was clearly observed when BMPO was added to a reaction system of xanthine oxidase-hypoxanthine (XOD/HYP); see Figure S4. The positive control spin trapping of O2•− by BMPO in the XOD/HYP O2•−-generating system in our lab was successful.
We then measured O2•− generation from nNOS using EPR spectroscopy and BMPO. Unlike eNOS, deficiency of l-Arg, but not H4B, primarily causes uncoupling in nNOS [12, 43]. Therefore, we focused on studying nNOS O2•− production as a function of l-Arg concentration in this work. To determine if Hsp90-associated nNOS protein production of O2•− differs from the nNOS protein alone under l-Arg-depleted conditions, EPR spin trapping was used to detect and compare the O2•− generation from nNOS and nNOS-Hsp90 systems. Saturated CaM (1 µM) existed in all the tested reaction systems along with 20 nM nNOS because O2•− generation from nNOS holoprotein takes place at the heme site [39], and CaM is required for the electron transport across the NOS domains to reduce the heme center. As expected, in the absence of l-Arg, nNOSµ protein alone produced a considerable amount of O2•− (Figure 2). The EPR signal was predominantly quenched by SOD (Figure S5), showing that at least 98 % of the adduct is from O2•−. The adduct signal of nNOSµ was also notably inhibited upon adding 100 µM l-Arg (Figure S5). This concentration is physiologically relevant and is also well above the Km value of l-Arg (2 µM) [44]. These observations are consistent with the previous reports on l-Arg-dependent O2•− production by nNOSα [12, 23], showing that these two nNOS splice variants behave similarly in O2•− production in the absence and presence of l-Arg.
Figure 2.
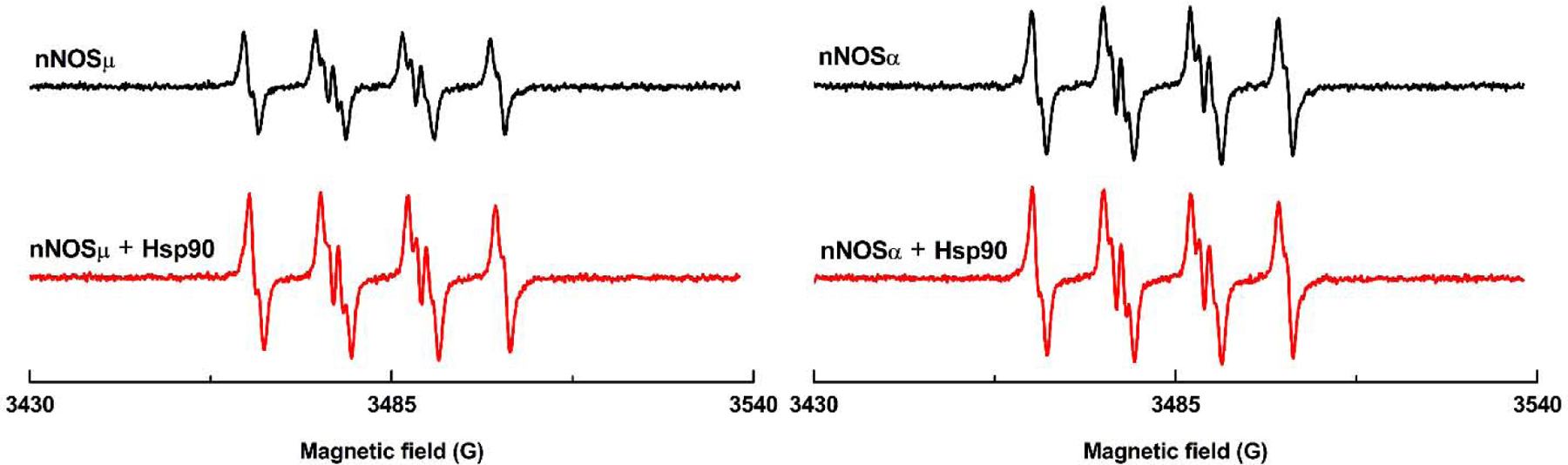
Human Hsp90α protein increases superoxide (O2•−) production by nNOSµ (left panel) and nNOSα (right panel) under l-Arg depleted conditions. nNOSα protein produces larger amount of O2•− than nNOSµ (top panels), while the nNOSα-Hsp90 system generates relatively comparable BMPO-•OOH as the nNOSµ-Hsp90 (bottom panels). Specifically, Hsp90α protein enhances O2•− production by the nNOSµ and nNOSα protein at varying proportions. The EPR spin trapping measurements were performed in phosphate buffer (50 mM, pH 7.4, 0.1 mM EDTA, 0.1 mM DTPA) containing 20 nM nNOS, 3 µM of Hsp90α, 1 µM CaM, 20 mM BMPO and 0.5 mM CaCl2. The reaction was initiated by adding NADPH. The depicted EPR signals were collected at 15 minutes from the beginning of the reaction. The concentrations of nNOSα and nNOSµ were the same (20 nM) in these experiments.
Surprisingly, the relative EPR peak height of the BMPO-•OOH adduct trapped for Hsp90-associated nNOSµ in the absence of l-Arg and under the same EPR spin trapping conditions was significantly higher than that of nNOSµ alone (Figure 2, left panels). In our study, measuring or displaying relative peak heights is an accepted method for comparing free radical production between samples under identical experimental conditions and acquired under the same EPR spectrometer settings. Such comparison of relative peak height is common in numerous EPR spectroscopy experiment models. The peak height is obtained peak-to-trough. The relative height of the 1st peak at the low magnetic field is 0.63 ± 0.05 and 0.87 ± 0.03 for nNOSµ alone and nNOSµ-Hsp90α, respectively. The values are statistically different according to the unpaired t test with a two-tailed P value of 0.0283. This contrasts with the previous reported inhibition of O2•− generation from nNOS at similar Hsp90:nNOS ratio, in which O2•− generation was suppressed over 80% using commercial Hsp90 [23]. We wish to stress that the EPR spin trapping experiments were repeated with different protein preparations on a separate date, with similar increasing trend observed.
To further ensure that our observation is reliable, O2•− productions by another nNOS splice variant, nNOSα, with and without added Hsp90α, was determined. The nNOSα protein in the absence of l-Arg also produced notable amount of O2•−, but at a higher level than that of nNOSµ (Figure 2, top panels). The relative height of the 1st peak at the low magnetic field is 0.84 ± 0.02 and 0.63 ± 0.05 for nNOSα and nNOSµ, respectively. Unpaired t test gives a two-tailed P value of 0.0313, indicating statistical significance difference between the relative peak heights. This observation is in good agreement with the reported difference between nNOSα and nNOSµ [32]. In that work, they used DMPO to trap O2•− [32] and SOD control experiments did show the adduct signal was predominantly attributed to DMPO-•OOH (i.e., O2•−). Therefore, our present work using BMPO, a much more stable spin trap, verifies the production of O2•− by nNOS in the previous study using DMPO [32]. In combination, results with two spin trapping systems and proper SOD control further solidify the significant difference in O2•− production between nNOSα and nNOSµ isoforms. Importantly, Hsp90α also enhanced O2•− production by nNOSα, which is similar to that of nNOSµ (Figure 2). Therefore, the noticeable Hsp90-induced increase in O2•− productions by nNOS (Figure 2) is substantiated. Note that the increase in the O2•− production by the nNOSα-Hsp90α is at a less degree, compared to that of nNOSµ-Hsp90α (Figure 2).
O2•− readily reacts with other chemicals, e.g., O2•− and NO react at a diffusion-controlled rate to form peroxynitrite [45]. The reaction rates between O2•− and NO with their respective spin traps are slower than that between NO and O2•− themselves. In the EPR spin trapping experiments millimolar concentrations of spin trap (e.g., 20 mM BMPO) is typically used to outcompete with O2•−/NO reaction in order to capture O2•−. Therefore, we can still measure O2•− from partially uncoupled NOS that produces both NO and O2•−. Clearly, the presence of high concentration levels of spin trap perturbs the native states of NOS protein. Therefore, researchers rarely make quantitative claims, which is a common practice in the field. It is more objective to report the phenomenon of NOS uncoupling. We elected to use EPR spectroscopy to define a reaction process and provide direct detection of changes in O2•− production since alternative approaches also present limitations and do not provide the sensitivity and definitive characterization of O2•− observed with EPR techniques. In fact, this kind of compromise is not rare in free radical research. Therefore, the reported difference in this work is rather qualitative, and we did not attempt to quantitively compare the EPR peak height values for all samples.
To exclude the possibility of Hsp90α alone generating O2•− in the reaction system, BMPO was added into the sample containing only the human Hsp90α protein. The EPR spectrum (Figure S6) shows no indication of BMPO-•OOH adduct formation. Therefore, the observed increase in the BMPO-•OOH adduct (Figure 2) is not from the Hsp90 protein itself. Furthermore, imidazole, a blocker of heme cofactor, was used to probe the site of O2•− production by nNOS-Hsp90α. The Ki value of imidazole is 263 µM for nNOS [46], and 5 mM imidazole was thus added into the reaction system. The imidazole addition dramatically suppressed O2•− production by the nNOS-Hsp90α system (Figure 3). Hence, it is the nNOS heme site that is responsible for the O2•− generation in the nNOS-Hsp90 system. Imidazole was also reported to inhibit O2•− formation from the heme center of nNOSα [12].
Figure 3.
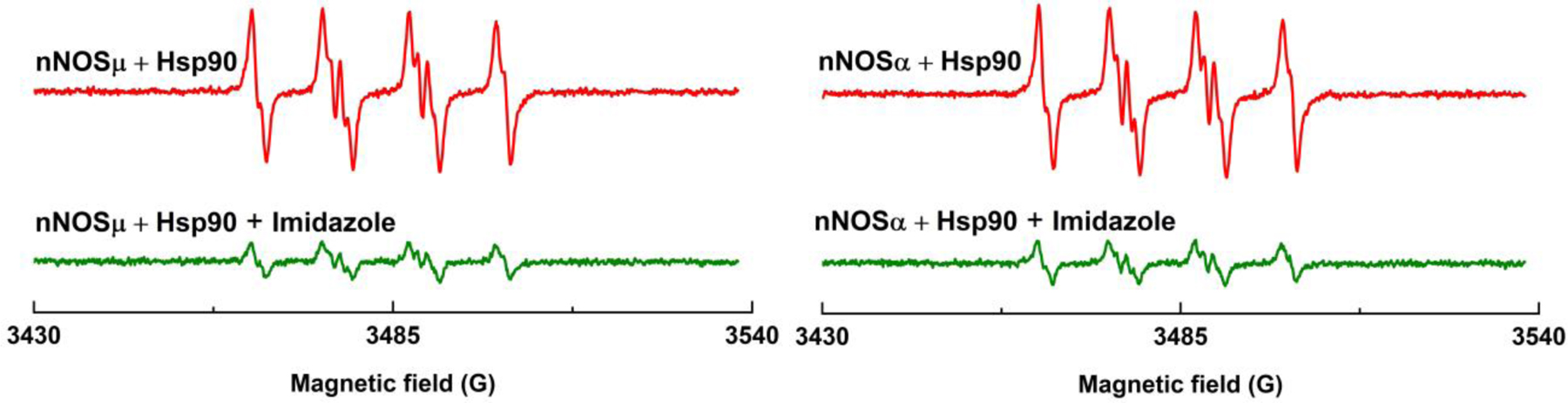
The heme blocker imidazole inhibits the O2•− production in Hsp90α associated nNOSµ (left panel) or nNOSα (right panel) protein under l-Arg depleted conditions. 5 mM imidazole was added into the reaction system. The experimental conditions were the same as described in Figure 2. EPR spectra were recorded continuously, and the spectra presented here were collected 15 minutes after adding NADPH into the reaction system.
Generation of O2•− production by nNOSµ or nNOSα alone and nNOS-Hsp90α system both decreased significantly upon addition of 100 µM l-Arg (Figure 4). Similar suppressing effects of l-Arg on the O2•− production have been reported for nNOS alone samples by other laboratories [3, 12, 32]. These results indicate that the nNOS proteins used, herein, behaved similarly and responded to l-Arg binding to the heme site. Notably, O2•− production by nNOS-Hsp90α in the presence of 100 µM l-Arg was still higher than that of nNOS alone under the same conditions (Figure S7), similar to what was observed for the l-Arg-depleted situation (Figure 2). Hsp90α enhances O2•− generation from nNOS in both l-Arg depleted and repleted conditions. Addition of 10 µM H4B further inhibited nNOS-generated O2•− to nearly complete levels (Figure 4). These effects agree with the reported coupling of l-Arg and H4B to NO production by nNOS [12].
Figure 4.
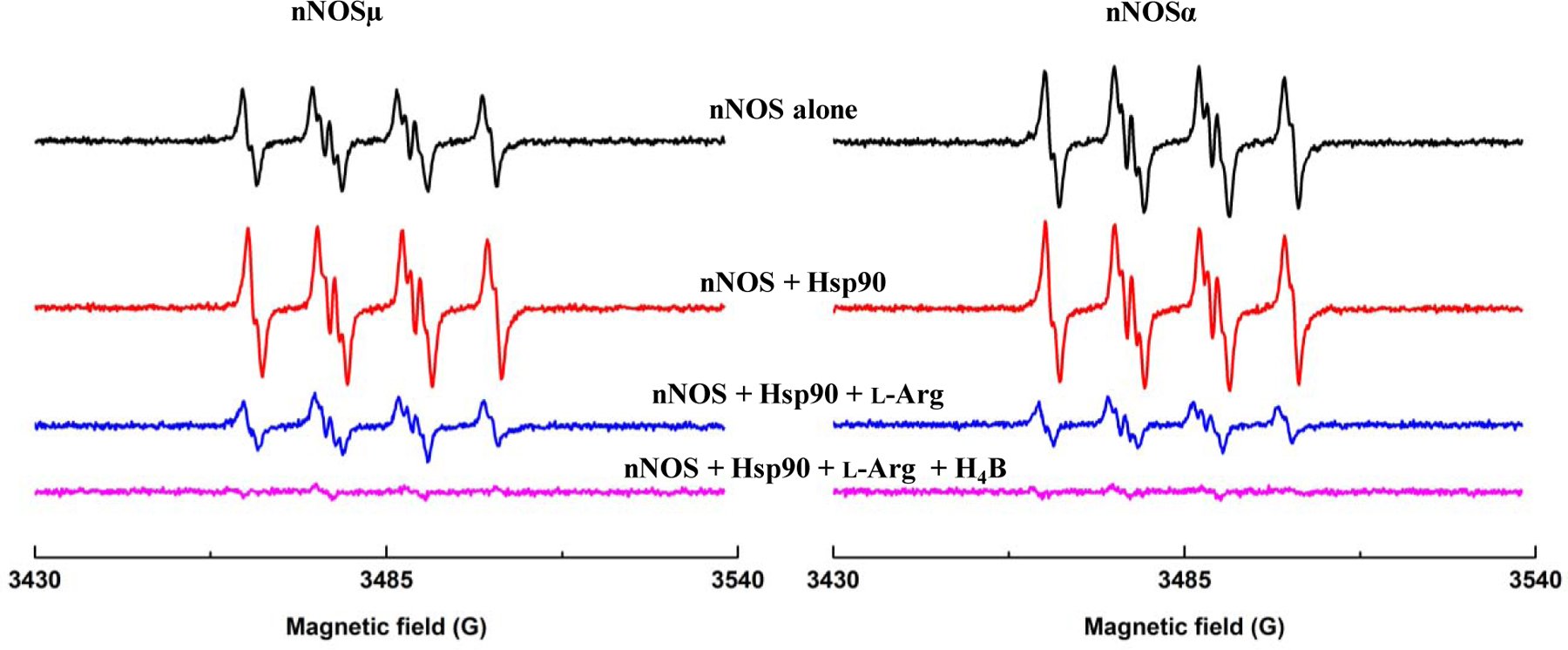
In the nNOS-Hsp90 system, the BMPO-•OOH adduct signal was significantly inhibited by 100 µM l-Arg. Additionally, the EPR signal was nearly completely abolished following addition of 10 µM H4B for both nNOSα and nNOSµ. The conditions were the same as described in Figure 2. The EPR spectra were collected 15 minutes after adding NADPH into the reaction system.
To investigate whether Hsp90 influences total electron flow through the NOS enzyme, the effect of Hsp90 on NADPH consumption was examined (Figure 5). The initial rates of NADPH oxidation by nNOS are listed in Table 1. The values for nNOSµ and nNOSα in the absence of l-Arg are 78.4 ± 0.6 and 100.1 ± 0.6 min−1, respectively. The slower NADPH consumption by nNOSµ in the absence of l-Arg is consistent with the observed smaller O2•−-adduct signal (Figure 2), compared to those of nNOSα. The NADPH oxidation rates were decreased upon l-Arg addition, which is also consistent with both the EPR spin trapping results (Figure 4) and the reported effect in the literature [47]. Moreover, the NADPH oxidation rate of nNOSµ was slower than that of nNOSα, regardless of l-Arg concentration. Importantly, the NADPH oxidation rate of Hsp90α-associated nNOS is significantly higher than that of nNOS alone (Table 1). For example, the initial rates of nNOSµ and nNOSµ-Hsp90α are 78.4 ± 0.6 and 86.8 ± 1.1 min−1, respectively. Unpaired t test confirms statistical significance with a two-tailed P value of 0.0109. Moreover, the total amount of NADPH consumed over 20 minutes of reaction, the same duration for the EPR spin trapping experiments, possessed similar trend/difference upon Hsp90 binding (Figure 5). In combination, Hsp90α augments O2•− generation from nNOS by increasing the rate of NADPH consumption. Taken together, the NADPH consumption and EPR spin trapping results for nNOSµ and nNOSα proteins provide strong evidence that human Hsp90α protein indeed enhances O2•− generation from the nNOS splice variants.
Figure 5.
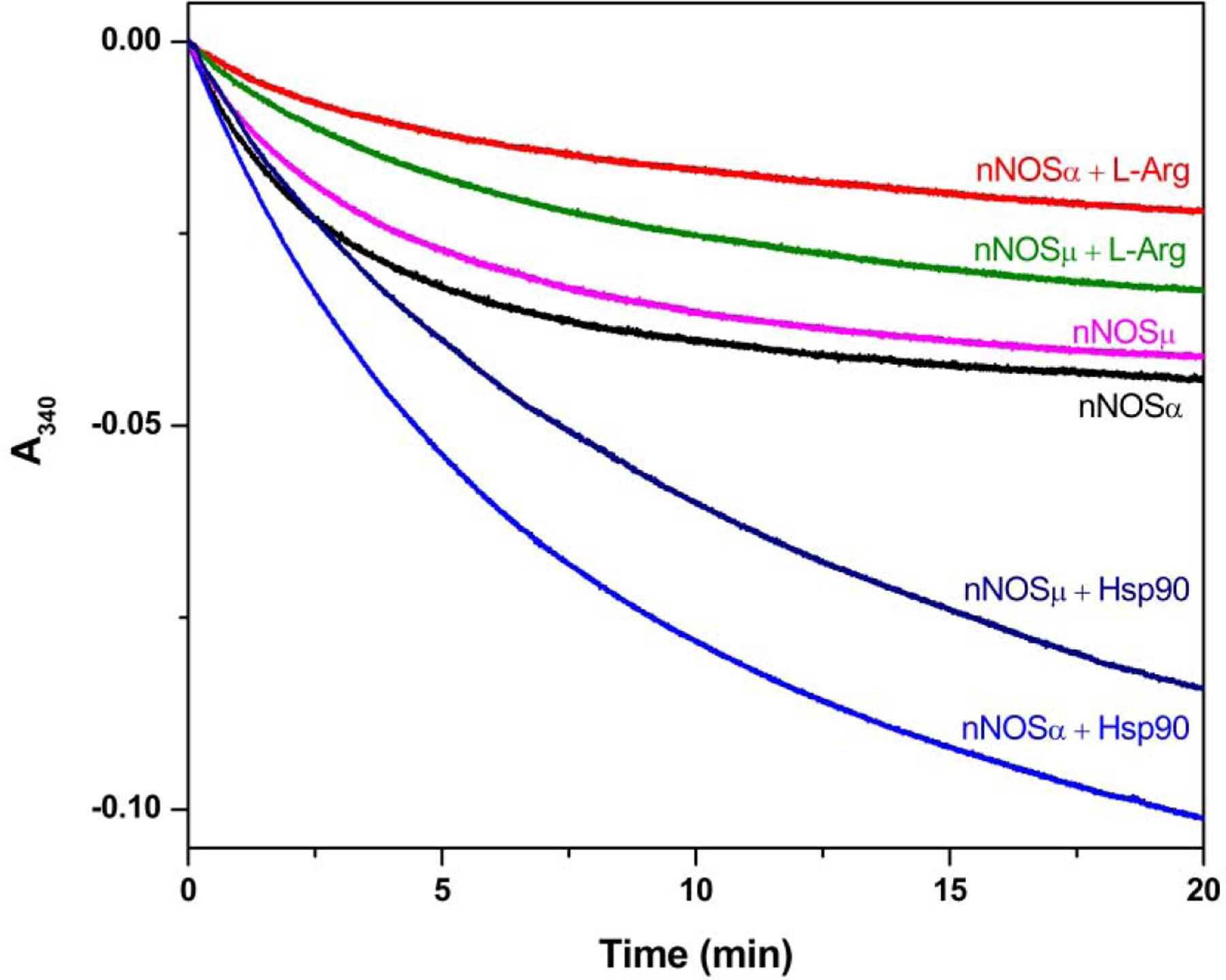
Effect of human Hsp90α on NADPH consumption by nNOS variants. NADPH oxidation was followed by decrease of absorbance at 340 nm. The initial rates of NADPH oxidation listed in Table 1 were obtained by fitting the linear decay within the first one minute.
Table 1.
NADPH oxidation rates of nNOS proteins under various conditions a
| NADPH oxidation (min−1) | |
|---|---|
| nNOSµ | 78.4 ± 0.6 |
| nNOSµ + l-Arg | 45.4 ± 5.1 |
| nNOSµ + Hsp90α | 86.8 ± 1.1 |
| nNOSα | 100.1 ± 0.6 |
| nNOSα + l-Arg | 30.9 ± 0.6 |
| nNOSα + Hsp90α | 119.8 ± 10.2 |
Initial rates of oxidation of NADPH by nNOS were determined in a pH 7.4 buffer containing 50 mM Tris, 100 mM NaCl, 200 µM CaCl2, and 100 µM NADPH. The final concentrations of nNOS and CaM were 20 nM and 1 µM, respectively. The concentrations of l-Arg and Hsp90 protein in the assay mixture, if added, were 100 µM and 3 µM, respectively.
Our results showed that the site of O2•− production in nNOS-Hsp90 is the nNOS heme. The heme iron needs to be reduced to ferrous state by the NADPH-derived electron in order to bind the dioxygen molecule, forming a ferrous-O2 species that is isoelectronic with ferric-superoxy (Scheme 1). At sub-optimal l-Arg concentration, the ferric-superoxy species rapidly decays to release O2•−. Bound l-Arg near the heme iron center stabilizes the [FeIII-O2•−] and disadvantages its decay to release O2•− [48] (Scheme 1), a major pathway in NOS uncoupling. This explains the observed decrease in O2•− production and NADPH consumption in the presence of l-Arg.
Scheme 1.
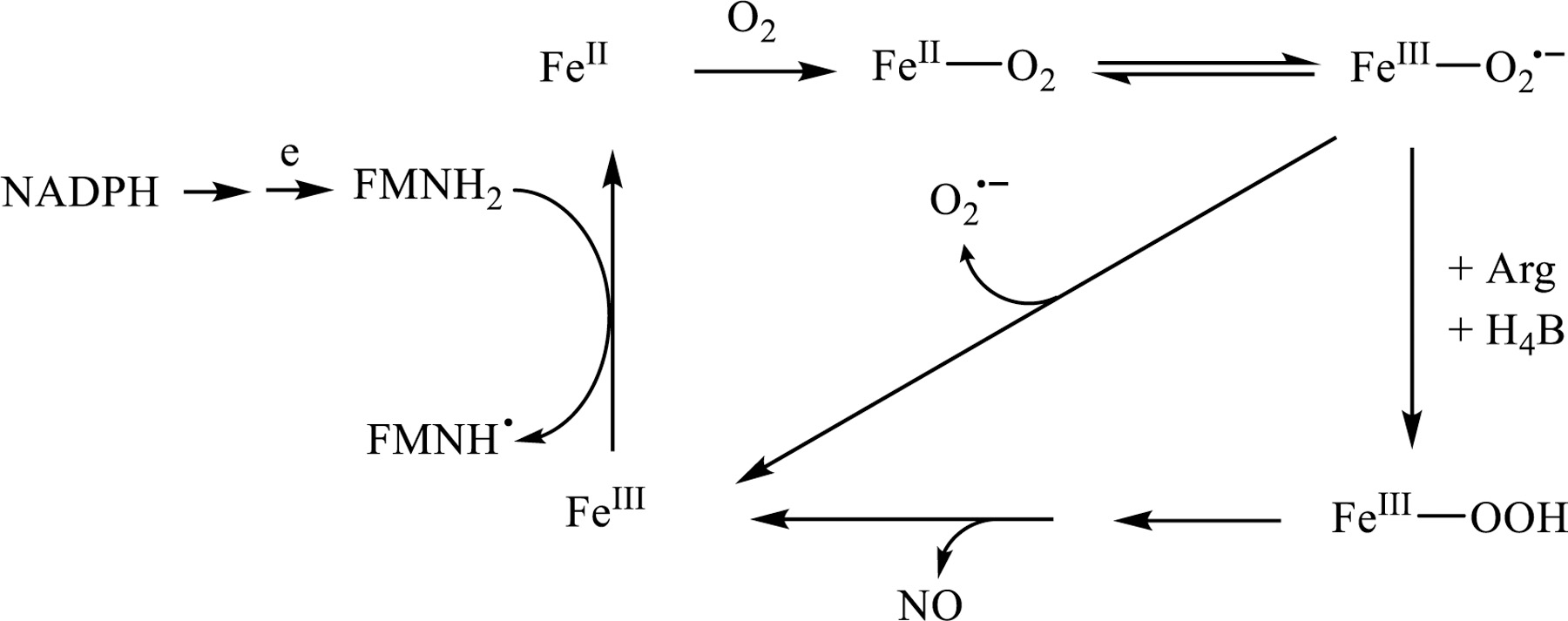
General mechanism of O2•− generation from nNOS.
Our observed increase in O2•− generation from nNOS by Hsp90α contrasts with the inhibition effect in the literature [23]. The difference between these two studies most likely arises from: (a) utilizing different nNOS preparations, and/or (b) the Hsp90 proteins used in our current study were unlike: the bovine Hsp90 protein was obtained from a commercial source [23]. Commercial Hsp90 protein contains unspecified Hsp90 forms and undefined oligomeric states (see above), while our work here used purified human Hsp90α protein in its native dimeric state. Thus, we have overcome limitations to unambiguously examine the effect of specific Hsp90 isoforms on O2•− production by nNOS. Another possibility for the contrast is the sequence or molecular differences between human and bovine Hsp90, which can be an issue with protein-protein nNOS-Hsp90 interactions. In fact, the protein sequences of bovine and human Hsp90 are 99.7% identical, excluding this possibility. We also wish to stress that the Hsp90α-induced increase in O2•− formation was observed for both nNOSα and nNOSµ proteins, and the experiments were conducted with different protein preparations on separate dates. Regardless of the origin of this difference, our EPR spin trapping results support our findings that Hsp90α enhances O2•− production by nNOS. Future studies should be performed with corroborating evidence from complementary techniques.
It is important to elaborate on the similarities and differences between nNOSα and nNOSµ without and with Hsp90α protein. nNOSµ is primarily found in differentiated skeletal muscle and heart [49], while most other tissues express nNOSα, another splice variant of nNOS. Almost all previous research on the structure-function aspect of nNOS in the literature was done using nNOSα. The expression of nNOSµ variant only in certain tissue types as well as the presence of additional 34 amino acids in the AR region (Figure 1) warrants further detailed studies of this variant in comparison to the much-studied variant, nNOSα. Herein we found that Hsp90 increased O2•− production in both nNOSα and nNOSµ (Figure 2). On the other hand, nNOSα produced more O2•− than nNOSµ in the absence of Hsp90, while the Hsp90-induced increasing effect on O2•− production was smaller in nNOSα than that of nNOSµ (Figure 2). This is an interesting difference because O2•− is a significant signaling molecule in muscle (where nNOSµ dominates) - much more so than in neurons (where nNOSα dominates). In other words, it makes sense that nNOSµ and nNOSα control O2•− production differently. Under stress conditions, Hsp90 level in the muscle and brain tissues can change rapidly [50, 51]. It would be interesting to further investigate the nNOS-Hsp90 system and O2•− level in related cellular and tissue models.
The biological implication of our new finding merits further investigation. Most of the in vivo and in vitro studies of NOS-Hsp90 system were on eNOS protein, which generally displayed an inhibition of eNOS-derived O2•− by Hsp90. Although an early study of purified nNOS protein showed that Hsp90 inhibits O2•− production by nNOS [23], more recent literature reported increased ROS from nNOS-Hsp90 complex, contributing to oxidative stress [52]. It is necessary to carry out additional in vitro cellular and in vivo animal studies to further elucidate the physiological and pathophysiological roles of Hsp90 in nNOS regulation. Based on our findings herein, several factors need to be considered for the study design and data interpretation: a) isoforms (α or β) of Hsp90 protein, b) oligomeric state of Hsp90 protein, and c) specific nNOS forms present in the studied tissues.
Why is there difference in O2•− production between the nNOSα and nNOSµ proteins (Figure 2)? nNOSµ contains 34 additional residues in an AR region compared to nNOSα, which is the only structural difference between these two nNOSs (Figure 1). Our results thus suggest that the AR insert itself influences O2•− production and may have important implications in the functional aspects of nNOS. The AR insert within the FMN domain may modulate the conformational equilibrium of the FMN domain, an important determinant of electron flux in NOS [31, 36]. O2•− production not only requires the ferric heme reduction and subsequent dioxygen binding to ferrous (Scheme 1), but also may involve other factors such as conformational states. The concept of pro-O2•− conformation of NOS has been speculated in the literature for a long time [4, 23, 25]. The challenge is how to prove or disprove such a model. There is no convenient way to directly monitor the NOS conformations and link a specific conformation to the O2•− production. In fact, we still know little about NOS conformations in solution to fully understand structural mechanism of O2•− production by NOS. Nonetheless, our study here should inspire further detailed investigation of factors (e.g., heme reduction, O2•− release, conformational equilibrium, and conformational states) governing O2•− generation from nNOS.
One may also ask how Hsp90α increases O2•− production by nNOS at the molecular level. Hsp90 itself does not produce O2•− (Figure S6). Hsp90 is a chaperon, and it is thus intriguing to assume that Hsp90 binding to nNOS might induce global structural changes in nNOS, rather than more local structural changes, to cause the enhancement of O2•− generation. One possibility is that Hsp90 dissociates the dimeric nNOS protein, making monomeric form. It is not likely, though, because of high affinity dimeric interaction in NOS that is due to > 3000 Å2 of dimer interface augmented by the Zn tetra-thiolate bonds between the monomers. Indeed, our recently reported kinetics data of the intersubunit electron transfer between the FMN and heme centers [37] indicate that the Hsp90-associated nNOS protein is still in its dimeric state, because the electron transfer would have been diminished in a monomeric nNOS. We thus speculate that Hsp90 more likely influences O2•− generation from nNOS through the NOS-inherent pathways (see above).
Conclusions
Our combined EPR spin trapping and NADPH oxidation results indicate that Hsp90α enhances O2•− production by nNOS proteins, particularly under l-Arg depleted conditions. Comparing the spin adduct and kinetic results for nNOSα and nNOSµ proteins showed that the Hsp90α enhancement effect differs between these two nNOS proteins, which may have interesting biological implications. The existence of two distinct isoforms of the same nNOS enzyme in different tissues has to have some biological significance, albeit unknown at this time. The information provided herein presents a new aspect of the effect of Hsp90 as a chaperone for nNOS isoform in various tissues, since both nNOSα and nNOSµ forms produce higher amounts of O2•− in its presence but they produce this ROS at different levels. These new findings should inspire further detailed mechanistical investigation of how O2•− production by nNOS is regulated. This work also provides useful general guidelines for the design of studying nNOS uncoupling in cellular and tissue models.
Supplementary Material
Supporting Information. Figure S1, SDS-PAGE analysis of purified nNOS proteins. Figure S2, absorption spectra of nNOSµ. Figure S3, absorption spectra of nNOSµ upon l-Arg removal and addition. Figures S4–S7: EPR detection of the BMPO-•OOH adduct formed in the XOD/HYP O2•− generating system; EPR signals of O2•−-BMPO adduct, BMPO-•OOH, observed in the reaction system containing 20 nM nNOSµ alone; EPR signal for the system containing only the human Hsp90α protein; comparing the BMPO-•OOH signal by the nNOSµ protein with and without Hsp90α under varying l-Arg concentrations.
Research Highlights.
Neuronal nitric oxide synthase, nNOS; heat shock protein 90, Hsp90; superoxide, O2•−.
O2•−-generation from nNOS was assessed in the presence of purified dimeric human Hsp90α.
O2•−-production by nNOSα and nNOSµ isoforms was compared.
The site of O2•−-production was determined using imidazole, a heme-directed blocker.
The work provides guidelines for studying the mechanism of nNOS regulation by Hsp90.
Acknowledgements.
This work was supported by the National Institutes of Health (GM081811 and GM133973).
Abbreviations:
- NO
nitric oxide
- NOS
nitric oxide synthase
- iNOS
inducible NOS
- nNOS
neuronal NOS
- eNOS
endothelial NOS
- AR
autoregulatory insert within the FMN domain of nNOS and eNOS, but not in iNOS
- CaM
calmodulin
- H4B
(6R)-5,6,7,8-tetrahydrobiopterin
- L-Arg
L-arginine
- Hsp90
heat shock protein 90
- BMPO
5-tert-butoxycarbonyl-5-methyl-1-pyrroline-N-oxide
- DMPO
5,5-Dimethyl-1-pyrroline N-oxide
- DTPA
Diethylenetriaminepentaacetic acid SOD superoxide dismutase
- O2•−
superoxide
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- EPR
electron paramagnetic resonance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Förstermann U, Sessa WC, Eur Heart J 33 (2012) 829–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Feng C, Coord. Chem. Rev 256 (2012) 393–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vasquez-Vivar J, Hogg N, Martasek P, Karoui H, Pritchard KA, Kalyanaraman B, J. Biol. Chem 274 (1999) 26736–26742. [DOI] [PubMed] [Google Scholar]
- [4].Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA Jr., Proc Natl Acad Sci U S A 95 (1998) 9220–9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Luo S, Lei H, Qin H, Xia Y, Curr Pharm Des 20 (2014) 3548–3553. [DOI] [PubMed] [Google Scholar]
- [6].Gao YT, Panda SP, Roman LJ, Martasek P, Ishimura Y, Masters BS, J Biol Chem 282 (2007) 7921–7929. [DOI] [PubMed] [Google Scholar]
- [7].Gebhart V, Reiss K, Kollau A, Mayer B, Gorren ACF, Nitric Oxide 89 (2019) 14–21. [DOI] [PubMed] [Google Scholar]
- [8].Schieber M, Chandel NS, Curr Biol 24 (2014) R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM, J. Biol. Chem 267 (1992) 24173–24176. [PubMed] [Google Scholar]
- [10].Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL, Proceedings of the National Academy of Sciences 93 (1996) 6770–6774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xia Y, Roman LJ, Masters BSS, Zweier JL, J. Biol. Chem 273 (1998) 22635–22639. [DOI] [PubMed] [Google Scholar]
- [12].Pou S, Keaton L, Surichamorn W, Rosen GM, J Biol Chem 274 (1999) 9573–9580. [DOI] [PubMed] [Google Scholar]
- [13].Hardy M, Zielonka J, Karoui H, Sikora A, Michalski R, Podsiadły R, Lopez M, Vasquez-Vivar J, Kalyanaraman B, Ouari O, Antioxid Redox Signal 28 (2018) 1416–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rosen GM, Tsai P, Pou S, Chem. Rev 102 (2002) 1191–1199. [DOI] [PubMed] [Google Scholar]
- [15].Li H, Förstermann U, Curr. Opin. Pharmacol 13 (2013) 161–167. [DOI] [PubMed] [Google Scholar]
- [16].Wu Y, Ding Y, Ramprasath T, Zou MH, Antioxid Redox Signal (2020. [DOI] [PMC free article] [PubMed]
- [17].Clapp KM, Peng HM, Jenkins GJ, Ford MJ, Morishima Y, Lau M, Osawa Y, J Biol Chem 287 (2012) 42601–42610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ghosh A, Chawla-Sarkar M, Stuehr DJ, The FASEB Journal 25 (2011) 2049–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen Y, Jiang B, Zhuang Y, Peng H, Chen W, PLoS One 12 (2017) e0179978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Averna M, Stifanese R, De Tullio R, Passalacqua M, Salamino F, Pontremoli S, Melloni E, J. Biol. Chem 283 (2008) 29069–29076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Pritchard KA, Ackerman AW, Gross ER, Stepp DW, Shi Y, Fontana JT, Baker JE, Sessa WC, J. Biol. Chem 276 (2001) 17621–17624. [DOI] [PubMed] [Google Scholar]
- [22].Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC, Nature 392 (1998) 821–824. [DOI] [PubMed] [Google Scholar]
- [23].Song Y, Cardounel AJ, Zweier JL, Xia Y, Biochemistry 41 (2002) 10616–10622. [DOI] [PubMed] [Google Scholar]
- [24].Garnier C, Lafitte D, Jorgensen TJ, Jensen ON, Briand C, Peyrot V, Eur J Biochem 268 (2001) 2402–2407. [DOI] [PubMed] [Google Scholar]
- [25].Cortes-Gonzalez C, Barrera-Chimal J, Ibarra-Sanchez M, Gilbert M, Gamba G, Zentella A, Flores ME, Bobadilla NA, Cell. Physiol. Biochem 26 (2010) 657–668. [DOI] [PubMed] [Google Scholar]
- [26].Moullintraffort L, Bruneaux M, Nazabal A, Allegro D, Giudice E, Zal F, Peyrot V, Barbier P, Thomas D, Garnier C, J. Biol. Chem 285 (2010) 15100–15110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Balke JE, Zhang L, Percival JM, Nitric Oxide 82 (2019) 35–47. [DOI] [PubMed] [Google Scholar]
- [28].Ihara H, Kuwamura M, Atsuta M, Nihonmatsu I, Okada T, Mukamoto M, Kozaki S, Nitric Oxide 15 (2006) 13–19. [DOI] [PubMed] [Google Scholar]
- [29].Silvagno F, Xia H, Bredt DS, J. Biol. Chem 271 (1996) 11204–11208. [DOI] [PubMed] [Google Scholar]
- [30].Brenman JE, Xia H, Chao DS, Black SM, Bredt DS, Dev Neurosci 19 (1997) 224–231. [DOI] [PubMed] [Google Scholar]
- [31].Panda SP, Li W, Venkatakrishnan P, Chen L, Astashkin AV, Masters BSS, Feng C, Roman LJ, FEBS Lett 587 (2013) 3973–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ihara H, Kitamura A, Kasamatsu S, Ida T, Kakihana Y, Tsutsuki H, Sawa T, Watanabe Y, Akaike T, Biochem J 474 (2017) 1149–1162. [DOI] [PubMed] [Google Scholar]
- [33].Chachlaki K, Garthwaite J, Prevot V, Nature Reviews Endocrinology 13 (2017) 521–535. [DOI] [PubMed] [Google Scholar]
- [34].Kellogg DL, McCammon KM, Hinchee-Rodriguez KS, Adamo ML, Roman LJ, Free Radical Biol. Med 110 (2017) 261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kar R, Kellogg Iii DL, Roman LJ, Biochem. Biophys. Res. Commun 459 (2015) 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Feng CJ, Roman LJ, Hazzard JT, Ghosh DK, Tollin G, Masters BSS, FEBS Lett 582 (2008) 2768–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zheng HY, Li JH, Feng CJ, FEBS Lett 594 (2020) 2904–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zhang M, Vogel HJ, J Biol Chem 269 (1994) 981–985. [PubMed] [Google Scholar]
- [39].Weaver J, Porasuphatana S, Tsai P, Pou S, Roman LJ, Rosen GM, Biochimica Et Biophysica Acta-General Subjects 1726 (2005) 302–308. [DOI] [PubMed] [Google Scholar]
- [40].Dipak Kumar Ghosh CC, McMury Jonathan L and Salerno John C, The FASEB Journal 26 (2012) 756. [Google Scholar]
- [41].Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS, Proc Natl Acad Sci U S A 92 (1995) 8428–8432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Zhao H, Joseph J, Zhang H, Karoui H, Kalyanaraman B, Free Radic Biol Med 31 (2001) 599–606. [DOI] [PubMed] [Google Scholar]
- [43].Xia Y, Tsai AL, Berka V, Zweier JL, J Biol Chem 273 (1998) 25804–25808. [DOI] [PubMed] [Google Scholar]
- [44].Furfine ES, Harmon MF, Paith JE, Knowles RG, Salter M, Kiff RJ, Duffy C, Hazelwood R, Oplinger JA, Garvey EP, J Biol Chem 269 (1994) 26677–26683. [PubMed] [Google Scholar]
- [45].Radi R, J Biol Chem 288 (2013) 26464–26472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mayer B, Klatt P, Werner ER, Schmidt K, Neuropharmacology 33 (1994) 1253–1259. [DOI] [PubMed] [Google Scholar]
- [47].Vasquez-Vivar J, Hogg N, Martasek P, Karoui H, Pritchard KA Jr., Kalyanaraman B, J Biol Chem 274 (1999) 26736–26742. [DOI] [PubMed] [Google Scholar]
- [48].Abu-Soud HM, Gachhui R, Raushel FM, Stuehr DJ, J. Biol. Chem 272 (1997) 17349–17353. [DOI] [PubMed] [Google Scholar]
- [49].Silvagno F, Xia H, Bredt DS, Journal of Biological Chemistry 271 (1996) 11204. [DOI] [PubMed] [Google Scholar]
- [50].Liu Y, Steinacker JM, Front Biosci 6 (2001) D12–25. [DOI] [PubMed] [Google Scholar]
- [51].Averna M, Stifanese R, De Tullio R, Salamino F, Pontremoli S, Melloni E, FEBS J 275 (2008) 2501–2511. [DOI] [PubMed] [Google Scholar]
- [52].Caito S, Zeng H, Aschner JL, Aschner M, PLoS One 9 (2014) e98161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information. Figure S1, SDS-PAGE analysis of purified nNOS proteins. Figure S2, absorption spectra of nNOSµ. Figure S3, absorption spectra of nNOSµ upon l-Arg removal and addition. Figures S4–S7: EPR detection of the BMPO-•OOH adduct formed in the XOD/HYP O2•− generating system; EPR signals of O2•−-BMPO adduct, BMPO-•OOH, observed in the reaction system containing 20 nM nNOSµ alone; EPR signal for the system containing only the human Hsp90α protein; comparing the BMPO-•OOH signal by the nNOSµ protein with and without Hsp90α under varying l-Arg concentrations.